

Highlights
-
•
The leading computational techniques for drug design and discovery are reviewed.
-
•
Successful applications of computational techniques are provided.
-
•
A novel drug–target binding kinetics calculation approach is introduced.
Abstract
In the past decades, China's computational drug design and discovery research has experienced fast development through various novel methodologies. Application of these methods spans a wide range, from drug target identification to hit discovery and lead optimization. In this review, we firstly provide an overview of China's status in this field and briefly analyze the possible reasons for this rapid advancement. The methodology development is then outlined. For each selected method, a short background precedes an assessment of the method with respect to the needs of drug discovery, and, in particular, work from China is highlighted. Furthermore, several successful applications of these methods are illustrated. Finally, we conclude with a discussion of current major challenges and future directions of the field.
Drug discovery in China
Since its emergence at the end of the 1970s, computational drug discovery and design (CDDD) has experienced a rapid increase in development, to which China has made significant contributions. With consistent financial support from the government and tremendous research effort, the overall research level of China in this field continues to advance. This trend can be partially revealed by a brief analysis of the research output over the past few years. Taking the structure-based drug design (SBDD) as an example, the publication of scientific papers from China during 2006 to 2010 ranked fifth (citation ranking is seventh), only behind the USA, the UK, Germany, France, and Italy. Among Asian countries, China ranked the top in both the number of publications and citations [1]. Currently, more than 80 universities and research institutions in China have established drug design research departments or centers and over 100 domestic research groups are engaged in drug design. The main reason for the rapid advancement of research should be ascribed to the major breakthrough in methodology and software development.
Currently, many technologies have been developed to boost the efficiency of the drug discovery process. Researchers in China, following the recent trend of scientific advances, have brought us a wealth of innovations, especially in the development of CDDD approaches. In this review, we first summarize leading computational techniques (Table 1 ) that include target prediction and drug repositioning approaches, docking and scoring algorithms, virtual screening (VS) and lead optimization techniques, as well as new absorption, distribution, metabolism, excretion, and toxicity (ADME/T) related frameworks, with a focus on how China has helped to drive new efforts in this field. In the second section, we show how these approaches have been successfully applied in drug discovery. In the final section, we discuss up-to-date research as the final symphony – a successful case of binding kinetics simulation, which is a paradigm of drug target binding kinetics calculation and is expected to inspire future research.
Table 1.
Summary of the highlighted computational techniques for drug design and discovery
| Name | Method | Features | Refs | |
|---|---|---|---|---|
| Drug target prediction | TarFisDock | Reverse docking | • An associated large database for potential drug targets (PDTD) • User-friendly web sever |
6, 8 |
| PharmMapper | Pharmacophore mapping | • Auto-generation of 3D structure for small molecules • User-friendly web sever • Fast (fit for batch processing) |
[12] | |
| miRTarCLIP | Sequence analysis | • miRNA target prediction using CLIP sequencing data • User-friendly web sever |
[20] | |
| Drug repositioning | STITCH | Search tool for interactions of chemicals | • Explore known and predict interactions of chemicals and proteins • User-friendly web sever • Visual presentation of results in network |
29, 30 |
| Drug repositioning by Li et al. | Cross-docking | • Large-scale docking to predict novel drug–target interactions | [31] | |
| SEA | Ligand-based similarity | • Compare targets by the similarity of the ligands that bind to them • User-friendly web sever • 3D structure of drug molecule is not required |
32, 33 | |
| NBI, EWNBI, and NWNBI | Network-based inference | • Interaction of novel protein or chemical can be predicted • 3D structure of drug molecule is not required |
34, 35 | |
| Protein–ligand interaction | GAsDock and MOSFOM | Docking | • Fast (time is in linear scale with the number of the rotatable bonds of ligand) | 39, 41 |
| Induced fit docking by Koska et al. | Flexible docking | • CHARMm-based docking/refinement • Protein flexibility can be considered • Computationally intensive |
[42] | |
| Induced fit docking by Sherman et al. | Flexible docking | • Iteration of rigid docking and protein structure prediction • Protein flexibility can be considered |
[43] | |
| IPMF | Knowledge-based scoring function | • ‘Enriched’ knowledge base by incorporating protein–ligand binding affinity data • Improved accuracy for protein–ligand binding affinity prediction |
[45] | |
| Virtual screening and lead optimization | Pocket v.2 | Structure-based pharmacophore modeling | • Derive pharmacophore models based on a given receptor or complex structure • Insensitive to minor conformational changes of protein upon binding of different ligands |
[50] |
| SHAFTS | 3D similarity calculation | • Fit for large-scale virtual screening • Single or multiple bioactive compounds as searching ‘templates’ • User-friendly web sever |
52, 80 | |
| LigBuilder | De novo drug design | • Detect and score potential binding sites of a protein • Build ligand molecule according to receptor-based pharmacophore • Analyze synthesis accessibility of designed molecule |
[53] | |
| LD1.0 | Target-focused library construction | • Comprehensive consideration of binding affinity, drug-likeness, and ADME/T properties | [54] | |
| AutoT&T | De novo drug design | • Fast (conformation searching is not required) • Fragment database is not required • Fit for fragment-based drug design |
[55] | |
| iScreen | Web service for TCM-related design | • Perform virtual screening and de novo drug design online • User-friendly web sever |
56, 57 | |
| ADME/T properties prediction | SOMEViz | Web service for SOM prediction | • Predict reaction-specific CYP450-mediated SOMs • User-friendly web server |
63, 64 |
| RS-WebPredictor | Web service for SOM prediction | • Predict isozyme-specific CYP450-mediated SOMs • User-friendly web sever • 3D structure of ligand is not required • Fast |
[65] | |
| hERG prediction by Wang et al. | Ligand-based SAR analysis | • Structural patterns favorable or unfavorable for hERG potassium channel blockage are highlighted • 3D structure of ligand is not required |
[69] | |
| hERG prediction by Di Martino et al. | Docking protocol | • Explain the structure–activity relationship for congeneric chemicals | [70] | |
| PKKB | Web service for ADME/T property searching | • High quality ADME/T data for drug molecules | [72] | |
| AdmetSAR | Web service for ADME/T property searching | • A user-friendly web server • Various ADME/T-related databases • Predefined SAR models allowing users to predict for chemicals not in database |
[73] | |
| MetaADEDB | Online web service for ADE property searching | • A user-friendly web server • Predefined SAR models allowing users to predict for chemicals not in database |
74, 75 |
Computational strategies and techniques in drug design
Drug target prediction
Identifying targets is the first key step in the drug discovery pipeline. As of 2006, there were only 324 identified molecular targets for FDA-approved drugs [2]. Although this total figure is under debate, it is significantly lower than the number of proteins unveiled by the completion of human and numerous pathogen genomes, of which many can be potentially druggable [3]. Moreover, many drugs elicit their therapeutic activities by modulating multiple targets, but the multi-target interactions are either largely unknown or insufficiently understood in most cases. The use of computational tools to predict protein targets of small molecules has been gaining importance in recent years. One of the computational approaches demonstrated to be efficient and cost effective in target identification is ‘reverse’ docking 4, 5, 6. Molecular docking is a method to predict the predominant binding mode(s) of a ligand with a protein of known 3D structure, which is routinely used in structure-based drug discovery for hit identification (VS) and lead optimization. More details about the method are provided in the section ‘Protein–ligand interaction’. Reverse docking, opposite to the conventional process of docking application, for example, in structure-based VS, is to dock a given small molecule into the predefined binding sites of a pool of protein structures. A comparison of these two paradigms is shown in Figure 1 .
Figure 1.
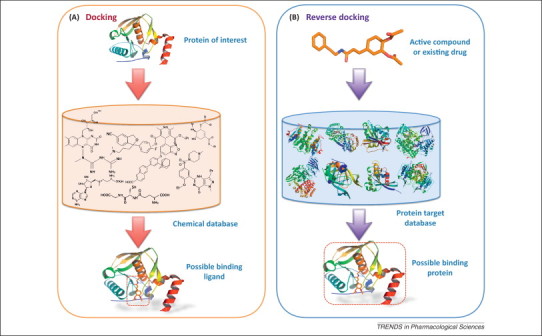
Schematic diagrams to compare (A) conventional docking and (B) reverse docking in ‘hit’ identification. Although conventional docking is used to screen libraries of compounds against one potential drug target, reverse docking is used to dock a given compound into the predefined binding sites of a pool of drug targets.
The identified protein ‘hit’ can then serve as a potential candidate target for experimental validation. Li et al. presented such a computational tool named ‘Target Fishing Dock’ (TarFisDock) [6], which consists of a front-end web interface and a back-end using DOCK [7] for reverse docking. A feature of this work is a companion potential drug target database (PDTD) containing 1207 entries and 841 potential drug targets [8]. The repository of drug targets can be categorized into 14 types according to their therapeutic areas, constituting a comprehensive and valuable source for identifying potential targets of small molecules. By searching through PDTD, TarFisDock can not only predict potential binding targets for a given small molecule but also provide valuable information about their related diseases, biological functions, and regulating (signaling) pathways. So far, TarFisDock has been used for evaluating over 5000 small molecules by more than 2497 users from over 30 countries and regions. Some of the predicted results have been verified by bioassay and crystallographic studies 9, 10, 11.
Despite increased efficiency and convenience, the docking-based methods still take a considerable amount of computational resources as a single job may involve executing hundreds or even thousands of molecular docking runs. Moreover, they heavily rely on the available 3D structures of the targets and a priori knowledge about the ligand binding sites. As a result, these methods are not appropriate for assessing a large batch of small molecules. Pharmacophore modeling is another frequently used technique in drug hit discovery, which usually requires less time to screen a ligand than docking. It measures to what extent a query molecule possesses the spatial arrangement of features essential for protein–ligand interaction. Similar to molecular docking, this approach can also be used ‘in reverse’ to screen a given small molecule against a collection of predefined pharmacophores representing different targets and binding sites. Recently, Liu et al. reported a free web server that hosts a large repertoire of pharmacophores essential for interaction modes between ligand probe and potential targets, entitled PharmMapper [12]. A total of 7302 pharmacophores were constructed by using a protein structure-based modeling approach [13]. Given the query of a small molecule, PharmMapper automatically starts the procedure of reverse mapping against the deposited models and outputs the top ranked hits. Owing to its high throughput ability, PharmMapper can be used for large-scale target screening to profile the regulation genomic network.
miRNAs are small non-coding RNA molecules capable of suppressing protein synthesis and regulating genes in many organisms [14]. This ability makes miRNA a promising technology for future therapeutic development because dysregulation of miRNA function and the changes in miRNA expression level are related to many disorders, including cancers, metabolic and neurodegenerative diseases. Therefore, it is of particular interest to reliably predict potential miRNA targets which might be involved in these diseases 15, 16. In mammals, miRNAs associate with members of the Argonaute (Ago) protein family, and function in multi-protein complexes to direct mRNA cleavage or repress the translation of complementary RNAs [17]. However, molecular details about the interactions between miRNAs and their target RNAs associating with Ago are lacking. Wang et al. analyzed RNA-bound and RNA-free Ago forms by performing molecular docking, molecular dynamics simulation, and binding free energy calculation [18]. Pang et al. investigated the role of miRNAs in choriocarcinoma, through the combined usage of in silico miRNA target prediction, luciferase functional assays, and western blotting analysis. Their study demonstrated that miR-34a is a tumor suppressive miRNA in choriocarcinoma cells that may be used as a therapeutic target for treating choriocarcinoma [19]. Recently, some computational approaches have been developed for the prediction of miRNA targets. Chou et al. reported a systematic approach to identify miRNA–target interactions (MTIs) by mining miRNA–target sites from crosslinking and immunoprecipitation (CLIP) and photoactivatable ribonucleoside-enhanced CLIP (PAR-CLIP) sequencing data, namely miRTarCLIP [20]. The workflow was integrated into a web-based browser to provide a user-friendly interface and detailed annotations of MTIs.
Drug repositioning
In addition to finding novel drug targets, another practice to boost the productivity of the current drug design process is to find new uses for an existing drug [21]. This concept is also known as drug repositioning (also known as drug repurposing or reprofiling) and has gained considerable attention recently 22, 23. Because the starting point in drug repositioning is usually approved drugs with known bioavailability and safety profiles, this approach can significantly reduce the failure risks associated with drug development and potentially facilitate the repositioned compounds to enter clinical phases more rapidly and less costly than new chemical entities [24]. Given that most of the repositioned drugs currently in the market are discovered by chance [25], it is of great interest to develop computational methods to rationalize and facilitate the process. The knowledge about interactions between proteins and small molecules is essential for developing drug repositioning tools. There are some databases that focus on the biological actions of drugs, for example, DrugBank [26], TTD [27], SuperTarget [28], and MATADOR [28]. Recently, Kuhn et al. reported on STITCH (search tool for interactions of chemicals), which integrates information about interactions from metabolic pathways, crystal structures, binding experiments, and drug–target relationships [29]. In the latest version, the database contains over 300 000 chemicals and 2.6 million proteins from 1133 organisms [30]. On the basis of these resources, many computational models have been developed to predict characteristic interactions between the existing drugs and novel molecular targets through target across assessment or ligand–target pathway analysis for prospective repositioning.
Li et al. performed a large-scale molecular docking of 4621 small molecule drugs from DrugBank against 252 human protein drug targets, in order to map the drug–target interaction space and find novel interactions. The method emphasizes removing false positive predictions using annotated ‘known’ interactions, consensus scoring, and ranking thresholds. The utility of the method was verified by predicting novel drug–target interactions. Through a literature search, 31 of the top ranked interactions found corresponding experimental evidence. The method was also used to identify the cancer drug nilotinib as a potent inhibitor of MAPK14, which is a target in inflammatory diseases [31].
On the basis of the central premise of medicinal chemistry that ‘structurally similar molecules have similar biological activity’, potential drug targets can be associated with small molecules or known pharmacological properties through chemical similarities. Shoichet and colleagues developed a statistics-based chemoinformatics method named SEA (Similarity Ensemble Approach), which compared targets by the similarity of the ligands that bind to them [32]. The approach was used to predict new off-targets for 878 purchasable FDA-approved small molecule drugs and 2787 pharmaceutical compounds. Overall, there were 6928 pairs of drug and ligand sets showing statistical significance. Subsequent pharmacological experiments confirmed 23 new drug–target associations and revealed the cross-activity of some drugs on G-protein-coupled receptors (GPCRs), ion channels, and HIV reverse transcriptase. These results suggested that polypharmacology widely exists, which is probably therapeutically essential for some types of drugs [33].
Besides classical target- and ligand-based computational methods, in recent years, many drug repositioning approaches based on systems biology have been developed. Cheng et al. proposed an unweighted network-based inference (NBI) method [34], which used the topology similarity of the ligand–target network to prioritize new targets for a given drug, or vice versa. New drug–target interactions for five known drugs were predicted and confirmed by in vitro assays. One restriction of NBI is that it is only suitable for those drugs having initial links to targets in the network. To tackle this problem, they further developed edge-weighted NBI (EWNBI) and node-weighted NBI (NWNBI) integrating both NBI and similarity strategies [35]. One significant advantage of these proposed approaches is that they are able to predict protein targets of small molecules even if the 3D structures of both the protein and the small molecule are unknown. Similar to the above-mentioned drug target prediction approaches, they have potential values in gaining insights into the promiscuous nature of the drug.
Protein–ligand interaction
Among various computational approaches, molecular docking plays a central role in predicting protein–ligand interactions, which has been extensively used for drug hit discovery and lead optimization [36]. Although detailed algorithms and their implementation may differ, docking involves a conformational sampling component to generate theoretical conformations in the binding pocket, and a binding affinity related scoring component to rank these conformations. Evaluation of some frequently used docking programs can be found elsewhere 37, 38. There are general trends that accuracy is system-dependent, suggesting that the tested methods are specific for particular protein families. Significant progress has been presented by several research groups in China recently. GAsDock developed by Li et al. is an ultra-fast and flexible docking program [39]. It features a conformation-sampling algorithm that utilizes an entropy-based genetic algorithm to optimize the binding poses of small molecules. To further increase the accuracy and speed of the program, Li et al. subsequently developed a new optimization model based on an improved adaptive genetic algorithm [40]. Some techniques including a multi-population genetic strategy and a new iteration scheme were adopted. The model showed reasonable predictive accuracy on reproducing the native binding poses and the docking time is in proportion to the number of rotatable bonds of the ligands. As a continuation of previous work, they also presented two multi-objective optimization methods (MOSFOM) [41], in which the solution will simultaneously satisfy energy and shape complementarity goals, effectively enhancing the performance as compared with those with a single score (GAsDock). Instead of accurately predicting the binding free energy, MOSFOM could yield reasonably ranked binding poses on eliminating the conformations that cannot satisfy the overall objective functions.
One of the factors influencing the reliability of molecular docking is protein flexibility [36]. There is increasing experimental evidence showing that many proteins may undergo significant conformation changes upon ligand binding. Traditional docking methods assume the rigidity of protein, and consequently their accuracy is limited for the ‘induced-fit’ protein–ligand systems. However, owing to the large size and many degrees of freedom of proteins, the computational complexity of algorithms accounting for protein flexibility increases dramatically. Several approximations have been frequently used to account for protein flexibility, including soft docking, side chain flexibility, molecular relaxation, and protein ensemble docking. Koska et al. proposed a two-stage protein flexible docking protocol that models flexibility of the protein before and after ligand placement [42]. The protein side chain conformations are firstly calculated, and low energy poses of ligands are obtained by docking and computing binding site hotspots. Then, side chains of the protein are refined and an energy minimization of the ligand pose in the presence of the protein is performed. Sherman et al. presented a novel procedure for modeling ligand–receptor induced fit effects. Both ligand and protein flexibility are accurately considered by iteratively combining rigid receptor docking (Glide) with protein structure prediction (Prime) techniques. Compared with traditional rigid receptor docking, this approach showed a significantly improved success rate in reproducing correct ligand binding poses on a test set containing 21 pharmaceutically relevant examples [43]. Although this flexible docking procedure mainly focuses on the flexibility of protein side chains, it can be extended to consider backbone motions by incorporating a loop prediction module.
Scoring poses another major obstacle for the reliability of molecular docking. The current scoring functions for docking are typically classified into force field-based, empirical, and knowledge-based [36]. Force-field based methods employ classical molecular mechanical models to compute the direct noncovalent interactions between the protein and ligand, such as van der Waals and electrostatic energies. Empirical scoring functions, by contrast, decompose the overall interactions into different energetic terms and obtain their weight factors by regression analyses on experimental protein–ligand binding affinities. Generally, force-field methods have wider applicability and empirical scoring functions can make a more accurate prediction within the range of regression model. For knowledge-based methods, ‘knowledge’ means structural information from experimentally determined protein–ligand complexes. This type of method uses the Boltzmann law to transform the atom pair preferences into distance-dependent pairwise potentials. The results usually show reasonable accuracy in reproducing the experimental binding poses, but are less accurate on approximating binding free energy [44]. Shen et al. reported an improved knowledge-based scoring function named IPMF [45]. Typically, the knowledge-based functions are developed by applying the Boltzmann law to transform the atom pair preferences into distance-dependent pairwise potentials. In addition to structural information, IPMF integrated experimental binding affinity information into the extracted potentials via an iteration optimization approach. It was found that the scoring function with the ‘enriched’ knowledge might achieve increased accuracy in binding affinity prediction. Compared with seven commonly used scoring functions on a third party test set containing 219 diverse protein–ligand complexes, IPMF performed best in predicting the binding affinity. With the rapid growing volume of high-quality structural and protein–ligand interaction data in the public domain, this work represents a positive step towards improving the accuracy of scoring in binding affinity prediction.
Virtual screening and lead optimization
From the perspective of computational drug design, the drug discovery process commonly involves two steps: hit identification and lead optimization. Hit identification related methods are frequently investigated for searching compounds with interesting biological activity, which is also known as VS 46, 47.
Pharmacophore-based VS is an established in silico tool that has resulted in the identification of many active compounds in drug discovery programs [48]. Generally, a pharmacophore model is deduced from a set of ligands with known activities when the 3D structure of a receptor is lacking. Several programs for automatic ligand-based pharmacophore derivation have been developed, which show different abilities to retrieve active compounds [49]. Alternatively, a pharmacophore model can be directly generated from a protein crystal structure, revealing the key elements required for protein–ligand binding. With fast advances in structural biology, the development of a reliable protein structure-based pharmacophore is gaining more and more attention. Chen and Lai developed a tool named Pocket v.2 for generating pharmacophore models based on given receptor or complex structures [50]. In this approach, hotspot analysis and grid-based scoring were used to identify essential pharmacophore features. As suggested by case study results, Pocket v.2 can yield consistent pharmacophore models for proteins with minor conformational changes upon binding of different ligands, indicating the robustness of the approach.
Shape-based VS is another useful tool for searching for novel lead compounds. Following the frequently used similarity concept, it assumes that molecules of shape and chemistry comparable to known active agents have a significant probability to show similar biological activities. The definition of shape similarity is usually derived from the alignment that achieves an optimal overlap of 3D structures of compounds, where the metric distance is the mismatch volume. Many application studies revealed that shape-based VS can result in good enrichment of active compounds and, meanwhile, sufficient diversity in the discovered compounds. As suggested by the comparative study of Hawkins et al., shape matching showed better VS performance than molecular docking in some test cases [51]. Liu et al. reported a useful VS tool named SHAFTS (SHApe FeaTure Similarity) [52], which is a hybrid approach for 3D molecular similarity searching by comparing both molecular shape and pharmacophore features. SHAFTS performs rigid body superimposition of 3D molecular models and can use single or multiple bioactive compounds as the query ‘templates’. On the basis of a feature triplet hashing algorithm, SHAFTS can efficiently perform rigid body superimposition of 3D molecular models for query and bioactive compounds. Compared with other ligand-based VS methods, it can also achieve a superior performance in enrichment assessment, which measures the number of active compounds found by employing a certain VS strategy as opposed to the number found by random sampling. These results suggest that SHAFTS is suitable for large-scale VS and prospectively exploring potential mechanisms for drug side effects.
Commercial chemical libraries for high-throughput screening (HTS) are primary sources for hit identification. Although increasing size and availability of libraries raises the possibility of finding useful compounds, it is of great interest to design new compounds independent of known chemical structures using de novo methods. Yuan et al. proposed a de novo structure-based drug design program LigBuilder, which can automatically build and screen ligand molecules within the binding pocket of a target protein [53]. In the updated version of the program, the synthesis accessibility of designed compounds can be analyzed with the aid of an embedded chemical reaction database and a retrosynthesis analyzer, which recursively checks if a target molecule or its precursor structures obtained by retrosynthetic rules are found in the database. LigBuilder can use Pocket v.2 to define the pharmacophore features within the binding pocket, which quantitatively calculates the ligandability of the binding site, and provides a visual presentation of the properties of the site. It also allows users to pick and remove fragments of a lead, and then uses ‘grow’ and ‘link’ strategies for optimization. In this way, it can also serve as an effective tool for fragment-based lead optimization.
One of the critical challenges of de novo drug design is to select fragment sets that have the best potential to be part of new drug leads for a given target. Virtual library construction has been suggested as one potential solution. Chen et al. established such an approach to build a target-focused library for finding hits towards studied targets by combining the scores of structural diversity, binding affinity, and drug-likeness assessment. On the test of constructing cyclooxygenase-2 (COX-2) and peroxisome proliferator-activated receptor γ (PPARγ) focused libraries, this model can not only successfully reproduce the key substructures of FDA-approved drugs but also generate some novel chemical classes with increased in vitro inhibition activities against COX-2 and PPARγ, respectively. At the same time, a software package, named LD1.0 [54], was developed for this new approach.
Active hits obtained from HTS typically exhibit low or medium level of activities, which are subsequently modified to improve their potency, selectivity, and to incorporate other favorable physical chemical properties. Recently, Li et al. devised a method called AutoT&T (automatic tailoring and transplanting) [55], which can capture strong binding fragments of hits from VS outputs and transplant them into a lead compound with novel and optimized chemical structure. It means that the approach does not need to perform sampling of the possible combinations of fragments and conformations of the resulting molecules during structural operations, making it more efficient than conventional de novo design methods. Another apparent advantage of AutoT&T is that it does not rely on a predefined library of building blocks but detects important fragments from VS hits. This feature expands the application of VS from hit discovery to lead optimization.
Traditional Chinese medicine (TCM), the quintessence of Chinese culture heritage, constitutes valuable sources for hit identification and lead optimization. Increasing effort has been devoted to study TCM and has resulted in a large number of isolated bioactive compounds. Recently, Chen constructed a 3D structure TCM database (Database@Taiwan) based on structures collected from Chinese medical literature and scientific publications [56]. Furthermore, based on this database, Tsai et al. constructed a web computing system for TCM intelligent screening system (iScreen). It provides users accession to the TCM database and performs VS and de novo drug design online [57].
In silico prediction of ADME/T properties
Most studies on ADME/T commence by highlighting the contribution of these properties to the failure rates of drug discovery and the resultant mounting cost of bringing a new drug to the market. Meanwhile, the number of marketed drug withdrawals continues to increase, mainly because of underlying ADME/T issues that could not have been detected earlier [58]. Currently, many solutions are proposed for identifying and addressing these issues before any leading compounds progress to clinical stages [59]. Among them, the role of early screening ADME/T properties with in silico tools has been widely appreciated 60, 61.
Metabolic biotransformation of a new chemical entity is of high interest because it may profoundly affect its bioavailability, activity, and toxicity profile. Cytochromes P450 (CYPs) are the major enzymes involved in the process responsible for the metabolism of ∼90% FDA-approved drugs [62]. Zheng et al. reported the CYP450-mediated site of metabolism (SOM) prediction for six most important metabolic reactions by incorporating the use of machine learning and semi-empirical quantum chemical calculation [63]. Both internal and external validation results suggest that the developed models are reasonably successful. Furthermore, Shen et al. developed a web-based SOM prediction service SOMEViz, which provides medicinal chemists a visual and easy-to-use interface for addressing some metabolism-related problems [64]. Although considerable overlap is present, individual CYP450 isoenzymes each have unique substrate specificity. Recently, Zaretzki et al. developed a server named RS-WebPredictor that can predict the isozyme-specific regioselectivity for CYP450-mediated reaction [65]. Given the genetic polymorphism of the enzyme, this valuable information may provide a basis for understanding and predicting individual differences in drug response.
Blockade of the human ether-a-go-go related gene (hERG) potassium channel has been identified as the most important mechanism of QT interval prolongation leading to severe cardiotoxicity, which has resulted in the withdrawal of many major drugs [66]. Therefore, it is important to assess hERG binding of compounds in the early phase of drug discovery 67, 68. On the basis of a diverse set of 806 compounds, Wang et al. developed a ligand-based classification model for hERG blockade prediction [69]. The model achieved over 85% accuracy for one internal and two external test sets. Moreover, some important structural fragments favorable or unfavorable for hERG channel inhibition were highlighted. Accurate prediction of hERG inhibitory potency can offer the possibility of screening and optimizing hit compounds to eliminate potentially lethal side effects. Although a full crystal structure for hERG is not yet available, considerable effort has been devoted to the development of structure-based models for hERG blockers. Recently, Di Martino et al. developed and validated an automated docking protocol that quantitatively characterizes the binding of a series of hERG channel blockers [70]. This approach turned out to be successful in both increasing the consistency of the docked binding modes and explaining the structure–activity relationships for the blockers. Clearly, as the 3D structures of some major ADME/T-related proteins become available, structure-based models will play an increasingly important role in this field [71].
Hou and colleagues have carried out extensive studies on in silico modeling of various ADME/T-related properties, including blood–brain barrier partitioning, Caco-2 permeability, human intestinal absorption (HIA), oral absorption, oral bioavailability, P-glycoprotein inhibition, etc. In a recent contribution, they developed a combined database PKKB (PharmacoKinetics Knowledge Base) [72], collecting structures, pharmacological information, important experimental or predicted physiochemical properties, and experimental ADME/T data for 1685 drugs. This database provides a useful resource for benchmarking pharmacokinetic studies, validating the accuracy of existing ADMET predictive models, and building new predictive models.
The web service AdmetSAR (http://www.admetexp.org) developed by Cheng et al. is a more comprehensive source of chemical ADME/T properties [73]. It provides a user-friendly interface to a database covering over 210 000 ADME/T annotated data points for more than 96 000 unique compounds with 45 types of ADME/T-associated properties. Moreover, the database includes classification and regression models with highly predictive accuracy, allowing users to predict the properties for chemicals that cannot be found in the database. Recently, Cheng et al. reported a database of adverse drug events (ADEs) named MetaADEDB, collected by data integration and text mining 74, 75. The database included more than 52 000 of drug–ADE associations among 3059 compounds and 13 200 ADE items. In addition, a computational module was developed for predicting potential ADEs based on the database and phenotypic network inference model (PNIM), which shows high prediction accuracy for an external validation set extracted from the US-FDA Adverse Events Reporting System.
Successful applications of computational drug discovery and design
As outlined above, the novel computational tools and strategies recently reported in China cover most aspects of drug discovery and development processes, and overall they have had a profound impact on rational drug design. Some approaches may have the potential to significantly accelerate and streamline all stages of drug discovery in conjunction with other techniques. In the following sections, we will demonstrate how these methods are successfully applied in different drug discovery phases.
Cases of drug target identification
Natural products provide a vast source for discovery of useful therapeutics. Because the molecular targets of many natural products remain unknown, unraveling the target or targets for a natural compound should provide insights into its molecular mechanism(s) and help in optimizing its potency and selectivity. The pathogenic bacterium Helicobacter pylori is a major causative factor for gastrointestinal illnesses. A natural product isolated from Ceratostigma willmottianum, a folk medicine used to remedy rheumatism, traumatic injury, and parotitis, was found effective in inhibiting the bacterium. However, the specific mechanism of action of the compound is unknown. Cai et al. used this compound as a probe to search PDTD with TarFisDock [9]. In total, 15 putative interacting proteins were found, among which diaminopimelate decarboxylase (DC) and peptide deformylase (PDF) showed a shared sequence homology to H. pylori. Particularly, the sequence of H. pylori PDF (HpPDF) shares 40% identity with that of Escherichia coli PDF. The interaction between the natural product and these two proteins was experimentally evaluated. Enzymatic assays demonstrated that the natural product and one of its analogs are potent inhibitors against HpPDF, with IC50 values of 10.8 and 1.25 μM, respectively. Moreover, the later determined X-ray crystal structures of apo–HpPDF and inhibitor–HpPDF complexes were confirmed at the atomic level. This study reveals that HpPDF is a potential target for anti-H. pylori agents [9]. Another interesting finding using TarFisDock includes revealing the mechanism of [6]-gingerol, the active constituent of fresh ginger, in cancer control and management. It exhibits considerable physiological effects such as antioxidant, anti-inflammatory, and antitumorigenic activity, but its exact molecular mechanism of action remains elusive. With TarFisDock, Jeong et al. ascertained that [6]-gingerol suppressed carcinogenesis by inhibiting the enzymatic activity of leukotriene A4 hydrolase (LTA4H). Given that leukotriene LTA4H is overexpressed in several human cancer cell lines, including colorectal cancer, the findings support the anticancer efficacy of [6]-gingerol [11].
Torcetrapib is a cholesteryl ester transfer protein (CETP) inhibitor suppressing the exchange of lowered high density lipoprotein (HDL) and triglyceride-rich lipoprotein in patients with hyperlipidemia. It is also documented that it may induce cardiac events associated with severe adverse effects. However, the detailed mechanism regarding the safety issue of torcetrapib is still lacking. Fan et al. established a systems biology approach by combining a human reassembled signaling network with microarray gene expression data to study drug–target interactions of torcetrapib. Furthermore, the obtained potential off-targets of torcetrapib were identified by employing the reverse docking strategy. The results suggested that platelet-derived growth factor receptor (PDGFR), hepatocyte growth factor receptor (HGFR), interleukin-2 (IL-2) receptor, and epidermal growth factor receptor (ErbB1) tyrosine kinase were highly relevant to unfavorable adverse effects [76].
Another case study of in silico target prediction include fibroblast growth factor receptors (FGFRs), which are a family of four structure-related receptor tyrosine kinases that are targets for the treatment of various human cancers. Qian and colleagues used the reverse pharmacophore mapping approach PharmMapper to identify target candidates for an active compound that they previously synthesized and showed great in vitro antiproliferative effects [10]. Among the predicted target candidates, tyrosine kinases were revealed as potential targets for the compound. This result was subsequently validated by enzyme-linked immunosorbent assay. After following structural optimization, the derivatives of the compound were found to be effective antiproliferative inhibitors against FGFR1-expressing cancer cell lines with micromolar to submicromolar IC50 values.
Cases of hit discovery
For hit discovery, molecular docking is one of the most widely employed techniques, and it is normally embedded in the workflow of different in silico as well as experimental approaches. Liu et al. reported a combinatorial computational strategy for discovering potential inhibitors against insulin-like growth factor-1 receptor (IGF-1R). IGF-1R belongs to the tyrosine kinase family and plays a pivotal role in the signaling pathway involving cell growth, proliferation, and apoptosis. The initial hit obtained from hierarchical VS was subsequently used as the query scaffold for the substructure search to build a focused library. The library was then screened against IGF-1R with an in-house pharmacophore-constrained docking protocol (PACDock). Eventually, 15 out of 39 purchased compounds exhibited inhibitory activity in enzymatic assessment. Strikingly, the two most potent inhibitors not only demonstrated excellent inhibitory potency (IC50 = 57 and 61 nM, respectively) but also presented considerable selectivity over the insulin receptor (IR) that is highly homological to IGF-1R [77]. Aside from being potential antitumor agents, the promising selective IGF-1R inhibitors can be investigated as molecular probes to differentiate the biological functions of IGF-1R and IR.
New Delhi metallo-β-lactamase-1 (NDM-1) has recently attracted extensive attention for its rapid dissemination and resistance to almost all known β-lactam antibiotics. Using the two-stage protein flexible docking protocol mentioned in the methodology section [43], Shen et al. established an interaction model for NDM-1 and the thiophene-carboxylic acid inhibitor. The results revealed that sulfur atoms of ligand might interfere with the water bridge and zinc ions in the active site of NDM-1, which provide useful clues for the rational design of effective NDM-1 inhibitors [78].
Another distinguished contribution made by Chinese researchers is the identification of a new indication for an old drug cinanserin, a well-characterized serotonin antagonist. During the severe acute respiratory syndrome (SARS) outbreak in China in the spring of 2003, by docking-based VS, Chen et al. identified that cinanserin is a potential inhibitor of the 3C-like (3CL) protease of SARS [79]. The later experimental tests showed that cinanserin can indeed inhibit 3CL protease (IC50 = 5 μM) at nontoxic drug concentrations and has the potential to kill the SARS virus. Because it is an old drug that is cheap and has an established safety record, cinanserin could be used as an emergency treatment or for stockpiling for future SARS pandemics. This is a paradigm for repurposing an existing drug, which provides a cost-effective new way of tackling a devastating disease.
Ligand-based VS is integral to the tool chest of computational chemists, as the effectiveness of docking in VS is highly variable owing to many factors, such as protein flexibility or scoring problems. Liu et al. reported applying SHAFTS to the discovery of novel inhibitors for p90 ribosomal S6 protein kinase 2 (RSK2), which plays a pivotal role in the regulation of diverse cellular processes. SHAFTS was used to perform 3D similarity searching by adopting two weak inhibitors as query templates. Sixteen compounds were discovered with IC50 < 20 μM, three of which showed low micromolar inhibitory activities against RSK2 and exhibited selectivity across a panel of related kinases [80]. In contrast to this, these new scaffolds were not identified by conventional 2D fingerprint methods. The result suggests that SHAFTS is an efficient and powerful tool in scaffold-hopping and hit identification endeavors.
Cases of lead optimization
The customized design of a library focused on a target of interest is expected to select and assemble fragment sets that have the best potential to be parts of new drug leads for the target. With the help of other technologies, the usefulness of designing a focused library in lead structure optimization has been well documented. Here, we illustrate the combined lead optimization strategies by three case studies and highlight some of the issues and successes observed when screening target-focused libraries.
The first successful application is the discovery of three series of potent inhibitors against β-Hydroxyacyl-Acyl Carrier Protein Dehydratase FabZ of H. pylori (HpFabZ) that is of intense interest in the treatment of gastric diseases. On the basis of fragments isolated from previously discovered HpFabZ inhibitors, a focused library was designed by the LD1.0 program. Through docking into binding pockets, 12 candidates were synthesized and tested with bioassays, among which eight compounds showed significant inhibitory activities with IC50 values ranging from 0.86 to 42.7 μM [81]. The high hit rate demonstrates the efficiency of the focused library. Moreover, by adopting a similar strategy, Liu et al. reported novel purine derivatives with potent and selective inhibitory activity against c-Src kinase. The most remarkable agent showed kinase inhibitory activity with an IC50 value of 0.02 μM, making it a potential lead for further development of c-Src kinase-related anticancer drugs [82].
The last excellent work to present is the design of inhibitors for Cyclophilin A (CypA) that is responsible for a variety of biological processes regulation via its peptidyl-prolyl isomerase (PPIase) activity. It is also reported to be a promising target for cardiovascular, immunosuppressive, and cancer therapy. Initially, Li et al. designed a focused library by LD1.0 on the basis of the previously reported CypA inhibitors and selected 16 compounds for synthesis. All 16 compounds were CypA binders with dissociation constants (K d) ranging from 0.076 to 41.0 μM, and five compounds showed PPIase inhibitory activities (IC50) of 0.25–6.43 μM [83]. The hit rates for binders and inhibitors were as high as 100% and 31.25%, respectively. Encouraged by the results, the authors continued the study with an acylurea linker that was previously identified crucial to CypA ligand binding. The de novo drug design program LigBuilder 2.0 was customized to generate inhibitors that incorporate the complementary properties and fit the shape of the CypA binding site. A potent inhibitor with IC50 of 31.6 nM was firstly obtained. Then, after two rounds of structure–activity analyses (SAR) and chemical modifications, two highly potent CypA inhibitors with nanomolar inhibitory potencies (2.59 nM and 1.52 nM) were obtained out of 19 synthesized compounds [84]. These promising results clearly demonstrate that the LD1.0 and the de novo method LigBuilder 2.0 are powerful tools for lead optimization.
Future directions
The year 2012 marked three decades of protein structure-based drug design, since the first docking program, DOCK, was published in 1982 by Kuntz et al. [85]. A special issue published in the Journal of Computer-Aided Molecular Design reflected on 25 years of molecular design and it was concluded that new compounds are ‘easy to design but challenging in evaluation’ [86]. Medicinal chemists use a variety of computational approaches to modify the chemical structure of a compound to maximize its in vitro activity. However, good in vitro activity cannot be extrapolated to good in vivo activity without the understanding of pharmacokinetics and drug metabolism properties. Even at the protein–ligand binding level, it is challenging to predict the efficacy of a ligand. Traditionally, drug discovery is driven by the idea that a ligand with higher binding affinity to a target should be more efficacious than that with lower binding affinity to the same target. It is clear that the efficacy of a drug is not only associated with thermodynamics but also related to the binding kinetics between the drug and a defined target. Numerous examples demonstrated that drug efficacy does not always linearly correlate with binding affinity. In particular, there is now a renewed emphasis to ligand–receptor binding kinetics in almost all steps along the drug research and development (R&D) pipeline. Similar to experimental approaches for drug discovery, the current computational drug design methods focus on maximizing ligand-binding affinity, which are either fast-but-inaccurate or slow-but-accurate. This situation tends to change with the development of a binding kinetics-emphasized paradigm. Drug efficacy mainly depends on the drug–target residence time t, calculated from k off – the dissociation rate constant (t = 1/k off). Despite more than 30 years of effort, predicting binding free energies for ligands interacting with targets with sufficient accuracy (within ± 3 kcal/mol) is still an unsolved problem due to the time-consuming free energy perturbation calculation and crude docking result, not to mention calculating k off.
Recently, our research team has made significant progress in this area. The energy landscape theory that was developed to understand protein folding and function can be extended to develop a generally applicable computational framework, making it possible to construct a complete ligand–target binding free energy landscape (BFEL) [87]. This enables both binding affinity and binding kinetics to be accurately estimated, and possible binding (unbinding) pathway(s) may be obtained as well. We applied this method to simulate the binding event of the anti-Alzheimer's disease drug (–)-Huperzine A (HupA) to its target acetylcholinesterase (AChE). The computational results are in excellent agreement with the concurrent experimental measurements. All predicted values of binding free energy and activation free energies of association and dissociation only deviate from the experimental data by less than 1 kcal/mol. The method also provides atomic resolution information for the HupA binding pathway, which may be useful in designing AChE inhibitors with higher potency.
Concluding remarks
CDDD is a multidisciplinary technology by exploiting state-of-art in silico models and algorithms to speed up the drug development process. Over the past decades, there has been significant progress in China in both development and application of novel methodologies. These methods streamline many aspects of drug discovery, from hit screening and lead optimization to drug target identification and ADME/T assessment. In future development, it is necessary to establish a research pattern that is oriented by computational analyses and brings into full play chemistry, biology, and other disciplines with complementary strengths. In particular, the philosophy embodied in CDDD is shifting from ‘one gene, one drug, and one disease’ to ‘multicomponent therapeutics, network targets’. Accordingly, the emerging discipline of network pharmacology [88], which combines network biology with chemogenomics, makes notable impact on the fundamental challenges of traditional medicines for complex diseases. Under the new paradigm, CDDD will play an even more pivotal role in discovering lead compounds with the desired polypharmacological profiles, and systematic data mining to elucidate synergistic effects, side effects, and to understand the promiscuity of drugs.
Acknowledgments
The authors gratefully acknowledge financial support from the National High Technology Research and Development Program of China (2012AA020302), the State Key Program of Basic Research of China grant (2009CB918502), the National Natural Science Foundation of China grants (21021063, 81230076, 81001399, and 21210003), and the National Science and Technology Major Project ‘Key New Drug Creation and Manufacturing Program’ (2013ZX09507-004).
References
- 1.Jiang H.L. Structure-based drug design. In: Han Y., Liang W.P., editors. Chemistry During 2001–2010: China in the World. National Natural Science Foundation of China; 2011. pp. 132–136. [Google Scholar]
- 2.Overington J.P. How many drug targets are there? Nat. Rev. Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 3.Hopkins A.L., Groom C.R. The druggable genome. Nat. Rev. Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 4.Chen Y.Z., Zhi D.G. Ligand–protein inverse docking and its potential use in the computer search of protein targets of a small molecule. Proteins. 2001;43:217–226. doi: 10.1002/1097-0134(20010501)43:2<217::aid-prot1032>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 5.Paul N. Recovering the true targets of specific ligands by virtual screening of the Protein Data Bank. Proteins. 2004;54:671–680. doi: 10.1002/prot.10625. [DOI] [PubMed] [Google Scholar]
- 6.Li H.L. TarFisDock: a web server for identifying drug targets with docking approach. Nucleic Acids Res. 2006;34:W219–W224. doi: 10.1093/nar/gkl114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ewing T.J.A. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 2001;15:411–428. doi: 10.1023/a:1011115820450. [DOI] [PubMed] [Google Scholar]
- 8.Gao Z.T. PDTD: a web-accessible protein database for drug target identification. BMC Bioinformatics. 2008;9:104. doi: 10.1186/1471-2105-9-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai J.H. Peptide deformylase is a potential target for anti-Helicobacter pylori drugs: reverse docking, enzymatic assay, and X-ray crystallography validation. Protein Sci. 2006;15:2071–2081. doi: 10.1110/ps.062238406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Z. Acenaphtho[1,2-b]pyrrole-based selective fibroblast growth factor receptor 1 (FGFR1) inhibitors: design, synthesis, and biological activity. J. Med. Chem. 2011;54:3732–3745. doi: 10.1021/jm200258t. [DOI] [PubMed] [Google Scholar]
- 11.Jeong C.H. [6]-Gingerol suppresses colon cancer growth by targeting leukotriene A4 hydrolase. Cancer Res. 2009;69:5584–5591. doi: 10.1158/0008-5472.CAN-09-0491. [DOI] [PubMed] [Google Scholar]
- 12.Liu X.F. PharmMapper server: a web server for potential drug target identification using pharmacophore mapping approach. Nucleic Acids Res. 2010;38:W609–W614. doi: 10.1093/nar/gkq300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolber G., Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005;45:160–169. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- 14.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 15.Lewis B.P. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 16.Krek A. Combinatorial microRNA target predictions. Nat. Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 17.Dueck A. microRNAs associated with the different human Argonaute proteins. Nucleic Acids Res. 2012;40:9850–9862. doi: 10.1093/nar/gks705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y.H. Mechanism of microRNA–target interaction: molecular dynamics simulations and thermodynamics analysis. PLoS Comput. Biol. 2010;6:e1000866. doi: 10.1371/journal.pcbi.1000866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pang R.T.K. MicroRNA-34a is a tumor suppressor in choriocarcinoma via regulation of Delta-like1. BMC Cancer. 2013;13:25. doi: 10.1186/1471-2407-13-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chou C.H. A computational approach for identifying microRNA–target interactions using high-throughput CLIP and PAR-CLIP sequencing. BMC Genomics. 2013;14(Suppl. 1):S2. doi: 10.1186/1471-2164-14-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chong C.R., Sullivan D.J. New uses for old drugs. Nature. 2007;448:645–646. doi: 10.1038/448645a. [DOI] [PubMed] [Google Scholar]
- 22.Tobinick E.L. The value of drug repositioning in the current pharmaceutical market. Drug News Perspect. 2009;22:119–125. doi: 10.1358/dnp.2009.22.2.1303818. [DOI] [PubMed] [Google Scholar]
- 23.Dudley J.T. Exploiting drug–disease relationships for computational drug repositioning. Brief. Bioinform. 2011;12:303–311. doi: 10.1093/bib/bbr013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boguski M.S. Repurposing with a difference. Science. 2009;324:1394–1395. doi: 10.1126/science.1169920. [DOI] [PubMed] [Google Scholar]
- 25.Sardana D. Drug repositioning for orphan diseases. Brief. Bioinform. 2011;12:346–356. doi: 10.1093/bib/bbr021. [DOI] [PubMed] [Google Scholar]
- 26.Wishart D.S. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36:D901–D906. doi: 10.1093/nar/gkm958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu F. Therapeutic target database update 2012: a resource for facilitating target-oriented drug discovery. Nucleic Acids Res. 2012;40:D1128–D1136. doi: 10.1093/nar/gkr797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gunther S. SuperTarget and Matador: resources for exploring drug–target relationships. Nucleic Acids Res. 2008;36:D919–D922. doi: 10.1093/nar/gkm862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuhn M. STITCH: interaction networks of chemicals and proteins. Nucleic Acids Res. 2008;36:D684–D688. doi: 10.1093/nar/gkm795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuhn M. STITCH 3: zooming in on protein–chemical interactions. Nucleic Acids Res. 2012;40:D876–D880. doi: 10.1093/nar/gkr1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y.Y. A computational approach to finding novel targets for existing drugs. PLoS Comput. Biol. 2011;9:e1002139. doi: 10.1371/journal.pcbi.1002139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keiser M.J. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007;25:197–206. doi: 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- 33.Keiser M.J. Predicting new molecular targets for known drugs. Nature. 2009;462:175–181. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng F.X. Prediction of drug–target interactions and drug repositioning via network-based inference. PLoS Comput. Biol. 2012;8:e1002503. doi: 10.1371/journal.pcbi.1002503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng F.X. Prediction of chemical–protein interactions network with weighted network-based inference method. PLoS ONE. 2012;7:e41064. doi: 10.1371/journal.pone.0041064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitchen D.B. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discov. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 37.Cross J.B. Comparison of several molecular docking programs: pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2009;49:1455–1474. doi: 10.1021/ci900056c. [DOI] [PubMed] [Google Scholar]
- 38.Warren G.L. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006;49:5912–5931. [Google Scholar]
- 39.Li H.L. GAsDock: a new approach for rapid flexible docking based on an improved multi-population genetic algorithm. Bioorg. Med. Chem. Lett. 2004;14:4671–4676. doi: 10.1016/j.bmcl.2004.06.091. [DOI] [PubMed] [Google Scholar]
- 40.Kang L. An improved adaptive genetic algorithm for protein–ligand docking. J. Comput. Aided Mol. Des. 2009;23:1–12. doi: 10.1007/s10822-008-9232-5. [DOI] [PubMed] [Google Scholar]
- 41.Li H.L. An effective docking strategy for virtual screening based on multi-objective optimization algorithm. BMC Bioinformatics. 2009;10:58. doi: 10.1186/1471-2105-10-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koska J. Fully automated molecular mechanics based induced fit protein–ligand docking method. J. Chem. Inf. Model. 2008;48:1965–1973. doi: 10.1021/ci800081s. [DOI] [PubMed] [Google Scholar]
- 43.Sherman W. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 44.Vaque M. Protein–ligand docking: a review of recent advances and future perspectives. Curr. Pharm. Anal. 2008;4:1–19. [Google Scholar]
- 45.Shen Q.C. Knowledge-based scoring functions in drug design: 2. Can the knowledge base be enriched? J. Chem. Inf. Model. 2011;51:386–397. doi: 10.1021/ci100343j. [DOI] [PubMed] [Google Scholar]
- 46.Shoichet B.K. Virtual screening of chemical libraries. Nature. 2004;432:862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walters W.P. Virtual screening – an overview. Drug Discov. Today. 1998;3:160–178. [Google Scholar]
- 48.Leach A.R. Three-dimensional pharmacophore methods in drug discovery. J. Med. Chem. 2010;53:539–558. doi: 10.1021/jm900817u. [DOI] [PubMed] [Google Scholar]
- 49.Sanders M.P. Comparative analysis of pharmacophore screening tools. J. Chem. Inf. Model. 2012;52:1607–1620. doi: 10.1021/ci2005274. [DOI] [PubMed] [Google Scholar]
- 50.Chen J., Lai L.H. Pocket v.2: further developments on receptor-based pharmacophore modeling. J. Chem. Inf. Model. 2006;46:2684–2691. doi: 10.1021/ci600246s. [DOI] [PubMed] [Google Scholar]
- 51.Hawkins P.C.D. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007;50:74–82. doi: 10.1021/jm0603365. [DOI] [PubMed] [Google Scholar]
- 52.Liu X.F. SHAFTS: a hybrid approach for 3D molecular similarity calculation. 1. Method and assessment of virtual screening. J. Chem. Inf. Model. 2011;51:2372–2385. doi: 10.1021/ci200060s. [DOI] [PubMed] [Google Scholar]
- 53.Yuan Y.X. LigBuilder 2: a practical de novo drug design approach. J. Chem. Inf. Model. 2011;51:1083–1091. doi: 10.1021/ci100350u. [DOI] [PubMed] [Google Scholar]
- 54.Chen G. Focused combinatorial library design based on structural diversity, druglikeness and binding affinity score. J. Comb. Chem. 2005;7:398–406. doi: 10.1021/cc049866h. [DOI] [PubMed] [Google Scholar]
- 55.Li Y. Automatic tailoring and transplanting: a practical method that makes virtual screening more useful. J. Chem. Inf. Model. 2011;51:1474–1491. doi: 10.1021/ci200036m. [DOI] [PubMed] [Google Scholar]
- 56.Chen C.Y.C. TCM Database@Taiwan: The world's largest traditional Chinese medicine database for drug screening in silico. PLoS ONE. 2011;6:e15939. doi: 10.1371/journal.pone.0015939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsai T.Y. iScreen: world's first cloud-computing web server for virtual screening and de novo drug design based on TCM database@Taiwan. J. Comput. Aided Mol. Des. 2011;25:525–531. doi: 10.1007/s10822-011-9438-9. [DOI] [PubMed] [Google Scholar]
- 58.van de Waterbeemd H., Gifford E. ADMET in silico modelling: towards prediction paradise? Nat. Rev. Drug Discov. 2003;2:192–204. doi: 10.1038/nrd1032. [DOI] [PubMed] [Google Scholar]
- 59.Li A.P. Screening for human ADME/Tox drug properties in drug discovery. Drug Discov. Today. 2001;6:357–366. doi: 10.1016/s1359-6446(01)01712-3. [DOI] [PubMed] [Google Scholar]
- 60.Valerio L.G., Jr In silico toxicology for the pharmaceutical sciences. Toxicol. Appl. Pharmacol. 2009;241:356–370. doi: 10.1016/j.taap.2009.08.022. [DOI] [PubMed] [Google Scholar]
- 61.Butina D. Predicting ADME properties in silico: methods and models. Drug Discov. Today. 2002;7:S83–S88. doi: 10.1016/s1359-6446(02)02288-2. [DOI] [PubMed] [Google Scholar]
- 62.Wilkinson G.R. Drug metabolism and variability among patients in drug response. N. Engl. J. Med. 2005;352:2211–2221. doi: 10.1056/NEJMra032424. [DOI] [PubMed] [Google Scholar]
- 63.Zheng M.Y. Site of metabolism prediction for six biotransformations mediated by cytochromes P450. Bioinformatics. 2009;25:1251–1258. doi: 10.1093/bioinformatics/btp140. [DOI] [PubMed] [Google Scholar]
- 64.Shen Q.C. SOMEViz: a web service for site of metabolism estimating and visualizing. Protein Pept. Lett. 2012;19:905–909. doi: 10.2174/092986612802084537. [DOI] [PubMed] [Google Scholar]
- 65.Zaretzki J. RS-WebPredictor: a server for predicting CYP-mediated sites of metabolism on drug-like molecules. Bioinformatics. 2013;29:497–498. doi: 10.1093/bioinformatics/bts705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sanguinetti M.C., Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- 67.Roche O. A virtual screening method for prediction of the hERG potassium channel liability of compound libraries. Chembiochem. 2002;3:455–459. doi: 10.1002/1439-7633(20020503)3:5<455::AID-CBIC455>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 68.Recanatini M. QT prolongation through hERG K+ channel blockade: current knowledge and strategies for the early prediction during drug development. Med. Res. Rev. 2005;25:133–166. doi: 10.1002/med.20019. [DOI] [PubMed] [Google Scholar]
- 69.Wang S. ADMET evaluation in drug discovery. 12. Development of binary classification models for prediction of hERG potassium channel blockage. Mol. Pharm. 2012;9:996–1010. doi: 10.1021/mp300023x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Di Martino G.P. An automated docking protocol for hERG channel blockers. J. Chem. Inf. Model. 2013;53:159–175. doi: 10.1021/ci300326d. [DOI] [PubMed] [Google Scholar]
- 71.Moroy G. Toward in silico structure-based ADMET prediction in drug discovery. Drug Discov. Today. 2012;17:44–55. doi: 10.1016/j.drudis.2011.10.023. [DOI] [PubMed] [Google Scholar]
- 72.Cao D.Y. ADMET evaluation in drug discovery. 11. PharmacoKinetics Knowledge Base (PKKB): a comprehensive database of pharmacokinetic and toxic properties for drugs. J. Chem. Inf. Model. 2012;52:1132–1137. doi: 10.1021/ci300112j. [DOI] [PubMed] [Google Scholar]
- 73.Cheng F.X. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012;52:3099–3105. doi: 10.1021/ci300367a. [DOI] [PubMed] [Google Scholar]
- 74.Cheng F.X. Adverse drug events: database construction and in silico prediction. J. Chem. Inf. Model. 2013;53:744–752. doi: 10.1021/ci4000079. [DOI] [PubMed] [Google Scholar]
- 75.Cheng F.X. Prediction of polypharmacological profiles of drugs by the integration of chemical, side effects and therapeutic space. J. Chem. Inf. Model. 2013;53:753–762. doi: 10.1021/ci400010x. [DOI] [PubMed] [Google Scholar]
- 76.Fan S.J. Clarifying off-target effects for torcetrapib using network pharmacology and reverse docking approach. BMC Syst. Biol. 2012;6:152. doi: 10.1186/1752-0509-6-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu X.F. Discovery and SAR of thiazolidine-2,4-dione analogues as insulin-like growth factor-1 receptor (IGF-1R) inhibitors via hierarchical virtual screening. J. Med. Chem. 2010;53:2661–2665. doi: 10.1021/jm901798e. [DOI] [PubMed] [Google Scholar]
- 78.Shen B. Inhibitor discovery of full-length New Delhi metallo-β-lactamase-1 (NDM-1) PLoS ONE. 2013;8:e62955. doi: 10.1371/journal.pone.0062955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen L.L. Cinanserin is an inhibitor of the 3C-like proteinase of severe acute respiratory syndrome coronavirus and strongly reduces virus replication in vitro. J. Virol. 2005;79:7095–7103. doi: 10.1128/JVI.79.11.7095-7103.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lu W.Q. SHAFTS: a hybrid approach for 3D molecular similarity calculation. 2. Prospective case study in the discovery of diverse p90 ribosomal S6 protein kinase 2 inhibitors to suppress cell migration. J. Med. Chem. 2011;54:3564–3574. doi: 10.1021/jm200139j. [DOI] [PubMed] [Google Scholar]
- 81.He L.Y. Discovering potent inhibitors against the β-hydroxyacyl-acyl carrier protein dehydratase (FabZ) of Helicobacter pylori: structure-based design, synthesis, bioassay, and crystal structure determination. J. Med. Chem. 2009;52:2465–2481. doi: 10.1021/jm8015602. [DOI] [PubMed] [Google Scholar]
- 82.Huang H. Discovery of novel purine derivatives with potent and selective inhibitory activity against c-Src tyrosine kinase. Bioorg. Med. Chem. 2010;18:4615–4624. doi: 10.1016/j.bmc.2010.05.032. [DOI] [PubMed] [Google Scholar]
- 83.Li J. Strategy for discovering chemical inhibitors of human cyclophilin A: focused library design, virtual screening, chemical synthesis and bioassay. J. Comb. Chem. 2006;8:326–337. doi: 10.1021/cc0501561. [DOI] [PubMed] [Google Scholar]
- 84.Ni S.S. Discovering potent small molecule inhibitors of cyclophilin A using de novo drug design approach. J. Med. Chem. 2009;52:5295–5298. doi: 10.1021/jm9008295. [DOI] [PubMed] [Google Scholar]
- 85.Kuntz I.D. A geometric approach to macromolecule–ligand interactions. J. Mol. Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 86.Blaney J. A very short history of structure-based design: how did we get here and where do we need to go? J. Comput. Aided Mol. Des. 2012;26:13–14. doi: 10.1007/s10822-011-9518-x. [DOI] [PubMed] [Google Scholar]
- 87.Bai F. Free energy landscape for the binding process of Huperzine A to acetylcholinesterase. Proc. Natl. Acad. Sci. U.S.A. 2013;110:4273–4278. doi: 10.1073/pnas.1301814110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hopkins A.L. Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol. 2008;4:682–690. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]