
Fig. 4.
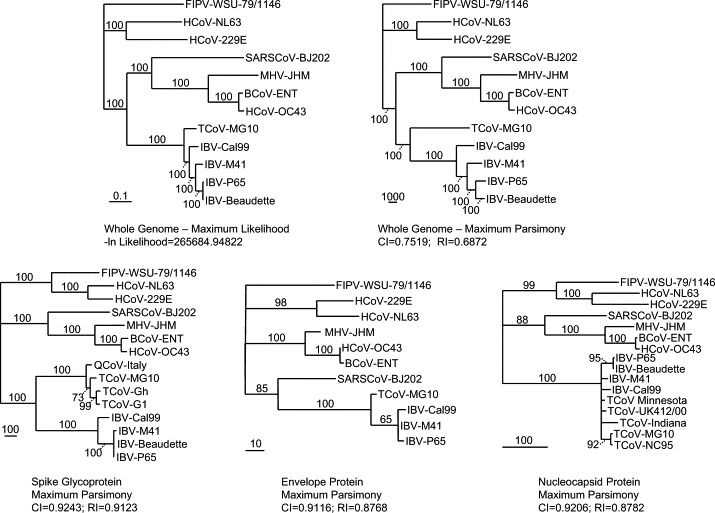
Phylogenetic trees for TCoV and other coronaviruses. Aligned nucleotide sequences of complete coronavirus genomes representing coronavirus groups I, II, and III were utilized to construct maximum likelihood (ML) and maximum parsimony (MP) trees using the PAUP software package followed by bootstrap analysis using a heuristic search method. For each tree, the bootstrap support for each branch is indicated for each branch and the horizontal lengths of branches are proportional to the amount of hypothesized evolutionary change. All trees are rooted using the group I coronaviruses as a functional outgroup. The ln likelihood for the ML tree and the consistency index (CI) and retention index (RI) are provided for the MP trees. For ingroup analyses of relationships among the group III coronaviruses, aligned amino acid sequences for the spike glycoprotein (S), envelope (E) and nucleocapsid (N) proteins were analyzed using maximum parsimony (branch and bound search algorithm). HCoV, human coronavirus; FIPV, feline infectious peritonitis virus; BCoV, bovine coronavirus; MHV, murine hepatitis virus; IBV, infectious bronchitis virus; TCoV, turkey coronavirus.