

Highlights
► We present crystal structures of 6 peptide aldehydes in complex with SARS-CoV Mpro. ► The aldehydes form thiohemiacetals with the catalytic cysteine of the enzyme. ► Two different configurations of the thiohemiacetal can be observed for one aldehyde. ► Asp and Ser side-chains of the aldehydes bind to the hydrophobic S2 pocket. ► P2-Asp interacts with two methionines of the enzyme via O...S nonbonded interactions.
Abbreviations: SARS-CoV Mpro, SARS coronavirus main protease; Cm-FF-H, Cinnamoyl-Phe-Phe-H; Boc, tert-butyl oxycarbonyl; IBX, 2-iodoxybenzoic acid; FRET, fluorescence resonance energy transfer; PEG, polyethylene glycol; MPD, 2-methyl-2,4-pentanediol; MES, 2-(N-morpholino)ethanesulfonic acid; DMSO, dimethylsulfoxide
Keywords: Aldehyde inhibitor, Antiviral drug design, Cysteine protease, Methionine–aspartic acid interaction, X-ray crystallography
Abstract
SARS coronavirus main protease (SARS-CoV Mpro) is essential for the replication of the virus and regarded as a major antiviral drug target. The enzyme is a cysteine protease, with a catalytic dyad (Cys-145/His-41) in the active site. Aldehyde inhibitors can bind reversibly to the active-site sulfhydryl of SARS-CoV Mpro. Previous studies using peptidic substrates and inhibitors showed that the substrate specificity of SARS-CoV Mpro requires glutamine in the P1 position and a large hydrophobic residue in the P2 position. We determined four crystal structures of SARS-CoV Mpro in complex with pentapeptide aldehydes (Ac-ESTLQ-H, Ac-NSFSQ-H, Ac-DSFDQ-H, and Ac-NSTSQ-H). Kinetic data showed that all of these aldehydes exhibit inhibitory activity towards SARS-CoV Mpro, with Ki values in the μM range. Surprisingly, the X-ray structures revealed that the hydrophobic S2 pocket of the enzyme can accommodate serine and even aspartic-acid side-chains in the P2 positions of the inhibitors. Consequently, we reassessed the substrate specificity of the enzyme by testing the cleavage of 20 different tetradecapeptide substrates with varying amino-acid residues in the P2 position. The cleavage efficiency for the substrate with serine in the P2 position was 160-times lower than that for the original substrate (P2 = Leu); furthermore, the substrate with aspartic acid in the P2 position was not cleaved at all. We also determined a crystal structure of SARS-CoV Mpro in complex with aldehyde Cm-FF-H, which has its P1-phenylalanine residue bound to the relatively hydrophilic S1 pocket of the enzyme and yet exhibits a high inhibitory activity against SARS-CoV Mpro, with Ki = 2.24 ± 0.58 μM. These results show that the stringent substrate specificity of the SARS-CoV Mpro with respect to the P1 and P2 positions can be overruled by the highly electrophilic character of the aldehyde warhead, thereby constituting a deviation from the dogma that peptidic inhibitors need to correspond to the observed cleavage specificity of the target protease.
1. Introduction
SARS coronavirus main protease (SARS-CoV Mpro) is essential for the replication of the virus and regarded as a major antiviral drug target (Anand et al., 2003, Yang et al., 2003, Tan et al., 2005, Steuber and Hilgenfeld, 2010). Peptide aldehyde inhibitors are widely used as research tools to characterize the substrate specificity of serine and cysteine proteases. In complex with the crystalline target enzyme, they provide a wealth of information on the specificity-defining subsites of the substrate-binding site. For this purpose, we have previously described peptide aldehydes as inhibitors of the SARS-coronavirus main protease (SARS-CoV Mpro) (Al-Gharabli et al., 2006). A major advantage of aldehydes over other peptide electrophiles is their reversible binding to the active-site sulfhydryl of cysteine proteases. We have previously used this property to screen a small library of non-peptide nucleophiles (mostly amines) for molecules that would efficiently enhance the inhibition of the aldehyde by formation of a stronger binding aldehyde-nucleophile ligation product. In a second step of this process that we named “Dynamic Ligation Screening” (Schmidt et al., 2008), the most efficient compound in this process had its amino group replaced by an aldehyde moiety and was reacted with the target enzyme, and the resulting covalent adduct was subsequently used to screen the same library of nucleophiles for those that would react with the aldehyde. This way, starting from a substrate-like peptide aldehyde, we obtained a low-μM non-peptidic, reversible inhibitor from two fragments which by themselves had no or only very low inhibitory activity (Schmidt et al., 2008).
Interestingly, it turned out that some peptide aldehydes with an amino-acid sequence deviating from the consensus sequence embracing the cleavage sites of polyprotein substrates were surprisingly efficient as inhibitors. In particular, peptide aldehydes with P2 = Asp or Ser inhibited the main protease with a relatively low IC50 (Al-Gharabli et al., 2006), although it is generally agreed that the S2 specificity subsite of the enzyme has a strong preference for large hydrophobic side-chains such as Leu, Phe, or Met (Fan et al., 2004, Fan et al., 2005, Lai et al., 2006). We therefore assumed that the hydrophilic P2 side-chain of these inhibitors would be oriented towards the solvent, rather than occupy the S2 pocket, and that the hydrophobic P3 residue would be binding to that pocket (Al-Gharabli et al., 2006, Schmidt et al., 2008). This model was based on a crystal structure of a peptidyl chloromethyl ketone bound to the SARS-CoV main protease, in which this arrangement had been observed (Yang et al., 2003). Here we show by X-ray crystallography of a number of peptide aldehyde complexes with the protein that this is not the case. Instead, the hydrophilic Ser or Asp residues in the P2 position bind to the hydrophobic S2 subsite. In order to understand these binding modes, the atomic interactions were analyzed in detail. Also, we report the inhibition kinetics of these compounds and re-evaluate the cleavage specificity of the enzyme as far as the P2 position is concerned. In addition, we determined a crystal structure of SARS-CoV Mpro in complex with the aldehyde Cm-FF-H (cinnamoyl-Phe-Phe-H), which has a phenylalanine residue in the P1 position and exhibits high inhibitory activity against SARS-CoV Mpro, with Ki = 2.24 ± 0.58 μM. These results show that the stringent substrate specificity of the SARS-CoV Mpro with respect to the P1 and P2 positions can be overruled by the highly electrophilic character of the aldehyde warhead.
2. Materials and methods
2.1. Synthesis of aldehydes with various P2 residues
Chemical synthesis of peptide aldehydes Ac-ESTLQ-H, Ac-NSFSQ-H, Ac-DSFDQ-H, and Ac-NSTSQ-H was performed employing a solid-state method (Al-Gharabli et al., 2006). Briefly, protected glutamine aldehyde obtained by racemization-free oxidation of the corresponding amino alcohol with Dess–Martin periodinane was immobilized on a threonyl resin as oxazolidine. Following N-tert-butyl oxycarbonyl (Boc)-protection of the ring nitrogen to yield the N-protected oxazolidine linker, peptide synthesis was performed on the resin.
2.2. Synthesis of Cm-FF-H (Scheme 1)
Scheme 1.

Synthesis of Cm-FF-H. Reagents and conditions: (a) EDCI, HOBt, Boc-Phe-OH, DMF, 0–25 °C, 18 h, 78%; (b) (i) TFA, CH2Cl2, 0–25 °C, 2 h; (ii) cinnamoyl chloride, N-methylmorpholine, THF, DMF, 0–25 °C, 12 h, 50%; (c) NaBH4, CaCl2, EtOH, THF (1:1), 0–25 °C, 14 h, 35%; (d) IBX, DMSO, 0–25 °C, 5 h, 47%.
Synthesis of Cm-FF-H was carried out by amidation of Boc-l-phenylalanine with l-phenylalanine methyl ester followed by deprotection of the Boc group and acylation of the corresponding product with cinnamoyl chloride to provide N-cinnamoyl-l-phenylalanyl-l-phenylalanine methyl ester. Cm-FF-H was then obtained by reduction of the ester with NaBH4/CaCl2 and alcohol oxidation with IBX.
2.3. Enzyme kinetics
The recombinant production and purification of SARS-CoV Mpro with authentic N and C termini were performed as described previously (Xue et al., 2007, Verschueren et al., 2008). The substrate Dabcyl-KTSAVLQ↓SGFRKME-(Edans)-amide (95% purity; Biosyntan GmbH, Berlin, Germany), which contains a main-protease cleavage site (indicated by the arrow), was used as the substrate in the fluorescence resonance energy transfer (FRET)-based cleavage assay. The substrate stock was prepared by dissolving 1 mM of the peptide in DMSO. The dequenching of the Dabcyl fluorescence due to the cleavage of the substrate as catalyzed by the SARS-CoV Mpro was monitored at 490 nm with excitation at 340 nm, using a Cary Eclipse fluorescence spectrophotometer. The experiments were performed in the buffer consisting of 20 mM Tris-HCl (pH 7.3), 100 mM NaCl, and 1 mM EDTA. The reaction was initiated by adding different final concentrations of the FRET peptide (10–50 μM) to a solution containing SARS-CoV Mpro (final concentration 0.5 μM). Kinetic constants (V max and K m) were derived by fitting the data to the Michaelis–Menten equation, V = V max × [S]/(K m + [S]). Then k cat was calculated according to the equation, k cat = V max/[E]. In order to estimate the population of catalytically active Mpro dimers, the respective monomer and dimer concentrations were calculated according to Graziano et al. (2006). The dimerization of the SARS-CoV Mpro follows the scheme
The equilibrium dissociation constant, KD, is defined by
where [M] and [D] are the molar concentrations of the monomer and dimer, respectively. The total protein concentration [MT] expressed in terms of molar monomer equivalents is
Since
substituting the above equation into the expression for the KD yields
Solving the above quadratic equation for [M] gives
It then follows that
2.4. Aldehyde inhibition assay
Aldehyde stock solutions were prepared by dissolving the compounds in DMSO at 1 mM. All aldehydes form covalent bonds between the aldehyde (–CHO) group of the inhibitor and the sulfhydryl (–SH) group of Cys145 of SARS-CoV Mpro. The binding of these inhibitors is reversible (Schmidt et al., 2008), although it is strong. Therefore, they were treated as reversible tight-binding inhibitors. For the measurement of the inhibition constant Ki, SARS-CoV Mpro was incubated with the aldehyde inhibitor in reaction buffer at room temperature for 30 min. Then the FRET peptide substrate, Dabcyl-KTSAVLQ↓SGFRKME-Edans, was added and the reaction velocity was calculated according to substrate cleavage. Values of the intrinsic () and apparent (, ) catalytic parameters for SARS-CoV Mpro catalyzing the hydrolysis of peptide substrate were determined in the absence and presence of aldehyde, respectively. The apparent inhibition constants () for aldehyde binding to SARS-CoV Mpro were obtained from the dependence of on the inhibitor concentration ([I]) at fixed substrate concentration ([S]), according to the equation (Ascenzi et al., 1987, Copeland, 2000). Values of the intrinsic inhibition constant (Ki) for aldehyde binding to SARS-CoV Mpro were calculated according to the equation = Ki × (1 + [S]/K m) (Ascenzi et al., 1987, Copeland, 2000).
2.5. Peptide cleavage assay
Twenty peptide substrates harboring alternative amino-acid residues in P2 (=X; SWTSAVXQ↓SGFRKWA) were purchased from GL Biochemistry Ltd. (Shanghai, China). These peptides correspond to the N-terminal autocleavage site of the SARS-CoV Mpro, with the exception of the P7 Ile which had been replaced by Trp, and the P6′ Met which had also been replaced by Trp. All peptide substrate stocks were dissolved in DMSO at 10 mM. To determine the k cat/K m for the substrate, 0.2 mM substrate peptide was incubated with 10 μM SARS-CoV Mpro in 40 mM Tris-HCl buffer, pH 7.3. Aliquots of reactions were removed at different times, stopped by the addition of 1% trichloroacetic acid. Separation of products and substrate was carried out using a reverse-phase (RP) HPLC and a linear gradient (1–90%) of acetonitrile in 0.1% trifluoroacetic acid. The absorbance was determined at 280 nm, and peak areas were calculated by integration. The (k cat/K m)app ratio was determined by plotting the substrate peak area using the equation lnPA = C − (k cat/K m)app × C E × t, where PA is the peak area of the substrate peptide, C E is the total concentration of SARS-CoV Mpro, and C is an experimental constant (Fan et al., 2004, Fan et al., 2005).
2.6. Crystallization of the complexes
SARS-CoV Mpro with authentic chain termini was concentrated to 10 mg/ml and crystallized by vapor diffusion using sitting drops (Xue et al., 2007). The crystals grew overnight at 20 °C by equilibration against a reservoir containing 6–8% polyethylene glycol (PEG) 6000, 0.1 M MES (pH 6.0), 3% 2-methyl-2,4-pentanediol (MPD), and 3% DMSO. All aldehydes were dissolved in 8% PEG 6000, 0.1 M MES (pH 6.0), 3% MPD, and 10% DMSO to a concentration of 10 mM. Crystals of the aldehyde complexes of SARS-CoV Mpro were obtained either by adding a 4-μl aliquot of aldehyde solution to the drop and soaking of the crystals for 12 h, or by incubating the enzyme for 2 h at 20 °C with a 7-fold excess of the aldehyde solution and subsequent cocrystallizing at 20 °C against a reservoir containing 8% PEG 6000, 0.1 M MES (pH 6.0), 3% MPD, and 3% DMSO. In the latter case, nucleation was initiated by microseeding using crushed monoclinic (space group C2) crystals of the SARS-CoV Mpro. Prior to diffraction data collection, the aldehyde complex crystals obtained from soaking and cocrystallization were transferred for a few seconds to a cryoprotectant solution containing the crystallization ingredients and 20% MPD.
2.7. Crystallographic data collection and processing, and structure elucidation and refinement
All diffraction data were collected at 100 K at the Joint EMBL/University of Hamburg/University of Lübeck synchrotron beamline X13 at DESY (Hamburg, Germany), using a 165-mm MAR CCD detector (Mar Research, Hamburg, Germany), or at synchrotron beamline BL14.1 at BESSY (Berlin, Germany), using an MX225 CCD detector (Rayonics, Evanston, IL). Statistics of data collection, processing, and refinement are summarized in Table 1 . Data were processed with MOSFLM (Leslie, 1992), and scaled using the SCALA program from the CCP4 suite (Collaborative Computational Project, 1994, Evans, 2006).
Table 1.
Data collection and refinement statistics.
| Inhibitor | Ac-ESTLQ-H | Ac-DSFDQ-H | Ac-NSTSQ-H | Ac-NSFSQ-H | Ac-ESTLQ-H | Cm-FF-H |
|---|---|---|---|---|---|---|
| Data collection statistics | ||||||
| Wavelenth (Å) | 0.8123 | 0.8123 | 0.8123 | 0.8123 | 0.8123 | 0.9184 |
| Method | Soaking | Soaking | Soaking | Soaking | Cocrystallization | Soaking |
| Space group | C2 | C2 | C2 | C2 | P21 | C2 |
| Unit cell dimensions (Å, °) | 108.91, 81.36, 53.40 β = 104.35 |
108.74, 81.86, 53.21 β = 104.26 |
107.83, 82.46, 53.32 β = 104.65 |
107.87, 82.09, 53.08 β = 104.30 |
52.37, 96.79, 68.07 β = 102.49 |
107.75, 82.88, 53.50 β = 104.63 |
| NPa | 1 | 1 | 1 | 1 | 2 | 1 |
| Resolution range (Å) | 32.21–2.60 | 32.32–2.40 | 26.58–2.58 | 64.56–3.05 | 27.29–1.89 | 32.82–1.99 |
| Number of reflections | 13.957 | 16.009 | 13.938 | 8643 | 50.901 | 31.388 |
| Redundancyb | 3.8(3.8) | 3.4(2.8) | 3.0(2.7) | 3.4(3.4) | 2.8(2.6) | 3.6(3.4) |
| Completeness (%)b | 99.9(100.0) | 91.3(85.7) | 96.7(82.5) | 99.9(100.0) | 94.5(78.3) | 99.7(98.1) |
| Rmerge (%)b | 10.2(45.6) | 10.4(25.7) | 6.0(32.0) | 8.3(47.2) | 6.7(33.6) | 17.0(36.6) |
| I/σ(I)b | 8.7(2.4) | 5.8(2.3) | 10.1(2.8) | 10.2(2.3) | 10.3(2.7) | 4.6(2.2) |
| Refinement statistics | ||||||
| R/Rfree (%) | 18.5/22.5 | 21.2/23.9 | 19.8/26.0 | 19.0/25.5 | 18.7/23.8 | 21.7/26.9 |
| r.m.s deviation from idea geometry | ||||||
| Bonds (Å) | 0.010 | 0.009 | 0.017 | 0.015 | 0.009 | 0.018 |
| Angles (°) | 1.535 | 1.162 | 1.820 | 1.661 | 1.172 | 1.689 |
| Ramachandran plot | ||||||
| Most favored (%) | 88.0 | 90.3 | 83.9 | 80.6 | 90.6 | 89.8 |
| Allowed (%) | 10.9 | 8.6 | 14.2 | 18.3 | 8.3 | 9.1 |
| Generously allowed (%) | 0.4 | 0.7 | 1.1 | 0.4 | 0.4 | 0.8 |
| Disallowed (%) | 0.7 | 0.4 | 0.7 | 0.8 | 0.8 | 0.4 |
| PDB ID | 3SNE | 3SNB | 3SNC | 3SNA | 3SND | 3SN8 |
Number of protein molecules per asymmetric unit.
Numbers in parentheses are for the outermost resolution shell.
Structure elucidation and refinement were carried out using the CCP4 suite of programs (Collaborative Computational Project, 1994, Potterton et al., 2003). Crystal structures were determined by molecular replacement, using the original X-ray structure of the SARS-CoV Mpro with authentic chain termini (PDB ID: 2H2Z) (Xue et al., 2007) as the initial model. REFMAC (Murshudov et al., 1997) was employed for structure refinement. The computer graphics program Coot (Emsley and Cowtan, 2004) was used for interpretation of electron density maps and model building. Gap volumes between inhibitor and protein in the subsites of the enzyme were calculated using UCSF-Chimera (Pettersen et al., 2004). The molecular graphics package PyMOL (DeLano, 2002) was used to generate the figures.
3. Results and discussion
3.1. Inhibition of SARS-CoV Mpro by peptide aldehydes
The kinetic parameters of SARS-CoV Mpro with authentic chain termini were determined using the FRET substrate Dabcyl-KTSAVLQ↓SGFRKME-Edans amide. The K m value for this substrate was determined as 24.5 μM and the apparent k cat/K m value was 2359 M−1·19 s−1. However, the dissociation of the catalytically active SARS-CoV Mpro dimer into inactive monomers (Fan et al., 2004) has to be taken into account. A wide range of KD values has been reported for Mpro dimer dissociation (see Grum-Tokars et al. (2008) for an overview). For the enzyme with authentic chain termini, the latter authors reported a KD of 0.25 to 1.0 μM. As the Mpro dimer tends to be stabilized by the presence of substrate (Cheng et al., 2010), we used the lower limit of this range for estimation of the necessary corrections and obtained a k cat/K m value of 7863 M−1 s−1. The K m value determined in our study is of the same magnitude as other values derived from data not corrected for dissociation, which are typically in the range from 10 to 50 μM (Grum-Tokars et al., 2008). This suggests that much higher values reported previously (e.g., Verschueren et al., 2008) should probably be reconsidered.
Initially, four pentapeptide substrate-analogous aldehydes containing the canonical P1 residue, glutamine, namely Ac-ESTLQ-H, Ac-NSTSQ-H, Ac-DSFDQ-H, and Ac-NSFSQ-H, were tested for inhibition of SARS-CoV Mpro. Overall, the four analogues were found to be reversible tight-binding inhibitors. Harboring the canonical P2 Leu, aldehyde Ac-ESTLQ-H exhibited inhibition with a relatively low Ki of 8.27 ± 1.52 μM. Aldehydes Ac-NSTSQ-H, Ac-DSFDQ-H, and Ac-NSFSQ-H, all with a non-canonical P2 residue, exhibited moderate inhibition with Ki values of 40.98 ± 2.63, 41.24 ± 2.25, and 72.73 ± 3.60 μM. Surprisingly, aldehyde CmFF-H, carrying a cinnamoyl group in the P3 and a Phe residue in the P1 position, had an even higher inhibitory activity against SARS-CoV Mpro than the four pentapeptide aldehydes, with a Ki of 2.24 ± 0.58 μM.
3.2. Overall structures of the aldehyde complexes
The aldehydes Ac-ESTLQ-H, Ac-NSTSQ-H, Ac-DSFDQ-H, Ac-NSFSQ-H, and Cm-FF-H were separately soaked into crystals of SARS-CoV Mpro. The crystals were all of space group C2, which is often observed for SARS-CoV Mpro (Lee et al., 2005, Xue et al., 2007, Verschueren et al., 2008). These crystals contain one SARS-CoV Mpro monomer per asymmetric unit and the dimer (which is the enzymatically active species) is formed through the symmetry of the crystal. The four pentapeptide aldehydes Ac-ESTLQ-H, Ac-NSTSQ-H, Ac-DSFDQ-H, and Ac-NSFSQ-H are bound in extended conformations in the S6–S1 specificity subsites of SARS-CoV Mpro. Cm-FF-H occupies sites S3–S1. Remarkably, the P1 phenylalanine side chain of this inhibitor is bound deeply in the S1 pocket, which is generally considered to be specific for glutamine. 2F o − F c electron density maps of these aldehyde inhibitors are shown in Fig. 1 . In all complexes, continuous electron density between the aldehyde carbonyl C-atom of the inhibitor and Cys145-Sγ of SARS-CoV Mpro (Fig. 1) indicates the formation of a thiohemiacetal, as a result of the nucleophilic attack of the catalytic cysteine onto the C-terminal aldehyde of the inhibitor. The main-chain conformations of SARS-CoV Mpro in the five complexes are basically identical, with overall root mean-square deviations (RMSD) of 0.16 − 0.36 Å for Cα atoms. In addition to the crystal soaking experiments, we also tried to cocrystallize SARS-CoV Mpro with the aldehydes. The crystals obtained were predominantly of space group P21, with the exception of the complex with Ac-NSFSQ-H, which still displayed space group C2. However, in most of the P21 crystals, no electron density for the aldehyde could be observed; thus, while the presence of the inhibitors induced a change of space group in most cases, the compounds themselves were not detected in the crystals. An exception was Ac-ESTLQ-H. The crystals of its complex with the Mpro diffracted to 1.89 Å, but in both enzyme monomers in the asymmetric unit, electron density could only be seen for residues P1 (Gln) and P2 (Leu) of the inhibitor (Fig. 1E and F). Crystallographic data and refinement statistics for all six crystal structures are summarized in Table 1.
Fig. 1.

Inhibitor binding at the active site of SARS-CoV Mpro. 2Fo − Fc electron-density maps are shown for inhibitors (A) Ac-ESTLQ-H, (B) Ac-NSTSQ-H, (C) Ac-DSFDQ-H, (D) Ac-NSFSQ-H, (E) the visible portion of Ac-ESTLQ-H cocrystallized with the Mpro, molecule A, and (F) molecule B, (G) Cm-FF-H. The maps are contoured at a level of 1σ. The catalytic Cys145 of SARS-CoV Mpro forms a thiohemiacetal with the aldehyde group of the inhibitors.
3.3. Dual configurations of the thiohemiacetal in the complex with Ac-NSFSQ-H
In the SARS-CoV Mpro complex with aldehyde inhibitor Ac-NSFSQ-H, the thiohemiacetal adopts alternative configurations (Fig. 2 A and B). This is likely the result of nucleophilic attack by Cys145 onto the planar carbonyl from either side. In the (R)-isomer, the thiohemiacetal oxygen forms a hydrogen bond with the imidazole of His41 (3.30 Å) of the catalytic dyad (Fig. 2C), whereas in the (S)-isomer, the oxygen atom points away from His41 and into the oxyanion hole, forming H-bonds with the main-chain amides of Gly143 (2.71 Å) and Cys 145 (3.15 Å) (Fig. 2D). Previous X-ray and NMR studies of aldehyde binding to various cysteine and serine proteases demonstrated that one or both configurations were present in the structures of the complexes (Delbaere and Brayer, 1985, Webber et al., 1998, Robin et al., 2009).
Fig. 2.

Dual configurations of the thiohemiacetal in the complex with Ac-NSFSQ-H. (A) and (B): Ac-NSFSQ-H binding at the active site in two alternative configurations. Hydrogen bonding interactions are represented by broken lines. Distances between atoms are shown in Å. (C) and (D): Schematic diagram of the aldehyde inhibitor Ac-NSFSQ-H attacked by the catalytic cysteine 145, leading to the formation of two possible diastereomeric products.
3.4. The S2 subsite
The S2 subsite is a large pocket lined by residues His41, Met49, Cys145, His164, Met165, Asp187, Arg188mc (mc: contribution from main-chain atoms only), and Gln 189 (Fig. 1). At the cleavage sites of the SARS-CoV Mpro in the viral polyproteins, the P2 position is mostly found to be occupied by Leu or Phe. Accordingly, almost all inhibitors designed for the enzyme carry a large hydrophobic group in the P2 position. We were therefore very surprised to find the Ser or Asp side chains in the P2 position of the aldehydes bound to this hydrophobic site (Fig. 1). In the SARS-CoV Mpro complexes with Ac-NSTSQ-H or Ac-NSFSQ-H, there is no hydrogen-bonding interaction between the side chain of P2-Ser and residues of the S2 subsite. As the Ser side chain is small, there is no steric barrier for Ser binding in the large S2 pocket. The serine does not quite reach the “bottom” of the S2 pocket, but the distance between its Oγ atom and the carbonyl oxygen of residue Gln192 is 4.0 Å, leaving no space for a water molecule to locate in between. Accordingly, we did not find any electron density suggesting the presence of a well-ordered water in the S2 pocket, although disordered water is probably present in the pocket on both sides of the serine side-chain, where free spaces with a total volume of >70 Å3 are present between the substrate and the protein. It should be noted that the side-chain of Gln189, which contributes to one wall of the S2 pocket, is quite flexible and adopts different conformations in the various inhibitor complexes, allowing or preventing the access of water to the pocket.
In case of the SARS-CoV Mpro complex with Ac-DSFDQ-H, the P2 Asp side-chain is situated between the side chains of Met49 and Met165, which form opposite walls of the S2 pocket. Most interestingly, its carboxylate oxygens undergo close interactions with the sulfur atoms of the two methionines. The distances (rS…O) between the Sδ atoms of Met49 and Met165 on the one hand and the Oδ2 and Oδ1 atoms, respectively, of P2-Asp on the other are 3.36 and 3.45 Å (Fig. 3 ). Then the relative distances (dS…O) can be calculated according to dS…O = rS…O − vdw(S) − vdw(O), where values of 1.80 and 1.52 Å are used as van der Waals radii (vdw) for S and O atoms, respectively. With dS…O = 0.04 and 0.13 Å, the distances between the P2-Asp and the two S2-Met side-chains fulfill the condition of dS…O ⩽ 0.2 Å (Iwaoka et al., 2002) for a non-bonded S…O contact. These interactions are not in the plane of the methionine sulfides; the θ values (θ is the polar angle between the normal to the sulfide plane and the S…O vector (Pal and Chakrabarti, 2001)) are 30.6° and 52.8°. Similar nonbonded interactions between the methionine sulfur atoms and main-chain carbonyl oxygens or carboxylate side-chains have been detected previously in the hydrophobic cores of proteins and were proposed to stabilize the protein fold (Pal and Chakrabarti, 2001). It has also been suggested that S…O interactions should be taken into account in protein engineering studies (Iwaoka et al., 2002, Pal and Chakrabarti, 2001), but to the best of our knowledge, we provide here the first description of a methionine-carboxylate interaction in a protein-ligand complex. The unexpected finding of Ser and Asp binding in the S2 subsite constitutes a deviation from the dogma that peptide inhibitors of proteases should contain amino-acid residues corresponding to the sequence specificity of the target enzyme.
Fig. 3.
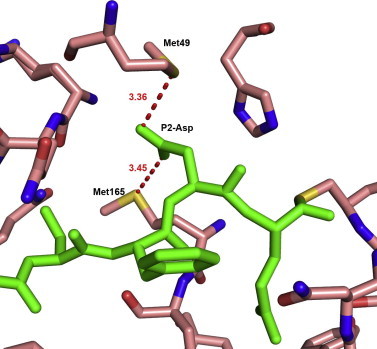
Interactions of P2-Asp in the S2 subsite in the complex structure SARS-CoV Mpro: Ac-DSFDQ-H. P2-Asp carboxylate oxygens interact with the sulfur atoms of Met49 and Met165. These non-bonded interactions are represented by dashed lines. Distances between atoms are shown in Å.
3.5. Analysis of peptide substrates harboring different amino-acid residues in P2
The unexpected observation of the P2-Asp residue of aldehyde Ac-DSFDQ-H binding to the hydrophobic S2 pocket prompted us to re-determine the cleavage specificity of the SARS-CoV Mpro with respect to the P2 position. Within the context of the peptide substrate SWTSAVXQ↓SGFRKWA, all 20 proteinogenic amino acids were tested in position P2 (=X). In the HPLC-based assay, the protease concentration was kept constant at 0.5 μM. The canonical substrate with Leu in the P2 position was completely cleaved within two minutes and was therefore used as the standard to compare the proteolytic activities with other substrates. The results are listed in Fig. 4 . Our study confirmed that the most favored residues in the P2 position of the substrate (after Leu) are the hydrophobic amino acids Phe, Met, Val, and Ile. Substrates with polar amino acids in P2 are poorly cleaved. The cleavage efficiency for the substrate with P2 = Ser was 160-times lower than for the best substrate (P2 = Leu); furthermore, the peptide with P2 = Asp was not cleaved at all after incubation with SARS-CoV Mpro for 16 h.
Fig. 4.

Relative cleavage efficiencies of 20 peptide substrates harboring different amino-acid residues in P2.
3.6. S1 subsite
The S1 subsite is lined by residues Phe140, Asn142, Gly143, Ser144, Cys145, His163, Glu166, and His172. From the substrate specificity results (Fan et al., 2004, Fan et al., 2005, Lai et al., 2006), P1 has to be Gln. Moreover, in all SARS-CoV Mpro complex structures described so far (Lee et al., 2005, Xue et al., 2007, Yang et al., 2005, Yang et al., 2003, Yin et al., 2007), the S1 subsite is occupied by a polar group, i.e. the side chain of Gln or a five-membered lactam (used as a glutamine surrogate). Shie et al. synthesized a series of α,β-unsaturated esters containing both P1 and P2 phenylalanine residues which showed modest inhibitory activity against the SARS-CoV Mpro (IC50 = 11–39 μM) (Shie et al., 2005). The α,β-unsaturated ethyl ester of 4-(dimethylamino)cinnamoyl-Phe-Phe was reported to be a potent inhibitor, with an inhibition constant of 0.52 μM (Shie et al., 2005). The computer model for the complex of SARS-CoV Mpro with the compound proposed that the P1 and P2 phenyl groups occupy the S2 and S3 pockets, respectively (Shie et al., 2005). However, in the crystal structure of the complex with the aldehyde Cm-FF-H that we describe in this communication, the side chain of P1-Phe is inserted into the S1 subsite (Fig. 1G). In order to understand this unexpected deviation from the commonly observed specificity, we compared the interaction with the S1 subsite observed in our structures of the complexes with Cm-FF-H and Ac-ESTLQ-H, the latter of which corresponds exactly to the described cleavage specificity of the Mpro. The predominant S1 specificity of the enzyme for Gln is determined primarily by His163. In the complex formed with Ac-ESTLQ-H, Nε2 of His163 donates a 2.83-Å hydrogen bond to the side-chain carbonyl oxygen (Oε1) of P1-Gln (Fig. 1). In the complex with Cm-FF-H, the side chain of Asn142 undergoes an 83.8° rotation about its χ1 torsion angle compared to the conformation in the complex with Ac-ESTLQ-H (Fig. 5 ), leading to an opening of the S1 subsite towards the solvent. A similar movement of Asn142 has been observed in the crystal structure of SARS-CoV Mpro in complex with an inhibitor called “N1” (PDB ID: 1WOF), where both the regular conformation of this residue and the one observed in our structure were found (Yang et al., 2005). The P1-benzyl group of Cm-FF-H binds to the S1 subsite by making hydrophobic interactions with Phe140, Leu141, Asn142, and the P3 cinnamoyl group of Cm-FF-H. As the S1 subsite specificity does not seem to be as stringent as previously thought. It may well be possible that other chemical groups can be accommodated here, thereby expanding the opportunities for designing and synthesizing efficient inhibitors of the SARS-CoV Mpro.
Fig. 5.

Superimposition of the structures of SARS-CoV Mpro complexes with Cm-FF-H (red) and Ac-ESTLQ-H (green) showing the conformational changes in the S1 subsite. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
4. Conclusions
In this study, we report six crystal structures of SARS-Cov Mpro in complex with peptide aldehydes and the inhibition kinetics of these compounds. The crystal structures reveal the mechanism of aldehyde binding to the active site of SARS-Cov Mpro and provide detailed information on the atomic interactions. Since Asp or Ser were found to bind to the hydrophobic S2 subsite, and Phe was located in the hydrophilic S1 subsite of SARS-Cov Mpro, we conclude that the stringent substrate specificity of the SARS-CoV Mpro with respect to the P1 and P2 positions can be overruled by the highly electrophilic character of the aldehyde warhead. This constitutes a deviation from the dogma that peptidic protease inhibitors should comprise an amino-acid sequence corresponding to the cleavage specificity of the target enzyme. The observed non-bonded interaction of the carboxylate oxygen atoms of an aspartate residue of an inhibitor with the thioether moieties of two methionines forming part of an hydrophobic pocket of the target protein is remarkable and suggests that such S…O interactions should be added to the repertoire of computer-aided drug design.
4.1. Accession numbers
Atomic coordinates and structure factors have been deposited in the RCSB Protein Data Bank under ID codes 3SN8, 3SNA, 3SNB, 3SNC, 3SND, and 3SNE (see Table 1).
Acknowledgments
We thank Sebastian Schwarz for technical assistance during the re-determination of the cleavage rates of SARS-CoV Mpro with respect to the P2 position of the substrate, and Stefan Anemüller, Qingjun Ma, Jeroen R. Mesters as well as Jiajie Zhang for discussion. This work was initially supported by the Deutsche Forschungsgemeinschaft (Grants Hi611/4, Ra895/2), the Sino-German Center for the Promotion of Research, Beijing, and the Sino-European Project on SARS Diagnostics and Antivirals (SEPSDA) of the European Commission (contract number SP22-CT-2004-003831), and subsequently, by the SILVER project of the European Commission (contract number HEALTH-F3-2010-260644). RH and JR acknowledge continuous support by the Fonds der Chemischen Industrie. RH is also supported by a Chinese Academy of Sciences Visiting Professorship for Senior International Scientists, Grant No. 2010T1S6.
References
- Al-Gharabli S.I., Shah S.T., Weik S., Schmidt M.F., Mesters J.R., Kuhn D., Klebe G., Hilgenfeld R., Rademann J. An efficient method for the synthesis of peptide aldehyde libraries employed in the discovery of reversible SARS coronavirus main protease (SARS-CoV Mpro) inhibitors. Chembiochem. 2006;7:1048–1055. doi: 10.1002/cbic.200500533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Ascenzi P., Ascenzi M.G., Amiconi G. Enzyme competitive-inhibition – graphical determination of Ki and presentation of data in comparative studies. Biochem. Educ. 1987;15:134–135. [Google Scholar]
- Cheng S.C., Chang G.G., Chou C.Y. Mutation of Glu-166 blocks the substrate-induced dimerization of SARS coronavirus main protease. Biophys. J. 2010;98:1327–1336. doi: 10.1016/j.bpj.2009.12.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project No. 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D: Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Copeland R.A. John Wiley & Sons, Inc.; New York: 2000. Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis. [Google Scholar]
- DeLano, W.L., 2002. The PyMOL Molecular Graphics System Delano Scientific, San Carlos, CA, USA.
- Delbaere L.T., Brayer G.D. The 1.8 A structure of the complex between chymostatin and Streptomyces griseus protease A. A model for serine protease catalytic tetrahedral intermediates. J. Mol. Biol. 1985;183:89–103. doi: 10.1016/0022-2836(85)90283-9. [DOI] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D: Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Evans P.R. Scaling and assessment of data quality. Acta Crystallogr. D: Biol. Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Fan K., Wei P., Feng Q., Chen S., Huang C., Ma L., Lai B., Pei J., Liu Y., Chen J., Lai L. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J. Biol. Chem. 2004;279:1637–1642. doi: 10.1074/jbc.M310875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan K., Ma L., Han X., Liang H., Wei P., Liu Y., Lai L. The substrate specificity of SARS coronavirus 3C-like proteinase. Biochem. Biophys. Res. Commun. 2005;329:934–940. doi: 10.1016/j.bbrc.2005.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziano V., McGrath W.J., Yang L., Mangel W.F. SARS CoV main proteinase: the monomer-dimer equilibrium dissociation constant. Biochemistry. 2006;45:14632–14641. doi: 10.1021/bi061746y. [DOI] [PubMed] [Google Scholar]
- Grum-Tokars V., Ratia K., Begaye A., Baker S.C., Mesecar A.D. Evaluating the 3C-like protease activity of SARS-Coronavirus: recommendations for standardized assays for drug discovery. Virus Res. 2008;133:63–73. doi: 10.1016/j.virusres.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwaoka M., Takemoto S., Tomoda S. Statistical and theoretical investigations on the directionality of nonbonded S…O interactions. Implications for molecular design and protein engineering. Am. Chem. Soc. 2002;124:10613–10620. doi: 10.1021/ja026472q. [DOI] [PubMed] [Google Scholar]
- Lai L., Han X., Chen H., Wei P., Huang C., Liu S., Fan K., Zhou L., Liu Z., Pei J., Liu Y. Quaternary structure, substrate selectivity and inhibitor design for SARS 3C-like proteinase. Curr. Pharm. Des. 2006;12:4555–4564. doi: 10.2174/138161206779010396. [DOI] [PubMed] [Google Scholar]
- Lee T.W., Cherney M.M., Huitema C., Liu J., James K.E., Powers J.C., Eltis L.D., James M.N. Crystal structures of the main peptidase from the SARS coronavirus inhibited by a substrate-like aza-peptide epoxide. J. Mol. Biol. 2005;353:1137–1151. doi: 10.1016/j.jmb.2005.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie, A.G.W., 1992. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB, Newsletter Prot. Crystallogr. 26, 27–33.
- Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D: Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Pal D., Chakrabarti P. Non-hydrogen bond interactions involving the methionine sulfur atom. J. Biomol. Struct. Dyn. 2001;19:115–128. doi: 10.1080/07391102.2001.10506725. [DOI] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera – a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Potterton E., Briggs P., Turkenburg M., Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr. D: Biol. Crystallogr. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- Robin G., Chappell K., Stoermer M.J., Hu S.H., Young P.R., Fairlie D.P., Martin J.L. Structure of West Nile virus NS3 protease: ligand stabilization of the catalytic conformation. J. Mol. Biol. 2009;385:1568–1577. doi: 10.1016/j.jmb.2008.11.026. [DOI] [PubMed] [Google Scholar]
- Schmidt M.F., Isidro-Llobet A., Lisurek M., El-Dahshan A., Tan J., Hilgenfeld R., Rademann J. Sensitized detection of inhibitory fragments and iterative development of non-peptidic protease inhibitors by dynamic ligation screening. Angew. Chem., Int. Ed. Engl. 2008;47:3275–3278. doi: 10.1002/anie.200704594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shie J.J., Fang J.M., Kuo T.H., Kuo C.J., Liang P.H., Huang H.J., Wu Y.T., Jan J.T., Cheng Y.S., Wong C.H. Inhibition of the severe acute respiratory syndrome 3CL protease by peptidomimetic alpha, beta-unsaturated esters. Bioorg. Med. Chem. 2005;13:5240–5252. doi: 10.1016/j.bmc.2005.05.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steuber H., Hilgenfeld R. Recent advances in targeting viral proteases for the discovery of novel antivirals. Curr. Top. Med. Chem. 2010;10:323–345. doi: 10.2174/156802610790725470. [DOI] [PubMed] [Google Scholar]
- Tan J., Verschueren K.H., Anand K., Shen J., Yang M., Xu Y., Rao Z., Bigalke J., Heisen B., Mesters J.R., Chen K., Shen X., Jiang H., Hilgenfeld R. PH-dependent conformational flexibility of the SARS-CoV main proteinase (Mpro) dimer: molecular dynamics simulations and multiple X-ray structure analyses. J. Mol. Biol. 2005;354:25–40. doi: 10.1016/j.jmb.2005.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verschueren K.H., Pumpor K., Anemüller S., Chen S., Mesters J.R., Hilgenfeld R. A structural view of the inactivation of the SARS coronavirus main proteinase by benzotriazole esters. Chem. Biol. 2008;15:597–606. doi: 10.1016/j.chembiol.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber S.E., Okano K., Little T.L., Reich S.H., Xin Y., Fuhrman S.A., Matthews D.A., Love R.A., Hendrickson T.F., Patick A.K., Meador J.W., 3rd, Ferre R.A., Brown E.L., Ford C.E., Binford S.L., Worland S.T. Tripeptide aldehyde inhibitors of human rhinovirus 3C protease: design, synthesis, biological evaluation, and cocrystal structure solution of P1 glutamine isosteric replacements. J. Med. Chem. 1998;41:2786–2805. doi: 10.1021/jm980071x. [DOI] [PubMed] [Google Scholar]
- Xue X., Yang H., Shen W., Zhao Q., Li J., Yang K., Chen C., Jin Y., Bartlam M., Rao Z. Production of authentic SARS-CoV Mpro with enhanced activity: application as a novel tag-cleavage endopeptidase for protein overproduction. J. Mol. Biol. 2007;366:965–975. doi: 10.1016/j.jmb.2006.11.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Xie W., Xue X., Yang K., Ma J., Liang W., Zhao Q., Zhou Z., Pei D., Ziebuhr J., Hilgenfeld R., Yuen K.Y., Wong L., Gao G., Chen S., Chen Z., Ma D., Bartlam M., Rao Z. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005;3:e324. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J., Niu C., Cherney M.M., Zhang J., Huitema C., Eltis L.D., Vederas J.C., James M.N. A mechanistic view of enzyme inhibition and peptide hydrolysis in the active site of the SARS-CoV 3C-like peptidase. J. Mol. Biol. 2007;371:1060–1074. doi: 10.1016/j.jmb.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]