
Fig. 3.
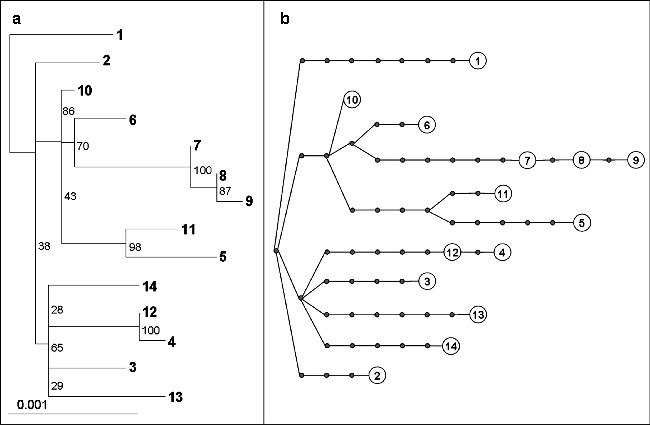
Phylogenetic analysis of FMDV genomes from the 2001 outbreak in the United Kingdom. (a) Maximum likelihood phylogenetic tree representing 14 FMDV complete genomes rooted to sequence “1,” constructed using PhyloWIN95 (33), incorporating the HKY model of nucleotide substitution with gamma distributed rate heterogeneity. Bootstrap values from 1,000 replicates are shown. (b) Statistical parsimony representation of the same 14 complete FMDV sequences, constructed using TCS (Version 1.21; 34). Each line represents a nucleotide substitution and each dot a putative ancestor virus.