

Graphical abstract
Keywords: Aminoarylazo compounds, Aryltriazene derivatives, Antiviral activity, BVDV RdRp targeting
Abstract
Twelve aminoarylazocompounds (A–C) and 46 aryltriazene 7 derivatives (D–G) have been synthesized and evaluated in cell-based assays for cytotoxicity and antiviral activity against a panel of 10 RNA and DNA viruses.
Eight aminoazocompounds and 27 aryltriazene derivatives exhibited antiviral activity, sometimes of high level, against one or more viruses. A marked activity against BVDV and YFV was prevailing among the former compounds, while the latter type of compounds affected mainly CVB-2 and RSV. None of the active compounds inhibited the multiplication of HIV-1, VSV and VV.
Arranged in order of decreasing potency and selectivity versus the host cell lines, the best compounds are the following; BVDV: 1 > 7 > 8 > 4; YFV: 7 > 5; CVB-2: 25 > 56 > 18; RSV: 14 > 20 > 55 > 38 > 18 > 19; HSV-1: 2. For these compounds the EC50 ranged from 1.6 μM (1) to 12 μM (18), and the S. I. from 19.4 (1) to 4.2 (2).
Thus the aminoarylazo and aryltriazene substructures appear as interesting molecular component for developing antiviral agents against ss RNA viruses, particularly against RSV and BVDV, which are important human and veterinary pathogens.
Finally, molecular modeling investigations indicated that compounds of structure A–C, active against BVDV, could work targeting the viral RNA-dependent RNA-polymerase (RdRp), having been observed a good agreement between the trends of the estimated IC50 and the experimental EC50 values.
1. Introduction
We have recently shown that some arylazoenamines (Fig. 1 ) are endowed with antiviral activity in vitro against RNA viruses, particularly CVB-2, RSV, BVDV, YFV and Sb-1, in many cases with EC50 in the range from 0.8 to 10 μM.1
Figure 1.
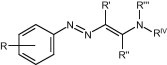
General structure of the previously studied arylazoenamines.
Arylazoenamines can be considered as substructures of ortho- (and para-) aminoazocompounds, many of which are endowed with antimicrobial and antiparasitic activities, but additional pharmacological activities have been also shown in particular cases (Fig. 2 ).
Figure 2.
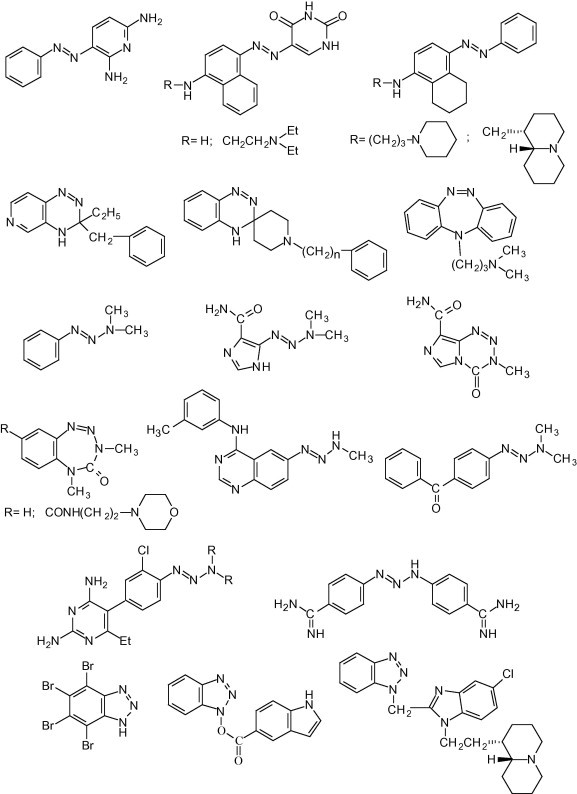
Some biologically active aminoarylazo compounds and aryltriazene derivatives.
2,6-Diamino-3-phenylazopyridine (Pyridium) inhibits various cocci and coli bacilli, 5-(4-amino-1-naphthylazo)uracil is highly effective against Schistosoma mansonii,2 while 1-[3-(4-phenylazo-5,6,7,8-tetrahydronaphth-1-yl)amino]propylpiperidine3 and 1-[3-(4-phenylazo-5,6,7,8-tetrahydro naphth-1-yl)amino]lupinane4 are very active against Mycobacterium tuberculosis. Antifungal and antiproliferative activities have been found in a peculiar class of cyclic aminoazocompounds (3,3-disubstituted-3,4-dihydro-1,2,4-benzo triazines), which, moreover, can display several other pharmacological activities depending on the substituents that are present on the bicyclic system.5, 6 Very potent relaxant activity on intestinal and uterine smooth muscles was displayed at nanomolar concentration by N-dialkylaminoalkyl derivatives of another cyclic aminoazo system, namely 11H-dibenzo-[1,2,5]-triazepine, which exhibited also local anesthetic and antithrombotic activities.7
Arylazoenamines could be also seen as vinylogues of aryltriazenes (Fig. 2), a class of compounds which hold an important position as carcinogens and/or anticancer agents. 3,3-Dimethyl-1-phenyltriazene and, even better, dacarbazine are able to methylate DNA; thus the former resulted a powerful carcinogen, while the latter is in clinical use for the treatment of malignant melanoma and Hodgkin’s lymphoma.8 Temozolamide,9 closely related to dacarbazine, is currently used to treat malignant glyomas. A recent development of this cyclic aryltriazene is represented by benzotriazepinones,10 that are very promising agents against breast cancer. It is worth noting that these compounds are weak alkylating agents and may damage DNA by a novel mechanism.
The triazene pharmacophore has been linked to other bioactive moieties in order to obtain chymeric compounds which should be able to exert a double cytotoxic mode of action. Thus, the triazene group was linked to 4-anilino-quinazoline backbone,11 which is known to inhibit the epidermal growth factor receptor tyrosine kinase (EGFR-TK), and also to a benzophenone residue,12 that is present in an inhibitor of pharnesyl-transferase and of tubulin-polymerization.
The introduction of a triazene group on the molecule of pyrimethamine, a dihydrofolate reductase inhibitor, generated a compound that combines antitumoral potential with inhibition of Pneumocystis jirovecii 13 (P. carinii). The insertion of triazene group between two benzamidine units produced diminazene (Berenil), a powerful agent against the African trypanosomiasis.
To the best of our knowledge, no antiviral activity has been seen with either amino arylazocompounds or aryltriazenes, with the only exception for a patent claiming the activity of 1-phenyl-3,3-dimethyltriazene against the tobacco mosaic virus.14 However, antiviral activity has been shown by several benzotriazole derivatives, which could be formally considered as cyclic triazenes, though the resonance stabilization of the heterocycle could imply even substantial differences in the chemical and hence the biological behavior of the two kinds of compounds. Interestingly, some halogenated benzotriazoles have been shown to inhibit the NTPase/helicase activities of hepatitis C and related viruses.15 Aromatic and heteroaromatic esters of 1-hydroxybenzotriazole were found to strongly inhibit (K i = 7.5 nM) the 3CL-protease, which is essential for the replication of the coronavirus responsible of SARS (severe acute respiratory syndrome).16 More recently, a set of benzotriazole derivatives have been found endowed with potent activity against RSV and moderate activity against YFV, BVDV and CVB-2.17
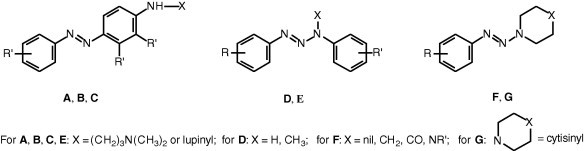
On this base we deemed interesting to investigate the possible antiviral activity of some assorted aminoarylazo compounds and aryltriazene derivatives, allotted in seven groups A–G (Fig. 3 ). On the whole 12 amino arylazo compounds (A–C) and 46 triazene derivatives (D–G) were evaluated in cell based assays for cytotoxicity and antiviral activity against a large panel of RNA and DNA viruses.
Figure 3.
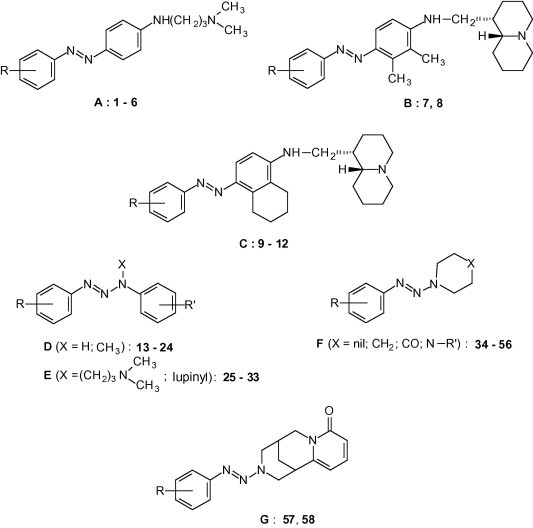
Structures of the investigated compounds.
2. Chemistry
Thirty out of the 58 compounds, that were evaluated for antiviral activity, were already known and were prepared according to the literature. Compounds 7–12 were already described by some of us.4 For 1,3-diaryltriazenes 13, 15–17, 20–24, see;a–h for arylazopyrrolidines 34, 36, 38–42, see;i–l for arylazopiperidines 45, 49–51, see;m,n for arylazopiperazine 52, 54, 55 see;o,p finally, for cytisine derivative 57 see.q 1-(3-Nitrophenylazo)piperidine (48) is reported as known,r but we failed to find out any data concerning its characterization.
References,a-r concerning the previously described compounds, are available as Supplementary data.
The 28 novel compounds were prepared according to the following Scheme 1, Scheme 2 .
Scheme 1.
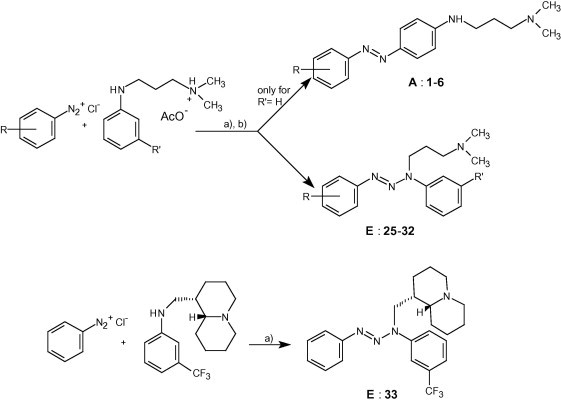
Reagents and conditions: (a) pH 7 (CH3COONa); Et2O; (b) CC (Al2O3/Et2O): E, yields 13–68%; then Et2O + 5%MeOH: A, yields 3–10%.
Scheme 2.
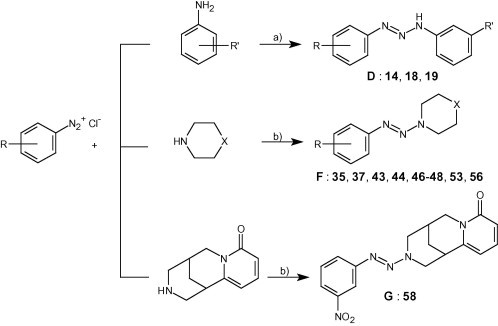
Reagents and conditions: (a) oily anilines added to the diazonium salt solution, followed by CH3COONa; yields: 30–92%; (b) pH 7–8 (1 N NaOH); yields: 25–90%.
All new compounds were characterized by elemental analyses and 1H NMR spectra.
The N-(3’-dimethylaminopropyl)-3-trifluoromethylaniline and the N-(lupinyl)-3-trifluoromethylaniline, required by Scheme 1, were already described by some of us.18
It is observed that the presence of a 3-trifluoromethyl group prevented completely the coupling of diazonium salt on the para position of the N-substituted anilines giving place only to the triazene derivatives E.
3. Results and discussion
3.1. Biological activity—general considerations
The prepared compounds (12 amino arylazocompounds A–C and 46 triazene derivatives D–G) were evaluated in vitro in parallel cell-based assays for cytotoxicity and antiviral activity (Table 3, Table 4, Table 5) against viruses representative of two of the three genera of the Flaviviridae family, that is, Flaviviruses (YFV) and Pestiviruses (BVDV), as Hepaciviruses can hardly be used in routine cell-based assays. Title compounds were also tested against representatives of other virus families. Among ssRNA+ were a Retrovirus (Human Immunodeficiency Virus type 1, HIV-1), two Picornaviruses (Coxsackie Virus type B2, CVB2, and Poliovirus type-1, Sabin strain, Sb-1); among ssRNA- were a Paramyxoviridae (Respiratory Syncytial Virus, RSV) and a Rhabdoviridae (Vesicular Stomatitis Virus, VSV) representative. Among double-stranded RNA (dsRNA) viruses was a Reoviridae representative (Respiratory Enteric Orphan Virus type-1, Reo-1). Two representatives of DNA virus families were also included: Herpes Symplex type 1, HSV-1 (Herpesviridae) and Vaccinia Virus, VV (Poxviridae).
Table 3.
Cytotoxicity against MT-4, MDBK, BHK and Vero-76 cell lines and BVDV, YFV, Reo-1, CVB-2, RSV and HSV-1 inhibitory activity of amino azocompounds of structure A–C
| Compda | R | MT-4 CC50b | MDBK CC50c | BVDV EC50d | BHK-21 CC50e | YFV EC50f | Reo-1 EC50g | VERO-76 CC50h | CVB-2 EC50i | RSV EC50j | HSV-1 EC50k |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | H | 17 | 58 | 1.6 | 31 | ⩾31 | >31 | 30 | >30 | 25 | >30 |
| 2l | 3-NO2 | 14 | 33 | 16 | 20 | 7 | >20 | 25 | 11 | >25 | 6 |
| 3 | 4-NO2 | 19 | >100 | >100 | >100 | >100 | >100 | 60 | >60 | >60 | >60 |
| 4l | 2,5-diF | 16 | 59 | 9 | 33 | 12 | >33 | 30 | >30 | >30 | 12 |
| 5 | 2,6-diF | 35 | 20 | ⩾20 | 43 | 10 | 12 | 65 | >65 | >65 | >65 |
| 6 | 4-CH3 | 17 | >100 | >100 | >100 | >100 | >100 | 50 | >50 | >50 | >50 |
| 7 | H | 31 | 48 | 2.5 | 47 | 9 | >47 | 60 | >60 | >60 | >60 |
| 8 | 3-CF3 | 77 | 52 | 7 | ⩾100 | >100 | >100 | 80 | >80 | >80 | >80 |
| 9 | H | 100 | >100 | 30 | >100 | ⩾100 | >100 | 80 | >80 | >80 | >80 |
| 10 | 3-CF3 | 15 | 30 | 7 | 14 | >14 | >14 | 50 | >50 | >50 | >50 |
| 11 | 2,5-diF | 40 | 64 | >64 | 38 | >38 | >38 | 80 | >80 | >80 | >80 |
| 12 | 2,4-diF | 51 | 11 | >11 | 9 | >9 | >9 | 15 | >15 | >15 | >15 |
| NM 108 (2′-C-methylguanosine) | >100 | >100 | 1.7 | 90 | 1.8 | 2.4 | >100 | 20 | >100 | >100 | |
| NM 299 (6-azauridine) | 2 | >100 | >100 | >100 | 26 | >100 | 20 | >20 | 1.2 | >20 | |
| ACG (acycloguanosine) | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 3 | |
| Ribavirin | 31 | >100 | 7 | >100 | >100 | >100 | >100 | >100 | 7 | >100 | |
| NM 176 (2′-C-ethynylcytidine) | ⩾100 | >100 | 38 | >100 | >100 | >100 | >100 | 24 | >100 | >100 | |
| M 5255 (mycophenolic acid) | 0.2 | 42 | >42 | >100 | >100 | >100 | ⩾13 | >13 | 0.6 | >13 | |
None of these compounds inhibited the multiplication of HIV-1, VSV, VV and Sb-1 viruses.
Compound concentration (μM) required to reduce the viability of mock-infected MT-4 (CD4+ human T cells containing an integrated HTLV-1 genome) cells by 50%, as determined by the MTT method.
Compound concentration (μM) required to reduce the viability of mock-infected MDBK (Bovine normal kidney) cells by 50%, as determined by the MTT method.
Compound concentration (μM) required to achieve 50% protection of MDBK cells from BVDV (Bovine Viral Diarrhea Virus) induced cytopathogenicity, as determined by the MTT method.
Compound concentration (μM) required to reduce the viability of mock-infected BHK (Hamster normal kidney fibroblast) monolayers by 50%, as determined by the MTT method.
Compound concentration (μM) required to achieve 50% protection of BHK cells (kidney fibroblast) from YFV (Yellow Fever Virus) induced cytopathogenicity, as determined by the MTT method.
Compound concentration (μM) required to achieve 50% protection of BHK cells (kidney fibroblast) from Reo-1 induced cytopathogenicity, as determined by the MTT method.
Compound concentration (μM) required to reduce the viability of mock-infected VERO-76 (monkey normal kidney) monolayers by 50%.
Compound concentration (μM) required to reduce the plaque number of CVB-2 (Coxsackie Virus B 2) by 50% in VERO-76 monolayers.
Compound concentration (μM) required to reduce the plaque number of RSV (Respiratory Syncytial Virus) by 50% in VERO-76 monolayers.
Compound concentration (μM) required to reduce the plaque number of HSV-1 (Herpes Simplex virus, Type-1) by 50% in VERO-76 monolayers.
Tested as hydrochloride.
Table 4.
Cytotoxicity against MT-4, MDBK, BHK and Vero-76 cell lines and BVDV, YFV, Reo-1, CVB-2, RSV, HSV-1 and Sb-1 inhibitory activity of 1,3-diaryltriazenes of structure D and E
| Compda | X | R | R′ | MT-4 CC50b | MDBK CC50c | BVDV EC50d | BHK-21 CC50e | YFV EC50f | Reo-1 EC50g | VERO-76 CC50h | CVB-2 EC50i | RSV EC50j | HSV-1 EC50k | Sb-1 EC50l |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 13 | H | H | H | 47 | >100 | >100 | >100 | >100 | >100 | 90 | >90 | ⩾90 | >90 | >90 |
| 14 | H | H | 3-CF3 | >100 | >100 | >100 | >100 | ⩾100 | >100 | 15 | 8 | 3 | >15 | >15 |
| 15 | H | H | CH2-Phm | 42 | >100 | >100 | >100 | >100 | >100 | 100 | >100 | >100 | >100 | >100 |
| 16 | H | 3-NO2 | H | 40 | 94 | 19 | 38 | >38 | >38 | 33 | >33 | >33 | >33 | >33 |
| 17 | H | 3-NO2 | 3-NO2 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 18 | H | 2,5-diF | H | 100 | >100 | >100 | >100 | >100 | >100 | 80 | 12 | 10 | 55 | >80 |
| 19 | H | 2,5-diF | 3-CF3 | >100 | >100 | >100 | >100 | 80 | >100 | 80 | >80 | 10 | >80 | >80 |
| 20 | H | 2,5-diF | 2,5-diF | ⩾100 | >100 | >100 | >100 | >100 | >100 | ⩾100 | 50 | 8 | >100 | >100 |
| 21 | CH3 | H | H | 55 | >100 | >100 | >100 | >100 | >100 | 50 | >50 | 20 | >50 | >50 |
| 22 | CH3 | H | CH2-Phm | ⩾100 | >100 | >100 | >100 | >100 | >100 | ⩾100 | >100 | >100 | >100 | >100 |
| 23 | CH3 | 3-NO2 | H | >100 | 82 | >82 | >100 | >100 | 33 | >100 | >100 | 15 | >100 | 29 |
| 24 | CH3 | 4-CH3 | H | 45 | >100 | >100 | >100 | >100 | >100 | 58 | >58 | >58 | >58 | >58 |
| 25 | (CH2)3-NMe2 | H | H | 33 | 95 | ⩾95 | 50 | >50 | >50 | 35 | 6 | >35 | 12 | >35 |
| 26 | (CH2)3-NMe2 | H | 3-CF3 | 14 | 38 | >38 | 49 | ⩾49 | >49 | 20 | ⩾20 | >20 | 14 | >20 |
| 27 | (CH2)3-NMe2 | 3-NO2 | H | 14 | 37 | >37 | 54 | 24 | >54 | 40 | >40 | >40 | 30 | >40 |
| 28 | (CH2)3-NMe2 | 4-NO2 | H | 20 | 39 | ⩾39 | 36 | ⩾36 | >36 | 40 | >40 | >40 | >40 | >40 |
| 29 | (CH2)3-NMe2 | 2,5-diF | H | 13 | 25 | >25 | 25 | >25 | >25 | 7 | >7 | >7 | >7 | >7 |
| 30 | (CH2)3-NMe2 | 2,5-diF | 3-CF3 | 16 | >100 | 90 | 54 | >54 | >54 | 30 | >30 | >30 | >30 | >30 |
| 31 | (CH2)3-NMe2 | 2,6-diF | H | 23 | 45 | >45 | 52 | 20 | >52 | 25 | 9 | ⩾25 | 12 | >25 |
| 32 | (CH2)3-NMe2 | 4-CH3 | H | 18 | >100 | >100 | >100 | >100 | >100 | 100 | >100 | >100 | >100 | >100 |
| 33 | Lupinyln | H | 3-CF3 | 17 | 22 | >22 | 40 | 23 | >40 | 75 | >75 | >75 | >75 | >75 |
| NM 108 | >100 | >100 | 1.7 | 90 | 1.8 | 2.4 | >100 | 20 | >100 | >100 | >100 | |||
| NM 299 | 2 | >100 | >100 | >100 | 26 | >100 | 20 | >20 | 1.2 | >20 | >20 | |||
| ACG | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 3 | >100 | |||
| Ribavirin | 31 | >100 | 7 | >100 | >100 | >100 | >100 | >100 | 7 | >100 | >100 | |||
| NM 176 | ⩾100 | >100 | 38 | >100 | >100 | >100 | >100 | 24 | >100 | >100 | 20 | |||
| M 5255 | 0.2 | 42 | >42 | >100 | >100 | >100 | ⩾13 | >13 | 0.6 | >13 | >13 |
None of compounds 13–33 inhibited the multiplication of HIV-1, VSV and VV viruses.
For the meaning see Table 3.
Compound concentration (μM) required to reduce the plaque number of Sb-1 (Poliovirus Type-1, Sabin strain) by 50% in Vero-76 monolayers.
In compounds 15 and 22 the R′-substituted phenyl ring is replaced by a benzyl residue.
(1S,9aR-Octahydro-2H-quinolizin-1-yl)methyl.
Table 5.
Cytotoxicity against MT-4, MDBK, BHK and Vero-76 cell lines and YFV, Reo-1, CVB-2, RSV, HSV-1 and Sb-1 inhibitory activity of triazene derivatives of structure F and G
| Compda | X | R | MT-4 CC50b | MDBK CC50c | BHK-21 CC50e | YFV EC50f | Reo-1 EC50g | VERO-76 CC50h | CVB-2 EC50i | RSV EC50j | HSV-1 EC50k | Sb-1 EC50l |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 34 | Nil | H | >100 | >100 | ⩾100 | >100 | >100 | >100 | >100 | ⩾100 | >100 | >100 |
| 35 | Nil | 3-Cl | >100 | >100 | >100 | >100 | >100 | ⩾100 | >100 | >100 | >100 | ⩾100 |
| 36 | Nil | 3-Br | >100 | >100 | >100 | >100 | >100 | ⩾100 | >100 | >100 | >100 | >100 |
| 37 | Nil | 3-CF3 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 38 | Nil | 3-NO2 | 66 | >100 | 42 | >42 | >42 | 80 | >80 | 8 | >80 | 30 |
| 39 | Nil | 4-Cl | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 42 |
| 40 | Nil | 4-Br | >100 | >100 | >100 | >100 | >100 | ⩾100 | >100 | >100 | >100 | 51 |
| 41 | Nil | 4-NO2 | ⩾100 | >100 | ⩾100 | >100 | >100 | 90 | >90 | 60 | >90 | 15 |
| 42 | Nil | 4-OCH3 | 68 | 60 | ⩾100 | >100 | >100 | 90 | >90 | >90 | >90 | >90 |
| 43 | Nil | 2,5-diF | >100 | >100 | >100 | >100 | >100 | >100 | 35 | >100 | >100 | 44 |
| 44 | Nil | 3,4-diCl | 75 | 70 | 50 | >50 | >50 | 58 | >58 | 20 | >58 | 25 |
| 45 | CH2 | H | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 46 | CH2 | 3-Cl | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 47 | CH2 | 3-Br | >100 | >100 | >100 | >100 | >100 | 85 | >85 | >85 | >85 | >85 |
| 48 | CH2 | 3-NO2 | >100 | >100 | >100 | >100 | >100 | 83 | >83 | >83 | >83 | >83 |
| 49 | CH2 | 4-Cl | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 50 | CH2 | 4-Br | >100 | >100 | >100 | >100 | >100 | >100 | 25 | 73 | >100 | >100 |
| 51 | CO | H | >100 | ⩾100 | 90 | >90 | >90 | 60 | 13 | >60 | >60 | >60 |
| 52 | N-CH3 | H | 65 | >100 | 70 | >70 | >70 | 45 | 14 | >45 | >45 | >45 |
| 53 | N-CH3 | 2,5-diF | >100 | >100 | >100 | 50 | 30 | ⩾100 | 71 | >100 | 64 | >100 |
| 54 | N-COOC2H5 | H | 100 | >100 | >100 | >100 | >100 | 90 | 30 | >90 | >90 | >90 |
| 55 | N-Ph | H | 33 | >100 | >100 | >100 | >100 | ⩾100 | >100 | 8 | >100 | >100 |
| 56 | N-2-Pyrimidinyl | H | 90 | >100 | >100 | >100 | >100 | 65 | 10 | >65 | >65 | >65 |
| 57 | 4-Cl | 46 | 97 | 85 | >85 | >85 | 55 | >55 | >55 | >55 | >55 | |
| 58 | 3-NO2 | >100 | >100 | >100 | >100 | 80 | 90 | >90 | ⩾90 | >90 | >90 | |
| NM-108 | >100 | >100 | 90 | 1.8 | 2.4 | >100 | 20 | >100 | >100 | >100 | ||
| NM 299 | 2 | >100 | >100 | 26 | >100 | 20 | >20 | 1.2 | >20 | >20 | ||
| ACG | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 3 | >100 | ||
| Ribavirin | 31 | >100 | >100 | >100 | >100 | >100 | >100 | 7 | >100 | >100 | ||
| NM 176 | ⩾100 | >100 | >100 | >100 | >100 | >100 | 24 | >100 | >100 | 20 | ||
| M 5255 | 0.2 | 42 | >100 | >100 | >100 | ⩾13 | >13 | 0.6 | >13 | >13 | ||
AZT (3′-azido-thymidine), NM 108 (2′-C-methyl-guanosine), NM 176 (2′-C-ethynyl-cytidine), Ribavirin, NM 299 (6-azauridine), M 5255 (mycophenolic acid) and ACG (acyclovir) were used as reference inhibitors of ssRNA+, ssRNA− and DNA viruses, respectively.
Interestingly, 35 (8 aminoazocompounds and 27 triazene derivatives) over 58 tested compounds exhibited antiviral activity against one or more viruses; in particular 16 compounds exhibited a selective activity against a single virus, while 13, 4 and 2 were, respectively, active against two, three and four viruses. On the other hand, 23 compounds (4 aminoarylazo and 19 triazene derivatives) were not able to inhibit the multiplication of any virus at concentrations up to 100 μM.
None of the active compounds inhibited the multiplication of HIV-1, VSV and VV, but an increasing number of them exhibited antiviral activity against, in the order, Reo-1 (4), Sb-1 (7), HSV-1 (8), BVDV (9), YFV (9), RSV (12) and CVB-2 (13) (Table 1, Table 2 ). Nineteen compounds have shown an EC50 ⩽ 10 μM, 28 had EC50 between 11 and 30 μM and only 15 had EC50 in the range 31–90 μM.
Table 1.
Number of active compounds of structures A–C on susceptible viruses and range of their EC50
| Virusa | No. of active aminoazocompounds (A–C) over 12 tested compoundsb | No. of active compounds (range of EC50, μM) | ||
|---|---|---|---|---|
| Reo-1c | 1 | / | 1 (12) | / |
| HSV-1d | 2 | 1 (6) | 1 (12) | / |
| BVDVe | 7 | 5 (1, 6–9) | 1 (16) | 1 (30) |
| YFVe | 4 | 3 (7–10) | 1 (12) | / |
| RSVf | 1 | / | / | 1 (25) |
| CVB-2e | 1 | / | 1 (11) | / |
HIV-1, VSV, VV and Sb-1 were unaffected by all tested compounds of structure A–C.
Compounds with EC50 > 100 μM, or higher than CC50 for the host cells are considered inactive.
Double stranded RNA virus.
DNA virus.
Single-stranded, positive RNA virus.
Single-stranded, negative RNA virus.
Table 2.
Number of active compounds of structures D–G on susceptible viruses and range of their EC50
| Virusa | No. of active triazene derivatives (D–G) over 46 tested compoundsb | No. of active compounds (range of EC50, μM) | ||||
|---|---|---|---|---|---|---|
| Reo-1c | 3 | / | / | 1 (30) | 1 (33) | 1 (80) |
| Sb-1d | 7 | / | 1 (15) | 3 (25–30) | 2 (42, 44) | 1 (51) |
| HSV-1e | 6 | / | 3 (12–14) | 1 (30) | / | 2 (55, 64) |
| BVDVd | 2 | / | 1 (19) | / | / | 1 (90) |
| YFVd | 5 | / | 1 (20) | 2 (23, 24) | 1 (50) | 1 (80) |
| RSVf | 11 | 6 (3–10) | 3 (15–20) | / | / | 2 (60, 73) |
| CVB-2d | 12 | 4 (6–10) | 3 (12–14) | 2 (25, 30) | 2 (35, 50) | 1 (71) |
HIV-1, VSV and VV were unaffected by all tested compounds of structure D–G.
Compounds with EC50 > 100 μM, or higher than CC50 for the host cells are considered inactive.
Double stranded RNA virus.
Single-stranded, positive RNA virus.
DNA virus.
Single-stranded, negative RNA virus.
Therefore, both the [(dialkylaminoalkyl)amino]azobenzene and the aryltriazene molecular patterns represent interesting pharmacophore for developing novel antiviral agents, particularly against ssRNA viruses.
Cytotoxicity and antiviral activities of tested and reference compounds are reported in Table 3, Table 4, Table 5 . In these Tables, viruses for which no active compounds have been found are not indicated.
3.2. Cytotoxicity on host cells
Test compounds showed different degrees of cytotoxicity against the confluent cell monolayers (in stationary growth) used to support the multiplication of the different viruses.
The most susceptible to toxicity were the exponentially growing lymphoblastoid human cells (MT-4) used to grow HIV-1, while the non-human host cell lines exhibited a progressively reduced sensitivity in the order Vero-76 > BHK > MDBK. On the other hand, it is observed that the toxicity is not evenly distributed among the different types of test compounds, being mainly shown by the triazene derivatives of structure E and by the amino azo compound A–C, which are characterized by the presence of the basic dimethylaminopropyl and lupinyl moieties. Indeed the most toxic compounds were 29 and 12, with a mean of the CC50 values for the host cells of 17.5 and 21.5 μM, respectively.
The other groups of triazene derivatives of structure D, F and G are relatively non toxic, exhibiting in most cases CC50 ⩾ 100 μM.
3.3. Structure–activity relationships
As already seen, eight [(dialkylaminoalkyl)amino]azobenzenes (A–C) and 27 triazene derivatives (D–G) have shown antiviral activity, sometimes of high level against one or more viruses.
From Table 1, Table 3 appears that compounds of structure A–C have a marked activity against BVDV and YFV, with only occasional, though still of high level, activity against HSV-1, Reo-1, CVB-2 and RSV.
On the whole the triazene derivatives D–G are characterized by the most frequent activity against CVB-2 and RSV, to which are associated, with decreasing frequency, the activity on Sb-1, HSV-1, YFV, Reo-1 and BVDV (Table 2, Table 4, Table 5).
However, considering separately each subgroup of triazene derivatives, it is observed that the activity against Sb-1 is mainly associated to the pyrrolidine-related triazenes F, while activity against HSV-1 is associated with the diaryltriazenes that bear a dimethylaminopropyl chain. The importance of a basic chain for the activity against HSV-1 is underlined by the high activity shown by compounds 2 and 4, bearing the same dimethylaminopropyl chain, even linked to the aminoazobenzene pharmacophore (A), but also by the moderate activity of triazene 53, which contains a basic N-methylpiperazine residue.
Conversely it is worth noting that the activity against BVDV, largely diffused among the amino azo compounds A–C, is very rarely present (2 over 46 compounds) among the triazene derivatives D–G, even among those (E) sharing the basic chain of the former groups.
Active and inactive compounds are found side by side in all groups A–G, thus their molecular patterns seem to address the activity against the different type of viruses, while the substituents on the aromatic nucleus are the determinants for activity or inactivity. However, attempts to define general structure-activity relationships resulted rather frustrating, since observations that hold for the activity against most viruses may not hold for some others.
Indeed the meta substitution with halogens, trifluoromethyl and nitro groups favors the display of activity in the groups A–E and G, but in compounds of group F is the para substitution that promotes the activity; however occasional exceptions are observed in both cases.
An unsubstituted aromatic ring can be found in active (1, 7, 9, 21, 25, 52, 54–56) and inactive (13, 34, 45) compounds, while the presence of electron-releasing groups as CH3 and OCH3 (32, 42) or of a benzyl residue (15, 22) is always detrimental for the expression of activity.
Despite the high antiviral activity of many compounds, the corresponding selectivity index (S. I.) are generally low, due to the rather high cytotoxicity exerted on the host cells. Only few compounds exhibited a S. I. higher than 10 for one or more viruses, while 13 compounds had a S. I. in the range from 9 to 5 and the remaining had S. I. below this value.
Taking into account the EC50 and S. I. values, the best compounds were the following, which are arranged in the order of decreasing potency and selectivity; BVDV: 1 > 7 > 8 > 4; YFV: 7 > 5; CVB-2: 25 > 56 > 18; RSV: 14 > 20 > 55 > 38 >18 > 19 > 23; Sb-1: 41; HSV-1: 2. For these compounds the EC50 ranged from 1.6 μM (1) to 15 μM (23, 41), while the corresponding S. I. values were in the range from 19.4 (1) to 4.2 (2).
Though the activity shown by the above compounds may appear as not particularly impressive, it must be noted that, at the present, very few experimental agents are known to act against those viruses and that, moreover, they often display high cytotoxicity.
A particular interest is deserved by compounds displaying good activity against RSV and BVDV. RSV, is responsible of serious respiratory tract infections in infants, elderly and immunocompromised patients with high mortality rate.
Very potent anti-RSV agents, interfering with the virus-host fusion process, have been developed in the last years, as the benzimidazole derivative BMS-433771 which exhibited an EC50 as low as 21 nM; however, attempts to demonstrate therapeutic efficacy were not successful,19 probably due to the rapid emergence of resistance.
Quite recently a chiral 1,4-benzodiazepine derivative, acting through a different mechanism, though much less potent than BMS-433771, showed very promising characteristics and is being examined in phase II clinical studies.20
Nevertheless the chirality-associated developability issue cannot be underestimate, since the antiviral activity was found to reside mainly in the S-enantiomer (RSV-604), that exhibited an EC50 = 0.9 μM, while racemate had IC50 = 3.5 μM, a value comparable with that of compound 14.
On the other hand, BVDV is the prototype of pestiviruses, which commonly affect cloven-hoofed animals (cattle, swine, sheep). BVDV infection is responsible of reduced dairy production and increased (even up to 30%) cattle mortality throughout the world.21
Thus, the availability of effective and inexpensive anti-pestivirus drugs is of importance to relieve such an heavy economic burden.
The development of anti-BVDV drugs and the understanding of their mechanism of action is of further interest because it may provide valuable informations for the design of agents active against hepatitis C virus (HCV).22 HCV, which like BVDV belongs to the Flaviviridae family, infects about 3% of world population. Since chronic HCV infection can progress to fatal cirrhosis and hepatocellular carcinoma, the development of effective and safe anti-HCV agents is urgently needed. The current combination therapy with pegylated interferon and ribavirin is effective in less than 60% of the treated patients and is associated with heavy side effects.
For the understanding of some aspects of virus replication, BVDV has been considered even more advantageous than the currently used HCV subgenomic replicon system.23
In the last years, several potent anti-BVDV agents have been developed and shown to target the RNA-dependent RNA-polymerase (RdRp), even if resulting almost inactive on the purified enzyme. Compounds VP32947 (3-[(dipropylaminoethyl)thio]-5H-1,2,4-triazino[5,6-b]indole)24 and BPIP (5-[4-bromobenzyl]-2-phenyl-5H-imidazo[4,5-c]pyridine)23 exhibited EC50 as low as 30–40 nM. Other compounds (as the thiazolylurea DPC-A69280-29,25 the benzimidazolone 145326 and 7-amino-1,3-dihydroxy-10-methyl-6-[4-(2-pyridinyl)-1-piperazinyl]-9(10H)acridone27) have EC50 in the range 0.6–1.5 μM, that are comparable with those of the presently described aminoaryl azocompounds 1 and 7 (1.6 and 2.5 μM, respectively). The structural simplicity of these compounds, which allows a wide range of easy and inexpensive molecular modifications, make them an attractive model to develop more active and less toxic anti-pestivirus agents.
In view of these considerations, we deemed interesting to perform some molecular modeling investigations to study wether the BVDV RdRp could be the target of also our compounds 1, 2, 4 and 7–10.
3.4. Modeling of the A–C series of compounds versus BVDV RdRp
BVDV, the best-studied pestivirus, has a genome that consists of an approximately 12.6-kb positive sense ssRNA. The BVDV genome is translated into a single polyprotein which is processed into at least four structural and six nonstructural (NS) proteins required for viral assembly and replication. Among the nonstructural proteins, the NS5B is an RNA-dependent RNA-polymerase (RdRp) enzyme responsible for genome replication as a part of a larger, membrane associated replicase complex. The crystal structure of RdRp from several families of single- or double-strand RNA viruses, including HCV28, 29, 30 and BVDV,31, 32 have been recently made available in the Protein Data Bank (PDB) repository.
Despite the low sequence identity between different polymerases, the crystal structures of these proteins (from BVDV, HCV and other families of ss and dsRNA viruses) all present the shape of a right hand with fingers, palm, and thumb domains. In particular, the BVDV RNA-dependent RNA-polymerase core domain (residues 139–679) has a dimension of approximately 74 × 60 × 58 Å around a central cavity,31 which serves as for RNA template binding, nucleotides recruitment, and polymerization reaction (see Fig. 4 a). In addition, there is an N-terminal region (residues 71–138) of which residues 71–91 are disordered in the relevant crystal structure. A thorough search of a putative binding site for our molecules onto BVDV RdRp was conducted following our successful recipe developed for studying allosteric inhibitors of Polio-virus helicase.33 The portion of the enzyme making up the binding site interacting with the inhibitors is located in the fingers domain (residues 139–313 and 351–410), consisting of 12 α-helices and 11 β-strands (see Fig. 4a).
Figure 4.

(a) Overview of the entire structure of the RdRp of BVDV. The protein domains are colored as follows: pink, N-terminal domain (residues 71–138); light green, fingers domain (residues 139–313 and 351–410); palm domain, kaki (residues 314–350 and 411–500); sienna, thumb domain (residues 501–679). (b) Overlay of the 3D models of RdRp of BVDV (purple) and HCV (red). (c) BVDV and HCV polymerase amino acid alignment: top line, BVDV (residues 92–679); bottom line, HCV (residues 1–531). The alignment of the fingers domain is highlighted in purple (BVDV) and red (HCV).
In BVDV RdRp, as in other viral RdRps, the N terminus of the fingers domain, together with a long insert in the fingers domain (residues 260–288), form the fingertip region that associates with the thumb domain. This region is characterized by a three-strand conformation, and since the fingers and the thumb domains are associated through this fingertip region, the conformational change induced by the RNA template binding into the central channel is somewhat limited. The remainder of the fingers domain is comprised of a β-strand rich region (β-fingers) and an α-helix rich region (α-fingers) close to the palm domain.
According to the procedure adopted, all compounds were characterized by a similar docking mode in the putative binding site of the BVDV RdRp, as exemplified by compound 1 in Figure 5 .
Figure 5.
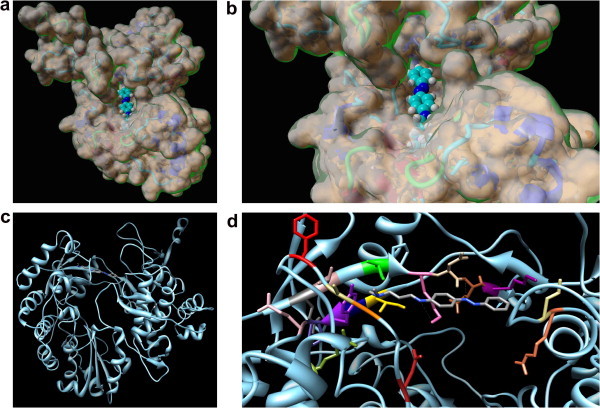
Overall (a, top left) and detailed (b, top right) space filling representation of the BVDV RdRp molecular surface and compound 1 docked into the protein putative binding site. The inhibitor is in CPK representation, with carbons in cyan, nitrogens in blue, and hydrogens in white. (c, bottom left) Ribbon diagram of BVDV RdRp/1 complex structure as resulting from the applied docking/MD procedure. The protein is colored light blue. The inhibitor 1 is represented as a stick model with carbons in gray and nitrogens in blue. (d, bottom right) Details of compound 1 (in a stick representation) in the binding pocket in the enzyme fingers domain. Color scheme as above. The side chains of all residues that form the primary binding pocket interacting with compound 1 are shown as stick models, and the atom color-coding is as follows: N217, firebrick; A221, orange; A222, dark kaki; F224, red; E258, dim gray; T259, rosy brown; I261, green; K263, hot pink; N264, tan; E265, sienna; K266, dark magenta; I287, gold; Q288, navy blue; Y289, purple; P290, dark slate blue; E291, pink; R295, olive drab; R529, coral; L530, kaki. Hydrogen bonds are highlighted as light gray broken lines. Hydrogen atoms, counterions, and water molecules are omitted for clarity.
In particular, the residues lining the pocket (see Fig. 5d) include the side chains of residues from N217 to F224 belonging to a loop between the two strands β3 and β4, and those of residues from K263 to E265 of a loop between strands β5 and β6. Amino acids from I287 to E291 contribute to binding from the β-sheet portion β8, whilst another loop, connecting two α-helical motifs (α19 and α20, respectively), concurs with residues from R529 and L530.
Going into details, the aminoalkyl part of the molecule is engaged in stabilizing nonbonded interactions with the apolar side chains of residues I261, I287, A221, and A222, while the diarylazo moiety is nicely encased in a subsite lined by the side chains of N217, N264, E265, K266, R529, and L530. More importantly, however, this molecular scaffold is anchored in place by three persistent hydrogen bonds (HBs). A first HB bridge involves the nitrogen atom of the N(CH3)2 substituent group on 1 and the –OH group of Y289 side chain, with an average dynamic length (ADL) of 3.08 ± 0.3 Å. The second HB interaction engages the terminal amino group of K263 and the N atom of the inhibitor secondary amino group (ADL = 3.18 ± 0.2 Å). Finally, one of the nitrogen atoms belonging to the azo group characterizing compound 1 is involved in the third HB interaction with the backbone –NH group of E265 (ADL = 2.92 ± 0.3 Å).
Importantly, quantitative information about the affinities of our ligands towards BVDV RdRp can be inferred by applying the MM/PBSA methodology to estimate the free energy of binding, ΔG bind, and its components. The calculated ΔG bind values for the dimethylaminopropyl derivatives 1, 2, 4, and the quinolizidinylmethyl derivatives 7–10 are listed in Table 6 . Generally speaking, and in harmony with our previous findings,33, 34 both the nonbonded mechanical energy components of ΔG bind, ΔE vdW and ΔE el, afford a substantial, favorable contribution to binding for both series of compounds. On the other hand, due the polar character of the azo group, the desolvation penalty paid by these molecules upon binding (ΔG PB) is also quite substantial, so that the net, resulting electrostatic contribution to the affinity of these inhibitors to their enzyme receptor are notably unfavorable. Specifically, for this series of compounds, the mean value of the electrostatic energy (ΔE el + ΔG PB) is 26 kcal/mol, whilst the corresponding mean values of the van der Waals and hydrophobic overall interaction energies (ΔE vdW + ΔG np) are −46 kcal/mol.
Table 6.
Free energy components and total binding free energies for compounds of the series A–C on BVDV RdRp
| 1 | 2 | 4 | 7 | 8 | 9 | 10 | |
|---|---|---|---|---|---|---|---|
| −40.2 ± 0.3 | −38.6± 0.2 | −39.5 ± 0.4 | −39.5 ± 0.3 | −39.8 ± 0.2 | −39.3 ± 0.3 | 39.4 ± 0.2 | |
| −32.5 ± 0.2 | −34.2 ± 0.3 | −33.9 ± 0.3 | −29.3 ± 0.3 | −29.8 ± 0.1 | −28.7 ± 0.3 | −29.4 ± 0.3 | |
| −72.7 ± 0.2 | −72.8 ± 0.2 | −73.4 ± 0.3 | −68.8 ± 0.3 | −69.6 ± 0.2 | −68.0 ± 0.2 | −68.8 ± 0.3 | |
| 57.5 ± 0.5 | 60.0 ± 0.4 | 52.6 ± 0.4 | 59.6 ± 0.3 | 55.7 ± 0.4 | 55.3± 0.4 | 55.6 ± 0.3 | |
| −6.3± 0.0 | −7.1 ± 0.0 | −6.7 ± 0.1 | −9.1 ± 0.0 | −8.7 ± 0.1 | −7.9 ± 0.1 | −7.8 ± 0.0 | |
| 51.2 ± 0.6 | 52.9 ± 0.4 | 52.9 ± 0.5 | 46.6 ± 0.2 | 48.4 ± 0.4 | 47.4 ± 0.3 | 47.8 ± 0.3 | |
| −21.5 ± 0.5 | −19.9 ± 0.4 | −20.5 ± 0.3 | −22.2 ± 0.2 | −21.2 ± 0.3 | −20.7± 0.4 | −21.0 ± 0.3 | |
| TΔSsolute | 14.3 | 14.0 | 14.3 | 15.2 | 14.8 | 15.0 | 14.7 |
| ΔGbind | −7.2 | −5.9 | −6.2 | −7.0 | −6.4 | −5.7 | −6.3 |
| IC50a | 5 | 38 | 28 | 7 | 22 | 66 | 25 |
Accordingly, it follows that the association between the ligands and the RdRp is mainly driven by more favorable nonpolar interactions in the complex than in the solution, in harmony with a proposed general scheme for noncovalent association.33, 34 However, as indicated for instance by the energy components of compound 2, this driving force can be weakened when the polar groups on the molecule do not find an adequate bonding pattern in the protein compared to water. The free energy penalty for this (ΔE el + ΔG PB) term is least for compound 1 and this, together with its substantial van der Waals contribution and moderate entropic unfavorable term, consequently leads to one of the highest binding affinity in this set of inhibitors.
One of the most important benchmark in this study, however, is the correspondence between the estimated free energies of binding and the experimental measured EC50 values. Indeed, we can observe a good agreement between the trend exhibited by the IC50 values reported in Table 6 and the corresponding biological activity determined for these compounds in BVDV infected cell line (see Table 3). Although we obviously cannot directly compare the computed binding free energy (and hence the corresponding IC50) with the EC50 values deriving from experiment, we can observe that, in most cases, the rank of the inhibitors with respect to their activity towards their putative targets, the RdRps of BVDV, is maintained, although with some discrepancies. In fact, all estimated IC50 values are systematically higher than those determined by in vitro experiments with infected cell lines.
The overall results obtained from molecular modeling deserve some more comments. First of all, the binding site identified by our procedure is very close to the putative binding site proposed for two allosteric inhibitors of BVDV RdRp, VP32947 and BPIP,23 and involves residue F224 (see Fig. 5d), a residue found mutated into S224 in BVDV BPIP-resistant clones. The binding pocket is located in a turn of the fingers domain between two β-sheets which is believed to be involved in finger flexibility for RNA template/product translocation,35 dimerization of the RdRp in the replication complex, or protein/protein interactions,36, 37 enabling the assembly of an active replication complex. According to our docking results, then, the hypothetical binding of our compounds to the polymerase could affect the finger flexibility or impair the capacity of the protein to translocate its template/product during RNA polymerization. Alternatively, the diarylazo moiety could possibly act by hampering the entrance of the template RNA in the template binding cavity.
Although our in silico data clearly show the same trend of the corresponding experimental EC50 values, at the same time they yield no evidence that binding of these inhibitors onto the BVDV RdRp result in a real allosteric inhibition of the enzyme or in a reduced template-binding ability. One of the explanations why we find IC50 values higher than the corresponding EC50 could be explained by the following rationale. Recently an attempt of co-crystallize the BVDV RdRp with the inhibitor VP32947 failed do to a dimer interface near the putative binding site of VP32947 in the BVDV RdRp crystal. The authors of the work proposed that the formation of this dimer could constitute an important point in the replication complex. Moreover, it was also hypothesized that the top of the fingers domain could be a protein-docking site pivotal to the interactions with other enzymes of the replicase complex. As the VP32947 binding site is part of the putative binding pocket for our molecular series, it could be that, upon binding, our compounds hinder the interactions among different proteins making up the replicase complex.
Last but not least, it is important to recall that RdRp functions in virus-infected cells in the context of membrane-bound replication complexes, that usually consist of several virus-encoded proteins, host proteins, and various forms of viral RNA. The RNA-dependent RNA polymerase may then have a role in interacting with these other components and/or in the stability of the replication complexes. If one or more of these putative actions of the RdRp in infected cells is sensitive to our inhibitors, viral RNA synthesis may be disrupted in a manner which is distinct from the one estimated by our in silico assays.
Now, given the similarity among the polymerases of pestiviruses, hepaciviruses, and flaviviruses, any knowledge gained from any one of these viral systems is likely to foster progress in the others. Given the high importance of the hepatitis C pandemic and the stringent need for efficient therapeutics for this pathology, it is expected that new molecular entities such as those described in this work can provide meaningful insights towards the identification of possible anti-HCV agents. Interestingly, the overall sequence identity between BVDV and HCV RdRp proteins is quite low, approximately 30%. If we consider the alignment of BVDV and HCV RdRp reported in Figure 4c, and in particular focus on the highlighted fingers domain part, we can immediately speculate that: (i) the F224 residue in the BVDV protein has no counterpart in the corresponding HCV enzyme; accordingly, if the drug would bind in the same binding pocket with the same binding mode, there should in principle be no problem of resistant mutant at this position. (ii) The important region of K263–K266 in the BVDV RdRp is highly conserved in the corresponding HCV polymerase (KNEK in BVDV and KNEV in HCV, respectively, see Fig. 4c); importantly, both K263 and E265 are engaged in fundamental stabilizing hydrogen bonds with the inhibitors in the BVDV case but, being conserved, they are able to establish the same interactions with the compounds also in the case of HCV. (iii) The residue Y289 of BVDV RdRp is not conserved in the corresponding HCV polymerase, being replaced by a phenylalanine. Therefore, the anchoring hydrogen bond between the Y289 –OH group and, for instance, the tertiary nitrogen of 1 can no longer take place. Taken all together, these evidences can justify a minor potency of this molecular series towards the HCV RdRp as a target with respect to the BVDV counterpart. At the same time, in the case of both viruses, the binding pockets on the respective RdRps do share a sufficient degree of structural and sequence homology; thus, since these regions involve the fingers domains, which overhang the palm domains of the enzymes, they might be involved in RNA substrate recognition and, hence, the inhibitor molecules may affect the RdRps interactions with the incoming RNA molecule.
4. Conclusions
Twelve aminoarylazocompounds of structures A–C, and 46 aryltriazene derivatives D–G have been synthesized and assayed for antiviral activity against a panel of 10 RNA and DNA viruses.
Thirty-five (8 aminoazocompounds and 27 triazene derivatives) over 58 tested compounds exhibited antiviral activity against one or more viruses and many of them had shown an EC50 ⩽ 10 μM against at least one virus. None of the active compounds inhibited the multiplication of HIV-1, VSV and VV.
Aminoazocompounds A–C have a marked activity against BVDV and YFV, while the triazene derivatives D–G are characterized by the most frequent activity against CVB-2 and RSV.
Despite the high antiviral activity of many compounds, the corresponding selectivity index (S. I.) are generally low, due the rather high cytotoxicity exerted on the host cell lines.
Taking into account the EC50 and S. I. values, the best compounds are the following, arranged in order of decreasing potency and selectivity; BVDV: 1 > 7 > 8 > 4; YFV: 7 > 5; CVB-2: 25 > 56 > 18; RSV: 14 > 20 > 55 > 38 > 18; HSV-1: 2. For these compounds the EC50 ranged from 1.6 μM (1) to 12 μM (18), while the corresponding S. I. values were in the range from 19.4 (1) to 4.2 (2).
A particular interest is deserved by compounds displaying good activities against RSV (as 14 and 20) and BVDV (as 1 and 7). The former virus is responsible of serious respiratory tract infections in humans, with high mortality rate, while the latter is responsible of severe epidemic outbreaks in livestock, but is also a surrogate model for the evaluation of novel, urgently needed agents against HCV, an emerging threat to human health worldwide.
The above compounds represent valuable starting models for developing better (more potent and less toxic) and inexpensive antiviral agents through the variation of the dialkylaminoalkyl chain and/or the substituents on the aromatic rings.
In view of these considerations, molecular modeling investigations were performed to study wether the active compounds of structures A-C could target the BVDV RNA-dependent RNA-polymerase (RdRp), which shares some structural similarity with HCV RdRp.
Indeed a good agreement was observed between the trend exhibited by the IC50 (calculated from the estimated free energies of binding) and the corresponding biological activities determined for these compounds in BVDV infected MDBK cell line.
The overall sequence identity between BVDV and HCV RdRp proteins is approximately 30%, but the important region K263-K266 in the BVDV RdRp is highly conserved in the corresponding HCV polymerase, thus targeting of HCV RdRp by the A-C molecular series could be expected, even if with a minor potency in respect to the BVDV counterpart.
5. Experimental
5.1. General
Chemicals, solvents and reagents used for syntheses were purchased from Sigma–Aldrich, Fluka or Lancaster, and were used without any further purification.
Column chromatography (CC): neutral alumina (Al2O3), activity 1 (Merck). Mps: Büchi apparatus, uncorrected. 1H NMR spectra: Varian Gemini-200 spectrometer; CDCl3; δ in ppm rel. to Me4Si as internal standard. J in hertz. Elemental analyses were performed on a Carlo Erba EA-1110 CHNS-O instrument in the Microanalysis Laboratory of the Department of Pharmaceutical Sciences of Genoa University.
5.2. Aminoazocompounds of structure A and triazenes of structure E. General method
A solution of arylamine (8 mmol) in 1.56 mL of conc. hydrochloridric acid and 1 mL of water was cooled to 0 °C and slowly diazotized with a solution of sodium nitrite (8 mmol) in 2 mL of water. The diazonium salt solution was added dropwise, during about 10–20 min, to a cold water solution of the N-(3-dimethylaminopropyl/lupinyl)aniline monoacetate (8 mmol), and maintaining carefully pH 7 with the simultaneous addition of a saturated solution of sodium acetate. After stirring the reaction mixture for 45 min at 0–5 °C, the oil product was extracted with Et2O, dried, then vacuum-distilled (0.1–0.2 torr) at 80–90 °C to remove the unreacted N-substituted aniline. The residue was fractionated by CC, eluting firstly with Et2O the triazene derivative (E) and then with Et2O + 5%MeOH the isomeric azocompound (A) in quite lower yield.
The isolated compounds were further purified either by crystallization or, when oily, by a second CC or distillation.
5.2.1. [N,N-Dimethyl-N′-(4-phenylazophenyl)]propane-1,3-diamine (1)
Yield: 6%. Oil (CC: Al2O3, Et2O). Bp (p = 0.1–0.2 torr) 100–105°. TLC: R f = 0.4 (Al2O3, Et2O + 2%Et2NH). 1H NMR (CDCl3): 1.74 (q, 2H, C(2)); 2.20 (s, N(CH3)2); 2.37 (t, J = 6.2, 2H, C(3)); 3.24 (t, J = 7.0, 2H, C(1)); 5.19 (s, NH); 6.58 (d, J = 10.0, 2 arom. H); 7.28–7.48 (m, 5 arom. H); 7.75 (d, J = 10.0, 2 arom. H). Anal. Calcd for C17H22N4: C, 72.31; H, 7.85; N, 19.84. Found: C, 72.15; H, 8.05; N, 19.74.
5.2.2. N,N-Dimethyl-N′-[4-(3-nitrophenylazo)phenyl]propane-1,3-diamine (2)
Yield: 4%. Mp 205–208 °C (EtOH abs.). TLC: R f = 0.3 (Al2O3, Et2O + 2%Et2NH). 1H NMR (CDCl3): 1.91 (q, 2H, C(2)); 2.07 (s, NH); 2.25 (s, N(CH3)2); 2.37 (t, J = 6.9, 2H, C(3)); 4.34 (t, J = 7.6, 2H, C(3)); 7.11–8.40 (m, 8 arom. H). Anal. Calcd for C17H21N5O2. HCl: C, 56.12; H, 6.09; N, 19.25. Found: C, 56.31; H, 6.20; N, 18.88.
5.2.3. N,N-Dimethyl-N′-[4-(4-nitrophenylazo)phenyl]propane-1,3-diamine (3)
Yield: 3%. Mp 116–119 °C (hexane). TLC: R f = 0.3 (Al2O3, Et2O + 2%Et2NH). 1H NMR (CDCl3): 1.76 (q, 2H, C(2)); 2.21 (s, N(CH3)2); 2.38 (t, J = 6.3, 2H, C(3)); 3.24 (t, J = 6.9, 2H, C(1)); 5.72 (s, NH); 6.54 (d, J = 10.0, 2 arom. H); 7.75–7.90 (m, 4 arom. H); 8.25 (d, J = 10.1, 2 arom. H). Anal. Calcd for C17H21N5O2: C, 62.37; H, 6.47; N, 21.39. Found: C, 62.06; H, 6.68; N, 21.00.
5.2.4. N′-[4-(2,5-Difluorophenylazo)phenyl]-N,N-dimethylpropane-1,3-diamine (4)
Yield: 11%. Oil (CC: Al2O3, Et2O). TLC: R f = 0.4 (Al2O3, Et2O + 2%Et2NH). 1H NMR (CDCl3): 1.76 (q, 2H, C(2)); 2.20 (s, N(CH3)2); 2.38 (t, J = 6.7, 2H, C(3)); 3.23 (t, J = 7.0, 2H, C(1)); 5.49 (s, NH); 6.55 (d, J = 9.0, 2 arom. H); 6.88–7.44 (m, 3 arom. H); 7.78 (d, J = 9.2, 2 arom. H). Anal. Calcd for C17H20F2N4·HCl: C, 57.54; H, 5.97; N, 15.79. Found: C, 57.43; H, 6.10; N, 15.73.
5.2.5. N′-[4-(2,6-Difluorophenylazo)phenyl]-N,N-dimethylpropane-1,3-diamine (5)
Yield: 15%. Oil (CC: Al2O3, Et2O). TLC: R f = 0.3 (Al2O3, Et2O + 2%Et2NH). 1H NMR (CDCl3): 1.70 (q, 2H, C(2)); 2.19 (s, N(CH3)2); 2.36 (t, J = 6.5, 2H, C(3)); 3.23 (t, J = 6.7, 2H, C(1)); 5.43 (s, NH); 6.56 (d, J = 10.0, 2 arom. H); 6.85–7.20 (m, 3 arom. H); 7.75 (d, J = 9.0, 2 arom. H). Anal. Calcd for C17H20F2N4: C, 64.14; H, 6.33; N, 17.60. Found: C, 63.90; H, 6.61; N, 17.52.
5.2.6. N,N-Dimethyl-N′-4-(4′-tolylazo)phenylpropane-1,3-diamine (6)
Yield: 10%. Mp 78–79 °C (Et2O). TLC: R f = 0.4 (Al2O3, Et2O + 2%Et2NH). 1H NMR (CDCl3): 1.84 (q, 2H, C(2)); 1.96 (s, CH3); 2.34 (s, N(CH3)2); 2.61 (t, J = 6.6, 2H, C(3)); 3.21 (t, J = 7.1, 2H, C(1)); 4.65 (s, NH); 6.56 (d, J = 10.1, 2 arom. H); 7.20 (d, J = 10.0, 2 arom. H); 7.62–7.78 (m, 4 arom. H). Anal. Calcd for C18H24N4: C, 72.94; H, 8.16; N, 18.90. Found: C, 72.68; H, 8.50; N, 19.20.
5.2.7. N,N-Dimethyl-1-(1,3-diphenyltriazen-3-yl)-3-propanamine (25)
Yield: 31%. Oil (CC: Al2O3, Et2O). Bp (p = 0.1–0.2 torr) 100–105 °C. TLC: R f = 0.7 (Al2O3, Et2O + 2%Et2NH). 1H NMR (CDCl3): 1.80 (q, 2H, C(2)); 2.20 (s, N(CH3)2); 2.31 (t, J = 7.1, 2H, C(3)); 4.27 (t, J = 7.3, 2H, C(1)); 6.52–7.83 (m, 10 arom. H). Anal. Calcd for C17H22N4: C, 72.31; H, 7.85; N, 19.84. Found: C, 72.31; H, 7.87; N, 19.88.
5.2.8. N,N-Dimethyl-1-[1-(3-trifluoromethylphenyl)-3-phenyltriazen-3-yl]-3-propanamine (26)
Yield: 13%. Oil (CC: Al2O3, Et2O). Bp (p = 0.1–0.2 torr) 80–85 °C. TLC: R f = 0.8 (Al2O3, Et2O + 2%Et2NH). 1H NMR (CDCl3): 1.83 (q, 2H, C(2)); 2.20 (s, N(CH3)2); 2.31 (t, J = 6.8, CH2(3)); 4.28 (t, J = 7.0, CH2(1)); 7.17–7.78 (m, 9 arom. H). Anal. Calcd for C18H21F3N4: C, 61.70; H, 6.04; N, 15.99. Found: C, 61.54; H, 6.08; N, 15.93.
5.2.9. 1-[1-(3-Nitrophenyl)-3-phenyltriazen-3-yl]-N,N-dimethyl-3-propanamine (27)
Yield: 58%. Oil (CC: Al2O3, Et2O). TLC: R f = 0.7 (Al2O3, Et2O + 2%Et2NH). 1H NMR (CDCl3): 1.86 (q, 2H, C(2)); 2.20 (s, N(CH3)2); 2.32 (t, J = 6.9, 2H, C(3)); 4.29 (t, J = 7.5, 2H, C(1)); 7.06–8.35 (m, 9 arom. H). Anal. Calcd for C17H21N5O2: C, 62.37; H, 6.47; N, 21.39. Found: C, 62.55; H, 6.61; N, 21.46.
5.2.10. N,N-Dimethyl-1-[1-(4-nitrophenyl)-3-phenyltriazen-3-yl]-3-propanamine (28)
Yield: 51%. Mp 74–75 °C (Et2O). TLC: R f = 0.8 (Al2O3, Et2O + 2%Et2NH). 1H NMR (CDCl3): 1.86 (q, 2H, C(2)); 2.18 (s, N(CH3)2); 2.32 (t, J = 7.1, 2H, C(3)); 4.30 (t, J = 7.8, 2H, C(1)); 7.10–7.50 (m, 5 arom. H); 7.59 (d, J = 10.2, 2 arom. H); 8.19 (d, J = 10.2, 2 arom. H). Anal. Calcd for C17H21N5O2: C, 62.37; H, 6.47; N, 21.39. Found: C, 62.27; H, 6.59; N, 21.56.
5.2.11. 1-[1-(2,5-Difluorophenyl)-3-phenyltriazen-3-yl]-N,N-dimethyl-3-propanamine (29)
Yield: 48%. Mp 61–64 °C (pentane). TLC: R f = 0.7 (Al2O3, Et2O + 2%Et2NH). 1H NMR (CDCl3): 1.89 (q, 2H, C(2)); 2.18 (s, N(CH3)2); 2.30 (t, J = 6.9, 2H, C(3)); 4.28 (t, J = 7.2, 2H, C(1)); 6.70–7.60 (m, 8 arom. H). Anal. Calcd for C17H20F2N4: C, 64.14; H, 6.33; N, 17.60. Found: C, 64.30; H, 6.55; N, 17.35.
5.2.12. 1-[1-(2,5-Difluorophenyl)-3-(3-trifluoromethylphenyl)triazen-3-yl]-N,N-dimethyl-3-propanamine (30)
Yield: 57%. Oil (CC: Al2O3, Et2O). TLC: R f = 0.7 (Al2O3, Et2O + 2%Et2NH). 1H NMR (CDCl3): 1.86 (q, 2H, C(2)); 2.18 (s, N(CH3)2); 2.29 (t, J = 6.7, 2H, C(3)); 4.30 (t, J = 7.2, 2H, C(1)); 6.77–7.80 (m, 7 arom. H). Anal. Calcd for C18H19F5N4: C, 55.96; H, 4.96; N, 14.50. Found: C, 55.95; H, 4.76; N, 14.66.
5.2.13. 1-[1-(2,6-Difluorophenyl)-3-phenyltriazen-3-yl]-N,N-dimethyl-3-propanamine (31)
Yield: 41%. Oil (CC: Al2O3, Et2O). TLC: R f = 0.7 (Al2O3, Et2O + 2%Et2NH). 1H NMR (CDCl3): 1.90 (q, 2H, C(2)); 2.19 (s, N(CH3)2); 2.33 (t, J = 7.0, 2H, C(3)); 4.26 (t, J = 7.6, 2H, C(1)); 6.81–7.47 (m, 8 arom. H). Anal. Calcd for C17H20F2N4: C, 64.14; H, 6.33; N, 17.60. Found: C, 63.98; H, 6.49; N, 17.55.
5.2.14. N,N-Dimethyl-1-[3-phenyl-1-(p-tolyl)triazen-3-yl]-3-propanamine (32)
Yield: 64%. Oil (CC: Al2O3, Et2O). TLC: R f = 0.8 (Al2O3, Et2O + 2%Et2NH). 1H NMR (CDCl3): 1.82 (q, 2H, C(2)); 2.19 (s, N(CH3)2); 2.24–2.37 (m, 2H, C(3) and 3H, CH3-Ph); 4.24 (t, J = 7.6, 2H, C(1)); 6.97–7.47 (m, 9 arom. H). Anal. Calcd for C18H24N4: C, 72.94; H, 8.16; N, 18.90. Found: C, 72.61; H, 8.24; N, 19.40.
5.2.15. 3-[(1S,9aR)-Octahydro-2H-quinolizin-1-ylmethyl]-1-phenyl-3-(trifluoromethylphenyl)triazene (33)
Yield: 47%. Mp 102–103 °C (pentane). 1H NMR (CDCl3): 0.66–2.20 (m, 14H of quinolizidine); 2.80–2.96 (m, 2Hα near N of quinolizidine); 4.23 (dd, J = 14.8, 4.0, 1H of CH2-Quinolizidine); 4.96 (dd, J = 15.0, 11.0, 1H of CH2-Quinolizidine); 7.23–7.76 (m, 9 arom. H). Anal. Calcd for C23H27F3N4: C, 66.32; H, 6.54; N, 13.45. Found: C, 66.58; H, 6.59; N, 13.03.
5.3. Aryltriazenes of structure D and F. General method
The aromatic amine (25 mmol) was dissolved in 7.3 mL of conc. hydrochloridric acid and 10 mL of water, cooling in a ice bath. The solution was kept at 0–5 °C, while adding drop by drop a solution of sodium nitrite (25 mmol) in 10 mL of water. The diazonium salt was then coupled with the relevant amine (25 mmol) dissolved in 45 mL of 1 N NaOH (pH 7–8). After 20 min of stirring, the precipitate was collected by filtration. The solid was crystallized from the suitable solvent.
In the case of compounds 14, 18 and 19 the proper oily aniline was added to the diazonium salt solution, followed by a solution of sodium acetate to reduce acidity.
When the coupling-product separated as an oil (compounds 37, 43, 53) it was extracted with Et2O and after removing the solvent the oil was purified by CC. The cytisine derivative 58 was extracted with CHCl3, and the solid obtained was crystallized from acetone.
5.3.1. 1-Phenyl-3-(3′-trifluoromethylphenyl)triazene (14)
Yield: 92%. Mp 95–96 °C (hexane). 1H NMR (CDCl3): 7.08–7.69 (m, 9 arom. H); 9.68 (s, NH). Anal. Calcd for C13H10F3N3: C, 58.87; H, 3.80; N, 15.84. Found: C, 58.66; H, 4.20; N, 15.97.
5.3.2. 1-(2,5-Difluorophenyl)-3-phenyltriazene (18)
Yield: 30%. Mp 125–126 °C (pentane). 1H NMR (CDCl3): 6.60–7.50 (m, 8 arom. H); 9.70 (s, NH). Anal. Calcd for C12H9F2N3 + 0.5H2O: C, 57.37; H, 3.58; N, 16.76. Found: C, 57.69; H, 3.52; N, 16.61.
5.3.3. 1-(2,5-Difluorophenyl)-3-(3′-trifluoromethylphenyl)triazene (19)
Yield: 61%. Mp 103–104 °C (hexane). 1H NMR (CDCl3): 6.64–7.72 (m, 7 arom. H); 9.71 (s, NH). Anal. Calcd for C13H8F5N3: C, 51.84; H, 2.68; N, 13.95. Found: C, 52.14; H, 2.66; N, 13.99.
5.3.4. 1-(3-Chlorophenyl)azopyrrolidine (35)
Yield: 90%. Mp 49–50.5 °C (hexane). 1H NMR (CDCl3): 1.96 (br s, 4H, C(3, 4)); 3.73 (br s, 4H, C(2, 5)); 6.96–7.37 (m, 4 arom. H). Anal. Calcd for C10H12ClN3: C, 57.28; H, 5.77; N, 20.04. Found: C, 57.30; H, 5.65; N, 20.25.
5.3.5. 1-(3-Trifluoromethylphenyl)azopyrrolidine (37)
Yield: 81%. Oil (CC: Al2O3, Et2O) 1H NMR (CDCl3): 1.97 (br s, 4H, C(3, 4)); 3.66 (br s, 2H); 3.80 (br s, 2H); 7.20–7.70 (m, 4 arom. H). Anal. Calcd for C11H12F3N3: C, 54.32; H, 4.97; N, 17.27. Found: C, 54.33; H, 5.14; N, 17.48.
5.3.6. 1-(2,5-Difluorophenyl)azopyrrolidine (43)
Yield: 89%. Oil (CC: Al2O3, Et2O). 1H NMR (CDCl3): 1.97 (br s, 4H, C(3, 4)); 3.64 (br s, 2H); 3.88 (br s, 2H); 6.60–7.20 (m, 3 arom. H). Anal. Calcd for C10H11F2N3: C, 56.87; H, 5.25; N, 19.89. Found: C, 56.76; H, 5.54; N, 20.19.
5.3.7. 1-(3,4-Dichlorophenyl)azopyrrolidine (44)
Yield: 65%. Mp 63–65 °C (hexane). 1H NMR (CDCl3): 1.97 (br s, 4H, C(3, 4)); 3.59 (br s, 2H); 3.83 (br s, 2H); 7.06–7.49 (m, 3 arom. H). Anal. Calcd for C10H11Cl2N4: C, 49.20; H, 4.54; N, 17.21. Found: C, 49.18; H, 4.57; N, 17.12.
5.3.8. 1-(3-Chlorophenyl)azopiperidine (46)
Yield: 30%. Mp 32–33°C (hexane). 1H NMR (CDCl3): 1.65 (br s, 6H, C(3, 4, 5)); 3.73 (br s, 4H, C(2, 6)); 7.00–7.44 (m, 4 arom. H). Anal. Calcd for C11H14ClN3: C, 59.00; H, 6.58; N, 19.12. Found: C, 59.06; H, 6.31; N, 18.78.
5.3.9. 1-(3-Bromophenyl)azopiperidine (47)
Yield: 28%. Mp 30 °C (hexane). 1H NMR (CDCl3): 1.65 (br s, 6H, C(3, 4, 5)); 3.73 (br s, 4H, C(2, 6)); 7.05–7.56 (m, 4 arom. H). Anal. Calcd for C11H14BrN3: C, 49.27; H, 5.26; N, 15.67. Found: C, 49.28; H, 4.99; N, 15.82.
5.3.10. 1-(3-Nitrophenyl)azopiperidine (48)
Yield: 40%. Mp 33–35 °C (hexane). 1H NMR (CDCl3): 1.66 (br s, 6H, C(3, 4, 5)); 3.74 (br s, 4H, C(2, 6)); 7.32–8.20 (m, 4 arom. H). Anal. Calcd for C11H14N4O2: C, 56.40; H, 6.02; N, 23.92. Found: C, 56.26; H, 6.16; N, 24.06.
5.3.11. 1-Methyl-4-(2,5-difluorophenylazo)piperazine (53)
Yield: 71%. Oil (CC: Al2O3, Et2O). 1H NMR (CDCl3): 2.31 (s, CH3); 2.50 (t, J = 5.2, 4H, C(2, 6)); 3.83 (t, J = 5.2, 4H, C(3, 5)); 6.65–7.22 (m, 3 arom. H). Anal. Calcd for C11H14F2N4: C, 54.99; H, 5.87; N, 23.32. Found: C, 54.88; H, 5.98; N, 23.61.
5.3.12. 4-Phenylazo-1-(pyrimidin-2′-yl)piperazine (56)
Yield: 48%. Mp 74–75 °C (hexane). 1H NMR (CDCl3): 3.84 (t, J = 5.5, 4H, C(2, 6)); 3.95 (t, J = 5.5, 4H, C(3, 5)); 6.48 (t, J = 4.8, 1H, C(5’)); 7.07–7.46 (m, 5 arom. H); 8.28 (d, J = 4.8, 2H, C(4′, 6′)). Anal. Calcd for C14H16N6: C, 62.67; H, 6.01; N, 31.32. Found: C, 62.39; H, 6.16; N, 31.63.
5.3.13. 3-(3-Nitrophenylazo)cytisine (58)
Yield: 25%. Mp 201–203 °C (acetone). 1H NMR (CDCl3): 1.95–2.19 (m, 2H, C(13)); 2.58–2.75 (m, 1H, C(5)); 3.12–3.27 (m, 5H, C(1,2,4)); 3.74–3.92 (m, 1H, C(6)); 4.20–4.30 (m, 1H, C(6)); 6.05 (d, J = 6.7, 1H, C(9)); 6.32 (d, J = 8.4, 1H, C(11)); 7.12–8.12 (m, 1H, C(10) and 4 arom. H). Anal. Calcd for C17H17N5O3: C, 60.17; H, 5.05; N, 20.64. Found: C, 60.27; H, 5.33; N, 20.31.
5.4. Cell-based assays
5.4.1. Compounds
Compounds were dissolved in DMSO at 100 mM and then diluted in culture medium.
5.4.2. Cells and viruses
Cell lines were purchased from American Type Culture Collection (ATCC). The absence of mycoplasma contamination was checked periodically by the Hoechst staining method. Cell lines supporting the multiplication of RNA and DNA viruses were the following: CD4+ human T-cells containing an integrated HTLV-1 genome (MT-4); Madin Darby Bovine Kidney (MDBK); Baby Hamster Kidney (BHK-21) and Monkey kidney (Vero 76) cells.
5.4.3. Cytotoxicity assays
For cytotoxicity tests, run in parallel with antiviral assays, MDBK and BHK cells were resuspended in 96 multiwell plates at an initial density of 6 × 105 and 1 × 106 cells/mL, respectively, in maintenance medium, without or with serial dilutions of test compounds. Cell viability was determined after 48–96 h at 37 °C in a humidified CO2 (5%) atmosphere by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) method.38 Vero 76 cells were resuspended in 24 multiwell plates at an initial density of 4 × 105 cells/mL. The cell number of Vero 76 monolayers was determined by staining with the crystal violet dye.
For cytotoxicity evaluations, exponentially growing cells derived from human hematological tumors [CD4+ human T-cells containing an integrated HTLV-1 genome (MT-4)] were seeded at an initial density of 1 × 105 cells/mL in 96 well plates in RPMI-1640 medium supplemented with 10% fetal calf serum (FCS), 100 units/mL penicillin G and 100 μg/mL streptomycin. Cell cultures were then incubated at 37 °C in a humidified, 5% CO2 atmosphere in the absence or presence of serial dilutions of test compounds. Cell viability was determined after 96 h at 37 °C by the MTT method.
5.4.4. Antiviral assay
Activity of compounds against Human Immunodeficiency virus type-1 (HIV-1) was based on inhibition of virus-induced cytopathogenicity in MT-4 cells acutely infected with a multiplicity of infection (m.o.i.) of 0.01. Briefly, 50 μL of RPMI containing 1 × 104 MT-4 were added to each well of flat-bottom microtitre trays containing 50 μL of RPMI, without or with serial dilutions of test compounds. Then, 20 μL of a HIV-1 suspension containing 100 CCID50 were added. After a 4-day incubation, cell viability was determined by the MTT method.
Activity of compounds against Yellow fever virus (YFV) and Reo virus type-1 (Reo-1) was based on inhibition of virus-induced cytopathogenicity in acutely infected BHK-21 cells. Activities against Bovine viral diarrhoea virus (BVDV), in infected MDBK cells, were also based on inhibition of virus-induced cytopathogenicity.
BHK and MDBK cells were seeded in 96-well plates at a density of 5 × 104 and 3 × 104 cells/well, respectively, and were allowed to form confluent monolayers by incubating overnight in growth medium at 37 °C in a humidified CO2 (5%) atmosphere. Cell monolayers were then infected with 50 μL of a proper virus dilution (in serum-free medium) to give a m.o.i = 0.01. One hour later, 50 μL of MEM Earle’s medium, supplemented with inactivated fetal calf serum (FCS), 1% final concentration, without or with serial dilutions of test compounds, were added. After 3–4 days of incubation at 37 °C, cell viability was determined by the MTT method.
Activity of compounds against Coxsackie Virus type B2 (CVB-2), Polio Virus type-1 Sabin strain (Sb-1), Vesicular Stomatitis Virus (VSV), Vaccinia Virus (VV), Herpes Virus 1 (HSV-1) and Respiratory Syncytial Virus (RSV), A-2 strain, in infected Vero 76 cells, was determined by plaque reduction assays in Vero 76 cell monolayers. To this end, Vero 76 cells were seeded in 24-well plates at a density of 2 × 105 cells/well and were allowed to form confluent monolayers by incubating overnight in growth medium at 37 °C in a humidified CO2 (5%) atmosphere. Then, monolayers were infected with 250 μL of proper virus dilutions to give 50–100 PFU/well. Following removal of unadsorbed virus, 500 μL of Dulbecco’s modified Eagle’s medium supplemented with 1% inactivated FCS and 0.75% methyl-cellulose, without or with serial dilutions of test compounds, were added. Cultures were incubated at 37 °C for 2 (Sb-1 and VSV), 3 (CVB-2, VV and HSV-1) or 5 days (RSV) and then fixed with PBS containing 50% ethanol and 0.8% crystal violet, washed and air-dried. Plaques were then counted. EC50 (50% effective concentration) was calculated by linear regression technique.
5.5. Molecular modeling
All molecular dynamics simulations were carried out using the sander and pmemd module within the amber 9 suite of programs,39 and the parm99 all-atom force field,40 working in parallel on 32 processors of the Tartaglia cluster at the University of Trieste (Trieste, Italy). The crystallographic coordinates of the RNA-dependent RNA-polymerase (RdRp) of BVDV (PDB entry 1S48.pdb)31 and HCV (PDB entry 1CSJ.pdb)28 were employed as starting geometries for protein simulations in complex with the most active compounds (1, 2, 4, 7–10). Missing hydrogen atoms were added to the protein backbone and side chains with LEaP module of amber 9. All ionizable residues were considered in the standard ionization state at neutral pH. The geometry of added hydrogens and ions was refined for 200 steps (steepest descent) in vacuum using the parm94 force field.40 Further protein geometry refinement was carried out using the sander module of amber 9 via a combined steepest descent—conjugate gradient algorithm, using as a convergence criterion for the energy gradient the root-mean-square of the Cartesian elements of the gradient equal to 0.01 kcal/(mol Å). The generalized Born/surface area (GB/SA) continuum solvation model41 was used to mimic a water environment. As expected, no relevant structural changes were observed between RdRp relaxed models and the original 3-D structure.
The putative binding site for our compounds on the BVDV RdRp was determined using the ActiveSite_Search option of the Binding Site module of InsightII (v. 2001, Accelrys, San Diego, USA).33 ActiveSite_Search identifies protein active sites or binding sites by locating cavities in the protein structure. According to the Site_Search algorithm employed, the protein is first mapped onto a grid which covers the complete protein space. The grid points are then defined as free points and protein points. The protein points are grid points, within 2 Å from a hydrogen atom or 2.5 Å from a heavy atom. Then, a cubic eraser moves from the outside of the protein toward the center to remove the free points until the opening is too small for it to move forward. Those free points not reached by the eraser will be defined as site points. After a site is located, it can be modified by expanding or contracting the site. One layer of grid points at the cavity opening site will be added or removed by each expand or contract operation, respectively.
The model structures of all ligand molecules were built and subjected to an initial energy minimization. The convergence criterion was set to 10−4 kcal/(mol Å). A conformational search was carried out using a well-validated, ad-hoc developed combined molecular mechanics/molecular dynamics simulated annealing (MDSA) protocol.33, 34 Accordingly, the relaxed structures were subjected to five repeated temperature cycles (from 310 K to 1000 K and back) using constant volume/constant temperature (NVT) MD conditions. At the end of each annealing cycle, the structures were again energy minimized to converge below 10−4 kcal/(mol Å), and only the structures corresponding to the minimum energy were used for further modeling. The atomic partial charges for the geometrically optimized compounds were obtained using the RESP procedure,42 and the electrostatic potentials were produced by single-point quantum mechanical calculations at the Hartree–Fock level with a 6-31G∗ basis set, using the Merz–Singh–Kollman van der Waals parameters.43 Eventual missing force field parameters for the compounds considered were generated as follows: AM1 geometry optimization of the structure was followed by RHF/6-31G∗ single point calculation to obtain the electrostatic potentials. Next, the RESP method was used for charge fitting. The missing bond, angle torsion or van der Waals parameters not included in the parm99 were generated using the antechamber module of amber 9.
The optimized structures of the inhibitors were docked into the putative enzyme allosteric binding site by applying a consolidated procedure;33, 34 accordingly, it will be briefly reported below. The software autodock 3.044 was employed to estimate the possible binding orientations of all compounds in the receptor. In order to encase a reasonable region of the protein surface and interior volume, centered on the binding site, the grids were 60 Å on each side. Grid spacing (0.375 Å), and 120 grid points were applied in each Cartesian direction so as to calculate mass-centered grid maps. amber 12-6 and 12-10 Lennard-Jones parameters were used in modeling van der Waals interactions and hydrogen bonding (N–H, O–H and S–H), respectively. In the generation of the electrostatic grid maps, the distance dependent relative permittivity of Mehler and Solmajer was applied.45 For the docking of each compound to the proteins, three hundred Monte Carlo/Simulated Annealing (MC/SA) runs were performed, with 100 constant temperature cycles for simulated annealing. For these calculations, the GB/SA implicit water model41 was used to mimic the solvated environment. The rotation of the angles φ and ϕ, and the angles of side chains were set free during the calculations. All other parameters of the MC/SA algorithm were kept as default. Following the docking procedure, the structure of all compounds were subjected to cluster analysis with a tolerance of 1 Å for an all-atom root-mean-square deviation from a lower-energy structure representing each cluster family. In the absence of any relevant crystallographic information for our compounds, the structure of each resulting complex characterized by the lowest interaction energy in the prevailing cluster was selected for further evaluation.
Each best substrate/RdRp complex resulting from the automated docking procedure was further refined in the amber 9 suite using the quenched molecular dynamics (QMD) method.33, 34 In this case, 1 ns MD simulation at 310 K were employed to sample the conformational space of the substrate–enzyme complex in the GB/SA continuum solvation environment. The integration step was equal to 1 fs and the parm99 force field parameters were applied in the simulations. After each ps, the system was cooled to 0 K, the structure was extensively minimized, and stored. To prevent global conformational changes of the enzyme, the backbone of the protein binding site was constrained by a harmonic force constant of 100 kcal/Å, whereas the amino acid side chains and the ligands were allowed moving without any constraint.
The best energy configuration of each complex resulting from the previous step was allowed to relax in a 55-Å radius sphere of TIP3P water molecules.46 The resulting system was minimized with a gradual decrease in the position restraints of the protein atoms. At the end of the relaxation process, all water molecules beyond the first hydration shell (i.e., at a distance > 3.5 Å from any protein atom) were removed. Finally, to achieve electroneutrality, a suitable number of counterions were added to neutralize the system using the leap module of amber 9, removing eventually overlapping water molecules. To reduce computational time to reasonable limits, all proteins residues with any atom closer than 20 Å from the center of mass of each bounded ligand were chosen to be flexible in the dynamic simulations. Subsequently, a spherical TIP3P water cap of radius equal to 20 Å was centered on each inhibitor in the corresponding RdRp complex, including the hydrating water molecules within the sphere resulting from the previous step. After energy minimization of the new water cap for 10 ps, keeping the protein, the ligand, and the pre-existing waters rigid, followed by a MD equilibration of the entire water sphere with fixed solute for 1 ns, further unfavorable interactions within the structures were relieved by progressively smaller positional restraints on the solute (from 25 to 0 kcal/(mol Å2) for a total of 2 ns. Each system was gradually heated to 310 K in three intervals, allowing a 1 ns interval per each 100 K, and then equilibrated for 5 ns at 310 K, followed by 10 ns of data collection runs, necessary for the estimation of the free energy of binding (vide infra). The MD simulations were performed at constant T = 310 K using the Berendsen et al. coupling algorithm47 with separate coupling of the solute and solvent to the heat, an integration time step of 2 fs, and the applications of the Shake algorithm to constrain all bonds to their equilibrium values, thus removing high frequency vibrations. Long-range nonbonded van der Waals interactions were truncated by using a dual cutoff of 9 and 13 Å, respectively, where energies and forces due to interactions between 9 and 13 Å were updated every 20 time steps. The particle mesh Ewald method was used to treat the long-range electrostatics. For the calculation of the binding free energy between the RdRp and each inhibitor in water, a total of 100 snapshots were saved during the MD data collection period described above, one snapshot per each 0.1 ns of MD simulation.
The binding free energy ΔG bind of each RdRp/drug complex in water was calculated according to the procedure termed Molecular Mechanic/Poisson–Boltzmann Surface Area (MM/PBSA), and originally proposed by Srinivasan et al.48 In the MM/PBSA framework of theory, the binding free energy of a given ligand to its receptor protein can be evaluated as
| (1) |
The individual terms of the MM/PBSA approach that contribute to the free energy of a molecule are
| (2) |
where E MM denotes the sum of intra- and intermolecular mechanical (MM) energies of a molecule in the gas phase, G solv is its solvation free energy, and –TSsolute represents an estimate of the solute entropy. E MM can be further divided into terms arising from electrostatic (E el), van der Waals (E vdW), and internal (E int) (i.e., bond, angle, and torsional energies):
| (3) |
The solvation free energy:
| (4) |
consists of a polar solvation energy component, G PB, which is calculated in a continuum solvent, usually a finite-difference Poisson-Boltzmann (PB) model, and a nonpolar term, G np, which is proportional to the solvent-accessible surface area (SA).
The ensemble of structures for the uncomplexed reactants are generated either running separate MD simulations for them, or by using the trajectory of the complex, simply removing the atoms of the protein or ligand. In this work we applied the latter variant. Accordingly, the term E int in Eq. 2 cancels out in the calculation of the free energy of binding. The calculations of the polar solvation term G PB were done with the DelPhi package,49 with interior and exterior dielectric constants equal to 1 and 80, respectively. A grid spacing of 2/Å, extending 20% beyond the dimensions of the solute, was employed. The non-polar component GNP was obtained using the following relationship: G NP = γSA + β, in which γ = 0.00542 kcal/(mol Å2), β = 0.92 kcal/mol, and the surface area was estimated by means of the msms software.50 The last parameter in Eq. 1, that is, the change in solute entropy upon association –TSsolute, was calculated through normal-mode analysis. In the first step of this calculation, an 8-Å sphere around the ligand was cut out from an MD snapshot for each ligand-protein complex. This value was shown to be large enough to yield converged mean changes in solute entropy. On the basis of the size-reduced snapshots of the complex, we generated structures of the uncomplexed reactants by removing the atoms of the protein and ligand, respectively. Each of those structures was minimized, using a distance-dependent dielectric constant ε = 4r, to account for solvent screening, and its entropy was calculated using classical statistical formulas and normal mode-analysis. To minimize the effects due to different conformations adopted by individual snapshots we averaged the estimation of entropy over 50 snapshots.
Acknowledgments
Financial support from Italian MIUR (FIRB RBNE01J3SK01) and BIOMEDICINE PROJECT is gratefully acknowledged. The authors thanks O. Gagliardo for performing the elemental analyses.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmc.2009.05.020.
Supplementary data
References concerning the previously described compounds.
References and notes
- 1.Tonelli M., Boido V., Canu C., Sparatore A., Sparatore F., Paneni M.S., Fermeglia M., Pricl S., La Colla P., Casula L., Ibba C., Collu D., Loddo R. Bioorg. Med. Chem. 2008;16:8447. doi: 10.1016/j.bmc.2008.08.028. [DOI] [PubMed] [Google Scholar]
- 2.Elslager E.F., Worth D.F. J. Med. Chem. 1963;6:444. doi: 10.1021/jm00340a024. [DOI] [PubMed] [Google Scholar]
- 3.Werbel L.M., Elslager E.F., Fisher M.V., Gavrilis Z.B., Phillips A.A. J. Med. Chem. 1968;11:411. doi: 10.1021/jm00309a001. [DOI] [PubMed] [Google Scholar]
- 4.Vazzana I., Sparatore F., Fadda G., Manca C. Farmaco. 1993;48:737. [PubMed] [Google Scholar]
- 5.Novelli F., Sparatore F. Farmaco. 2002;57:871. doi: 10.1016/s0014-827x(02)01293-4. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 6.Barbieri F., Sparatore A., Alama A., Novelli F., Bruzzo C., Sparatore F. Oncol. Res. 2003;13 doi: 10.3727/000000003108747974. [DOI] [PubMed] [Google Scholar]
- 7.Sparatore F., Sommovigo P.G., Boido V. Boll. Chim. Farm. 1974;113:219. [PubMed] [Google Scholar]
- 8.Gesher J.A., Hickman J.A., Simmonds R.G., Stevens M.F.G., Vaughan K. Biochem. Pharmacol. 1981;30:89. doi: 10.1016/0006-2952(81)90288-4. [DOI] [PubMed] [Google Scholar]
- 9.Prous J., Graul A., Castañer J. Drugs Fut. 1994;19:746. [Google Scholar]
- 10.Dumont-Hornebeck B., Strube Y.N., Vasilescu D., Jean-Claude B.J. Bioorg. Med. Chem. Lett. 2000;10:2325. doi: 10.1016/s0960-894x(00)00461-3. [DOI] [PubMed] [Google Scholar]
- 11.Matheson S.L., Mc Namee J., Jean-Claude B.J. Cancer Chemother. Pharmacol. 2003;51:11. doi: 10.1007/s00280-002-0525-4. [DOI] [PubMed] [Google Scholar]
- 12.Manolov J., Machulla H.-J., Momekov G. Pharmazie. 2006;61:6. doi: 10.1002/chin.200639088. [DOI] [PubMed] [Google Scholar]
- 13.Stevens M.F.G., Phillips K.S., Rathbone D.L., O’ Shea D.M., Queener S.F., Schwalbe C.H., Lambert P.A. J. Med. Chem. 1997;40:1886. doi: 10.1021/jm970050n. [DOI] [PubMed] [Google Scholar]
- 14.Yamada, Y.; Saito, J.; Kudamatsu, A.; Katsumata, O. Ger. Offen. 2147259, 1972; Chem. Abstr.1972, 77, 5145a.
- 15.Borowski P., Deinert J., Schalinski S., Bretner M., Ginalski K., Kulikowski T., Shugar D. Eur. J. Biochem. 2003;270:1645. doi: 10.1046/j.1432-1033.2003.03540.x. [DOI] [PubMed] [Google Scholar]
- 16.Wu C.-Y., King K.-J., Kuo C.-J., Fang J.-M., Wu J.-T., Ho M.-Y., Liao C.-L., Shie J.-J., Liang P.-H., Wong C.-H. Chem. Biol. 2006;13:261. doi: 10.1016/j.chembiol.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tonelli M., Paglietti G., Boido V., Sparatore F., Marongiu F., Marongiu E., La Colla P., Loddo R. Chem. Biodiversity. 2008;5:2386. doi: 10.1002/cbdv.200890203. [DOI] [PubMed] [Google Scholar]
- 18.Boido V., Sparatore F. Farmaco Ed. Sci. 1974;29:526. [PubMed] [Google Scholar]
- 19.Meanwell N.A., Krystal M. Drugs Fut. 2007;32:441. and references cited therein. [Google Scholar]
- 20.Henderson E.A., Alber D.G., Baxter R.C., Bithell S.K., Budworth J., Carter M.C., Chub A., Cockerill G.S., Dowdell V.C.L., Fraser I.J., Harris R.A., Keegan S.J., Kelsey R.D., Lumley J.A., Stables J.N., Weerasekera N., Willson L.J., Powell K.L. J. Med. Chem. 2007;50:1685. doi: 10.1021/jm060747l. [DOI] [PubMed] [Google Scholar]
- 21.Houe H. Biologicals. 2003;31:137. doi: 10.1016/s1045-1056(03)00030-7. [DOI] [PubMed] [Google Scholar]
- 22.Buckwold V.E., Beer B.E., Donis R.O. Antiviral Res. 2003;60:1. doi: 10.1016/s0166-3542(03)00174-8. [DOI] [PubMed] [Google Scholar]
- 23.Paeshuyse J., Leyssen P., Mabery E., Boddeker N., Vrancken R., Froeyen M., Ansari I.H., Dutartre H., Rozenski J., Gil L.H.V.G., Letellier C., Lanford R., Canard B., Koenen F., Kerkhofs P., Donis R.O., Herdewijn P., Watson J., De Clercq E., Puerstinger G., Neyts J. J. Virol. 2006;80:149. doi: 10.1128/JVI.80.1.149-160.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baginski S.G., Pevar D.C., Seipel M., Sun S.C., Benetatos C.A., Chunduru S.K., Rice C.M., Collett M.S. Proc. Natl. Acd. Sci. U.S.A. 2000;97:7981. doi: 10.1073/pnas.140220397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.King R.W., Scarnati H.T., Priestley E.S., De Lucca I., Bansal A., Williams J.K. Antivir. Chem. Chemother. 2002;13:315. doi: 10.1177/095632020201300507. [DOI] [PubMed] [Google Scholar]
- 26.Sun J.H., Lemm J.A., O’Boyle D.R., II, Racela J., Colonno R., Gao M. J. Virol. 2003;77:6753. doi: 10.1128/JVI.77.12.6753-6760.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tabarrini O., Manfroni G., Fravolini A., Cecchetti V., Sabatini S., De Clercq E., Rozenski J., Canard B., Dutartre H., Paeshuyse J., Neyts J. J. Med. Chem. 2006;49:2621. doi: 10.1021/jm051250z. [DOI] [PubMed] [Google Scholar]
- 28.Bressanelli S., Tomei L., Roussel A., Incitti I., Vitale R.L., Mathieu M., De Francesco R., Rey F.A. Proc. Natl. Acad. Sci. U.S.A. 1999;96:13034. doi: 10.1073/pnas.96.23.13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ago H., Adachi T., Yoshida A., Yamamoto M., Habuka N., Yatsunami K., Miyano M. Structure. 1999;7:1417. doi: 10.1016/s0969-2126(00)80031-3. [DOI] [PubMed] [Google Scholar]
- 30.Lesburg C.A., Cable M.B., Ferrari E., Hong Z., Mannarino A., Weber P.C. Nat. Struct. Biol. 1999;6:937. doi: 10.1038/13305. [DOI] [PubMed] [Google Scholar]
- 31.Choi K.H., Groarke J.M., Young D.C., Kuhn R.J., Smith J.L., Pevear D.C., Rossmann M.G. Proc. Natl. Acad. Sci. U.S.A. 2004;101:4425. doi: 10.1073/pnas.0400660101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi K.H., Gallei A., Becher P., Rossmann M.G. Structure. 2006;14:1107. doi: 10.1016/j.str.2006.05.020. [DOI] [PubMed] [Google Scholar]
- 33.Carta A., Loriga M., Paglietti G., Ferrone M., Fermeglia M., Pricl S., Sanna T., Ibba C., La Colla P., Loddo R. Bioorg. Med. Chem. 2007;15:1914. doi: 10.1016/j.bmc.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 34.Mazzei M., Nieddu E., Miele M., Balbi A., Ferrone M., Fermeglia M., Mazzei M.T., Pricl S., La Colla P., Marongiu F., Ibba C., Loddo R. Bioorg. Med. Chem. 2008;16:2591. doi: 10.1016/j.bmc.2007.11.045. [DOI] [PubMed] [Google Scholar]
- 35.Lai V.C., Kao C.C., Ferrari E., Park J., Uss A.S., Wright-Minogue J., Hong Z., Lau J.Y. J. Virol. 1999;73:10129. doi: 10.1128/jvi.73.12.10129-10136.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dimitrova M., Imbert I., Kieny M.P., Schuster C. J. Virol. 2003;77:5401. doi: 10.1128/JVI.77.9.5401-5414.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Piccininni S., Varaklioti A., Lardelli M., Dave B., Raney D.A., McCarthy J.E. J. Biol. Chem. 2002;277:45670. doi: 10.1074/jbc.M204124200. [DOI] [PubMed] [Google Scholar]
- 38.Pauwels R., Balzarini J., Baba M., Snoeck R., Schols D., Herdewijn P., Desmyter J., De Clercq E. J. Virol. Meth. 1988;20:309. doi: 10.1016/0166-0934(88)90134-6. [DOI] [PubMed] [Google Scholar]
- 39.Case, D. A.; Darden, T. A.; Cheatham, T. E., III; Simmerling, C. L.; Wang, J.; Duke, R. E.; Luo, R.; Merz. K. M.; Pearlman, D. A.; Crowley, M.; Walker, R. C.; Zhang, W.; Wang, B.; Hayik, S.; Roitberg, A.; Seabra, G; Wong, K. F.; Paesani, F.; Wu, X.; Brozell, S.; Tsui, V.; Gohlke, H.; Yang, L.; Tan, C.; Mongan, J.; Hornak, V.; Cui, G.; Beroza, P.; Mathews, D. H.; Schafmeister, C.; Ross, W. S.; Kollman, P. A., amber 9, University of California, San Francisco, CA USA, 2006.
- 40.Wang J., Cieplak P., Kollman P.A. J. Comput. Chem. 2000;21:1049. [Google Scholar]
- 41.Feig M., Onufriev A., Lee M., Im W., Case D.A., Brooks C.L., III J. Comput. Chem. 2004;25:265. doi: 10.1002/jcc.10378. [DOI] [PubMed] [Google Scholar]
- 42.Bayly C.I., Cieplak P., Cornell W.D., Kollman P.A. J. Phys. Chem. 1993;97:10269. [Google Scholar]
- 43.Besler B.H., Merz K.M., Kollman P.A. J. Comput. Chem. 1990;11:431. [Google Scholar]
- 44.Morris G.M., Goodsell D.S., Halliday R.S., Huey R., Hart W.E., Belew R.K., Olson A.J. J. Comput. Chem. 1998;19:1639. [Google Scholar]
- 45.Mehler E.L., Solmajer T. Protein Eng. 1991;4:903. doi: 10.1093/protein/4.8.903. [DOI] [PubMed] [Google Scholar]
- 46.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. J. Chem. Phys. 1983;79:926. [Google Scholar]
- 47.Berendsen H.J.C., Postma J.P.M., Van Gunsteren W.F., DiNola A., Haak J.R. J. Chem. Phys. 1984;81:3684. [Google Scholar]
- 48.Srinivasan J., Cheatham T.E., Cieplak P., Kollman P.A., Case D.A. J. Am. Chem. Soc. 1998;120:9401. [Google Scholar]
- 49.Gilson M.K., Sharp K.A., Honig B.H. J. Comput. Chem. 1988;9:327. [Google Scholar]
- 50.Sanner M.F., Olson A.J., Spehner J.C. Biopolymers. 1996;38:305. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
References concerning the previously described compounds.