
Fig. 1.
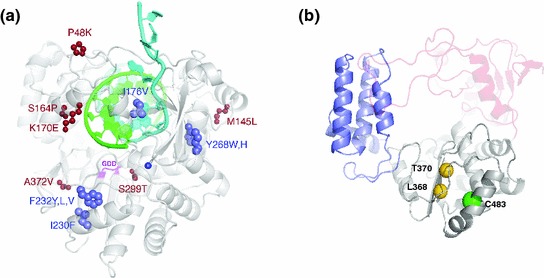
Viral polymerase structures. a Coxsackie virus B3 structure depicting the positions of all viable low-fidelity mutants (blue) likely to favor or alter different conformational states of the polymerase active site. The locations of compensatory mutations are shown in red. Adapted from (Liu et al. 2013). b Structural homology model of the CHIK nsp4 core polymerase showing the predicted locations of C483 (green sphere) and two nearby residues (L368 and T370, shown as gold spheres) that are the structural equivalents of known fidelity-altering sites in Coxsackie virus polymerase (positions I230 and F232, respectively, panel a) (Liu et al. 2013). Adapted from (Gnädig et al. 2012). Three domains are depicted in this figure: (1) the polymerase palm domain (gray), where the fidelity-altering mutations are located, is modeled with fairly high confidence because of the large number of conserved polymerase sequence motifs (motifs A–D); (2) the thumb domain (purple); (3) the fingers (red). Domains where the modeling is weak are shown as semitransparent