
Fig. 1.
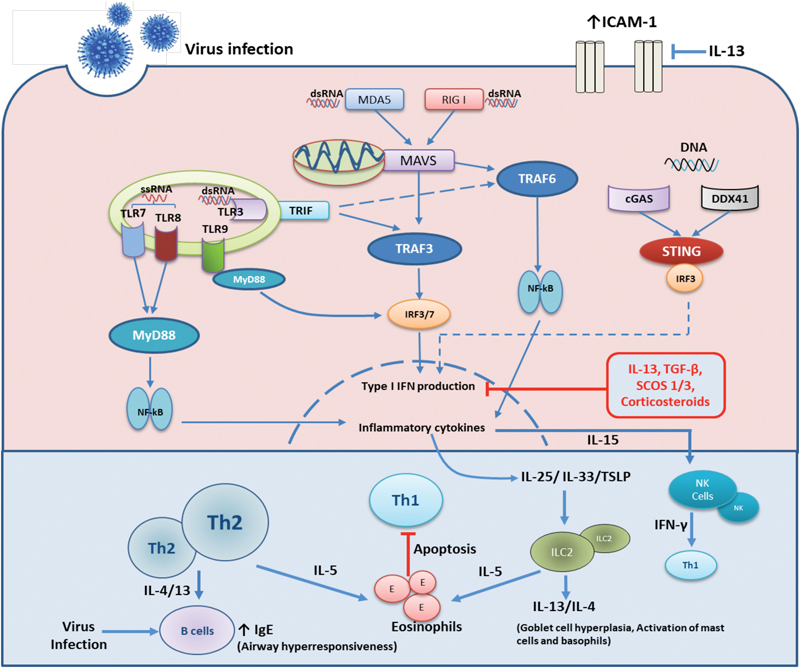
Innate and adaptive immune responses to respiratory virus infections in the airways. Under normal circumstances the innate immune response to virus infection is triggered by infection of airway epithelial cells and recognition of viral pathogen associated molecular patterns (PAMPs) by both the infected epithelium and resident immune cells. These cells contain various pathogen recognition receptors (PRRs) that recognise a diverse range of PAMPs and elicit innate then adaptive immune responses to eliminate virus infections. Viral dsRNA are recognised by RIG I, MDA5 or TLR3 receptors whiles sRNA are mainly sensed by TLR7/8. Various DNA viruses are recognised by cytoplasmic DNA sensors; cGAS, DDX41 or by endosomal TLR9. Upon recognition downstream signaling cascades are initiated to produce type I interferons via IRF3/7 activation that induces antiviral proteins that inhibit viral replication within infected cells and spread to neighboring cells. Inflammatory cytokines production via NF-kB activation, results in release of IL-6, CXCL8 and the recruitment and activation of neutrophils and macrophages. Infected cells release IL-15 that activates NK cells that produce IFN-β and that target infected cells. This supports a robust Th1 environment for recruitment of TH-1 and type 1 innate lymphoid cells (ILC-1), resulting in viral clearance. In asthma there is a pre-existent state of active airway inflammation. In the case of active type 2 inflammation, the presence of increased type 2 lymphocytes, both TH-2 cells and Ilc-2 cells promote this abnormal state. Following infection of the asthmatic epithelium there is heightened release of IL-25/IL-33 and TSLP that further activate ILC2 cells. Activated ILC2s and Th2 cells largely produce more IL-4/13 and IL-5 all of which activate other inflammatory cells such as eosinophils and this results in worsened inflammation of the airways. IL-4/13 also enhance the IgE production by B lymphocytes which may further impair activation of the innate immune cells such as dendritic cells to virus infection. Increased expression of type 2 cytokines, TGF-β and SOCS1/3 negatively regulates type I interferon production while type 2 cytokines also enhance ICAM-1 expression all of which results increased virus replication.