

Abstract
The nucleus is a highly organized and dynamic environment where regulation and coordination of processes such as gene expression and DNA replication are paramount. In recent years, noncoding RNAs have emerged as key participants in the regulation of nuclear processes. There are a multitude of functional roles for long noncoding RNA (lncRNA), mediated through their ability to act as molecular scaffolds bridging interactions with proteins, chromatin and other RNA molecules within the nuclear environment. In this review, we discuss the diversity of techniques that have been developed to probe the function of nuclear lncRNAs, along with the ways in which those techniques have revealed insights into their mechanisms of action. Foundational observations into lncRNA function have been gleaned from molecular cytology-based, single cell approaches to illuminate both the localization and abundance of lncRNAs in addition to their potential binding partners. Biochemical, extraction-based approaches have revealed the molecular contacts between lncRNAs and other molecules within the nuclear environment and how those interactions may contribute to nuclear organization and regulation. Using examples of well-studied nuclear lncRNAs, we demonstrate that the emerging functions of individual lncRNAs have been most clearly deduced from combined cytology and biochemical approaches tailored to study specific lncRNAs. As more functional nuclear lncRNAs continue to emerge, the development of additional technologies to study their interactions and mechanisms of action promise to continually expand our understanding of nuclear organization, chromosome architecture, genome regulation, and disease states.
Keywords: noncoding RNA, nuclear organization, genome regulation, molecular cytology, microscopy, biochemical methods
INTRODUCTION
The nucleus is a highly organized, complex environment consisting of chromatin, RNA and proteins in a dynamic meshwork, whose organization is critical for folding and regulation of the genome (Bonev and Cavalli, 2016; Nozawa and Gilbert, 2019; Pederson, 2011). 3-D folding of chromatin and its higher order organization into euchromatin (gene rich) and heterochromatin (gene poor) domains is specified early in development and maintained in a cell-type specific manner by processes that are still not well understood (reviewed in (Mattout et al., 2015). RNA has long been proposed to play a critical role in maintaining nuclear architecture (Nickerson et al., 1989), but only recently has RNA emerged as a potent regulator of genome organization and function. There are many distinct classes of RNA with defined regulatory roles, broadly classified into small RNAs (<200nt) including miRNAs, piRNAs, crasiRNAs, tRNA fragments (Carone et al., 2013; He and Hannon, 2019; Lee et al., 2009; Weick and Miska, 2014) and long non-coding RNAs (lncRNAs; >200nt) (Rinn et al., 2012). While small RNAs primarily have distinct roles in gene regulation, development and genome defense (Moazed, 2009) emerging roles for lncRNAs indicate this broad class is involved in all levels of nuclear organization and function, including chromatin organization, nuclear compartmentalization, gene regulation, and the formation of nuclear bodies. Furthermore, perturbation of individual lncRNA expression can lead to disease states, underscoring their critical roles in nuclear architecture and regulation.
As lncRNAs have become of increased interest to the study of nuclear organization and gene regulation, new methodologies have been developed to specifically study these long, non protein-coding RNAs. Significant insight into the diversity in sequence, length and processing (i.e. polyadenylation, splicing) has come from early microarray (Guttman et al., 2009) and RNA sequencing (RNA-seq) (Djebali et al., 2012) studies, revealing that lncRNAs, in general, can be both polyadenylated and non-polyadenylated and exist in both spliced and unspliced forms. They are often in low abundance and may only be expressed in certain cell types and developmental stages. This inherent diversity in the features of lncRNAs has required the development of new technologies targeted to analyze the structure and function of lncRNAs, separate from mRNAs and small RNAs. In this review, we highlight the technologies used to investigate nuclear-restricted long non-coding RNA properties, dynamics, and functions, with an emphasis on insights gained from molecular cytology-based single-cell studies in combination with emerging biochemical (extraction-based) techniques to more holistically understand the molecular interactions and structural properties of lncRNAs involved in chromosome and nuclear function. Following results obtained from these established and emerging technologies, it has become clear that lncRNAs play a wide variety of roles in both nuclear organization and gene regulation due to their ability to complex with DNA, proteins, and other RNA molecules. Though their functional roles are diverse, their mechanisms of action are beginning to be classified into broad molecular processes through the development of technologies aimed to understand the molecular functions of lncRNAs. Insights gained from studying the localization and abundance of lncRNAs via molecular cytology have allowed for the development of strong hypotheses as to their functions, while biochemical studies have been critical to identifying their interacting partners and structures in order to further refine their functional roles. The combined use of molecular cytology and biochemical techniques for individual lncRNAs represent a powerful toolkit that has revealed the most significant insight into their functions within the nuclear environment. We highlight here a few representative examples of nuclear-restricted lncRNAs roles’ as molecular scaffolds and in the formation and maintenance of genome regulation, and discuss the methods that have revealed these key insights.
lncRNA INSIGHTS GAINED FROM MOLECULAR CYTOLOGY
“Molecular cytology” can describe any of a number of techniques that provide a glimpse into the dynamic workings of a cell through the targeted visualization of biomolecules of interest. Labeling techniques have moved well beyond the initial autoradiographic emulsions used in the mid-1900s, and are now sufficiently advanced that it is possible to simultaneously tag a variety of biomolecules with high sensitivity through several labeling techniques, broadly described below.
Immuno-based detection can show the specific localization of a given molecule within a whole cell. A wide array of antibodies raised in any of several hosts may now be easily purchased with high specificity for most proteins, as well as other antigens (e.g., proteins, dNTPs, dsRNA, R loops, etc.). Protein products may also be complexed to a fluorescent marker protein to avoid the antibody-based staining process (Yu et al., 2015). Because lncRNAs commonly interact with many other molecules within the nucleus, fluorescent imaging, through either immunofluorescence or fusion proteins, provides a quick and reliable way to evaluate potential interaction partners of lncRNAs.
The technique of DNA fluorescence in situ hybridization (DNA FISH) emerged with radio-labeled complementary DNA and RNA probes initially developed by Pardue and Gall to hybridize to and detect specific DNA sequences in Xenopus cells (Gall and Pardue, 1969; Pardue and Gall, 1969). For ease of use and safety, the radio-labeled probes used in these initial autoradiographic emulsions were then replaced later with the fluorescently labeled probes still used today (Rudkin and Stollar, 1977). DNA FISH requires the denaturation of double stranded DNA to allow an oligonucleotide probe to access and hybridize to its complementary site in the nucleus. However, in non-denaturing conditions, similarly labeled probes can be utilized for the detection of available single-stranded RNA molecules, thus distinguishing detection of DNA and RNA based on the method of hybridization (Byron et al., 2013). Shortly after the development of DNA FISH, a similar technique was adapted for the detection of RNA molecules in muscle cells (Singer and Ward, 1982) and interphase nuclei (Lawrence et al., 1989). Importantly, both the DNA and transcribed RNA for any given locus can be visualized in a single sample through sequential RNA FISH, followed by DNA FISH, with a fixation step in between.
More recent innovations have further expanded FISH into an incredibly precise array of cytological tools for studying nucleic acid species in situ. Single molecule RNA FISH (smFISH), for example, allows for the visualization of individual RNA transcripts using probes of specified lengths labeled with a single fluorophore, providing unparalleled insight into transcription levels and quantification of transcript accumulations (Raj et al., 2008). Techniques such as Oligopaints target a broad array of sequences simultaneously using oligonucleotide libraries to generate tiling probes, making it possible to “paint” the DNA of whole chromosomes and visualize their position and organization within the nucleus (Beliveau et al., 2012). Together, FISH allows a means to clearly visualize the localization and measure the abundance of specific DNA and RNA species in single cells (Byron et al., 2013), which is essential for the study of lncRNAs, whose natural and pathological roles are intimately connected to their cytological location.
The use of halogenated thymidine analogues (e.g., 5-bromo or 5-iododeoxyuridine – BrdU or IdU for DNA; 5-bromo or 5-iodouridine – BrU or IU for RNA) that may be incorporated into replicating DNA (Eminaga et al., 2016; Gratzner, 1982; Lengronne et al., 2001) or nascent RNA (Haukenes et al., 1997; Sadoni and Zink, 2004; Wansink et al., 1993; Wei et al., 1999) and later detected through immunological means adds a temporal dimension to lncRNA research, by enabling the study of transcription dynamics and stability. A catalytically mediated direct fluorescence approach using EU and EdU (ethynyluridine and 5-ethynyl-2-deoxyuridine) provides an alternative to immunological detection (Darzynkiewicz et al., 2011; Horisawa, 2014; Jao and Salic, 2008; Johnsson et al., 2014; Salic and Mitchison, 2008). Though labeling techniques have been used to study the transcription and stability of many RNA transcripts (Fox et al., 2002; Hagemeijer et al., 2012), these techniques remain a broadly under-utilized yet potentially invaluable resource in the study of lncRNAs with the potential to provide key insights into their cell cycle dynamics.
Molecular cytology necessitates not only the faithful tagging and labeling of molecules of interest, but also the reliable detection of these same epitopes at sufficiently high resolution. There are many techniques available for imaging tagged biomolecules – beyond more standard techniques (in all their many variants) such as epifluorescence, confocal, light sheet, and two-photon microscopy, are newer super resolution techniques that go well beyond the 200 to 250 nm resolution of diffraction-limited microscopes.
These super resolution techniques can achieve resolutions as high as 10 nm through either the spatial (deterministic) or temporal (stochastic) coordination of fluorophore activation (Galbraith and Galbraith, 2011; Thorn, 2016). Deterministic techniques, including Structured Illumination Microscopy (SIM; ~100 nm resolution) and Stimulated Emission Depletion microscopy (STED; ~30–80 nm resolution), use forms of patterned illumination to more clearly distinguish the excitation of fluorophores within a given field of view in order to visualize objects below the diffraction limit (Galbraith and Galbraith, 2011; Sigal et al., 2018). Stochastic techniques, on the other hand, use single-molecule localization microscopy (SMLM) for techniques such as Stochastic Optical Reconstruction Microscopy (STORM; ~10–55 nm resolution) and Photo-Activated Localization Microscopy (PALM/FPALM; ~10–55 nm resolution), to sequentially activate subsets of photoswitchable dyes in a field of view to achieve high spatial resolution (Galbraith and Galbraith, 2011; Sigal et al., 2018).
Each imaging technique comes with unique advantages and disadvantages in the way of hardware cost, necessary technical expertise, and required dyes or other reagents, among others. For example, the high number of on-off switches for fluorophores in STED may lead to increased photobleaching, especially given the generally high light intensities required for the technique; likewise, the generation of a single image by STORM, through many individual frames of subsets of fluorescing molecules, necessitates a lengthy acquisition period along with extensive processing (Tam and Merino, 2015). A careful consideration of technical requirements along with experimental requirements is necessary for the selection of any one technique for an experiment. Significantly, all of the techniques mentioned above can be utilized in some form to generate 3D “stacks” of individual 2D images taken at multiple Z positions. This is especially important for the study of lncRNAs, where 3D imaging of RNA, DNA, and protein with a high XYZ-resolution can paint a picture of immense clarity to locate and study their interacting partners within a single cell or nucleus. The greatest strength of all if these molecular cytological methods is that the same cell may be stained for multiple elements of interest (e.g. a protein, DNA locus, and an RNA; a chromosome, a specific RNA, and all newly transcribed RNA; etc) in order to understand their 3D spatial interactions. Only the number of fluorophores that can be simultaneously visualized with a microscope limits these combinations. With the advent of new LED-based light sources alongside lasers and more traditional gas lamps, the combination of individual channels that can be reliably visualized for fluorescence microscopy is steadily increasing (Buchwalow et al., 2018).
For the study of nuclear lncRNAs, which may be involved in complicated interactional networks with their molecular partners, it is particularly essential to be able to visualize multiple components in the same cell. Below we highlight the utility of single cell cytological studies in the study of nuclear lncRNA function by discussing several case studies highlighting the functional insights that came from the combinatorial use of molecular cytological techniques in the study of three lncRNAs: NEAT1, XIST, and HSATII.
NEAT1
When confronted with a potential lncRNA with no previously described role, cytological methods provide an excellent starting point to begin to collect information in order to understand its biological significance. Hutchinson et al. first discovered the two most abundant non-coding NEAT1 (Nuclear-enriched abundant transcript 1) isoforms, 3.7 kb and 23 kb long, through an array-based screen searching for RNAs enriched in cell nuclear fractions (Hutchinson et al., 2007). A combined co-immunoFISH approach initially demonstrated NEAT1 RNA to be localized in paraspeckles (Fox, 2009; Clemson et al., 2009; Fox and Lamond, 2010; Sasaki et al., 2009), which were later found to consist of phase-separated RNA-protein complexes along the periphery of nuclear speckles that regulate gene expression. These paraspeckles retain highly edited inosine-rich mRNA transcripts within the nucleus (Fox and Lamond, 2010; Fox et al., 2018; Klec et al., 2019; Sasaki et al., 2009; Zhang and Carmichael, 2001).
Simply knowing that a lncRNA localizes within a specified nuclear territory allows for a more pointed study of its role and function and the identification of interacting partners. Indeed, this initial discovery of NEAT1 localization paved the way for a number of crucial studies, all of which contributed to the development of the current model for the action of NEAT1 in paraspeckle formation (Fig. 1A). A series of siRNA knockdown experiments followed by co-immunoFISH for core paraspeckle proteins in combination with NEAT1 RNA explored the dynamics of its localization, and subsequently found NEAT1 transcription to be essential to the formation and maintenance of these paraspeckles (Chen and Carmichael, 2009; Clemson et al., 2009; Sasaki et al., 2009; Sunwoo et al., 2009). Further co-immunoFISH of core paraspeckle proteins, this time combined with NEAT1 DNA, revealed that paraspeckles form near the Neat1 locus in early G1, and often cluster around the locus in interphase (Bond and Fox, 2009; Clemson et al., 2009; Hutchinson et al., 2007). What we now know of the nucleating role of NEAT1 transcripts to co-transcriptionally recruit structurally crucial paraspeckle proteins was built on these early insights (Bond and Fox, 2009; Clemson et al., 2009; Naganuma et al., 2012; West et al., 2016; Yamazaki and Hirose, 2015).
Fig 1.
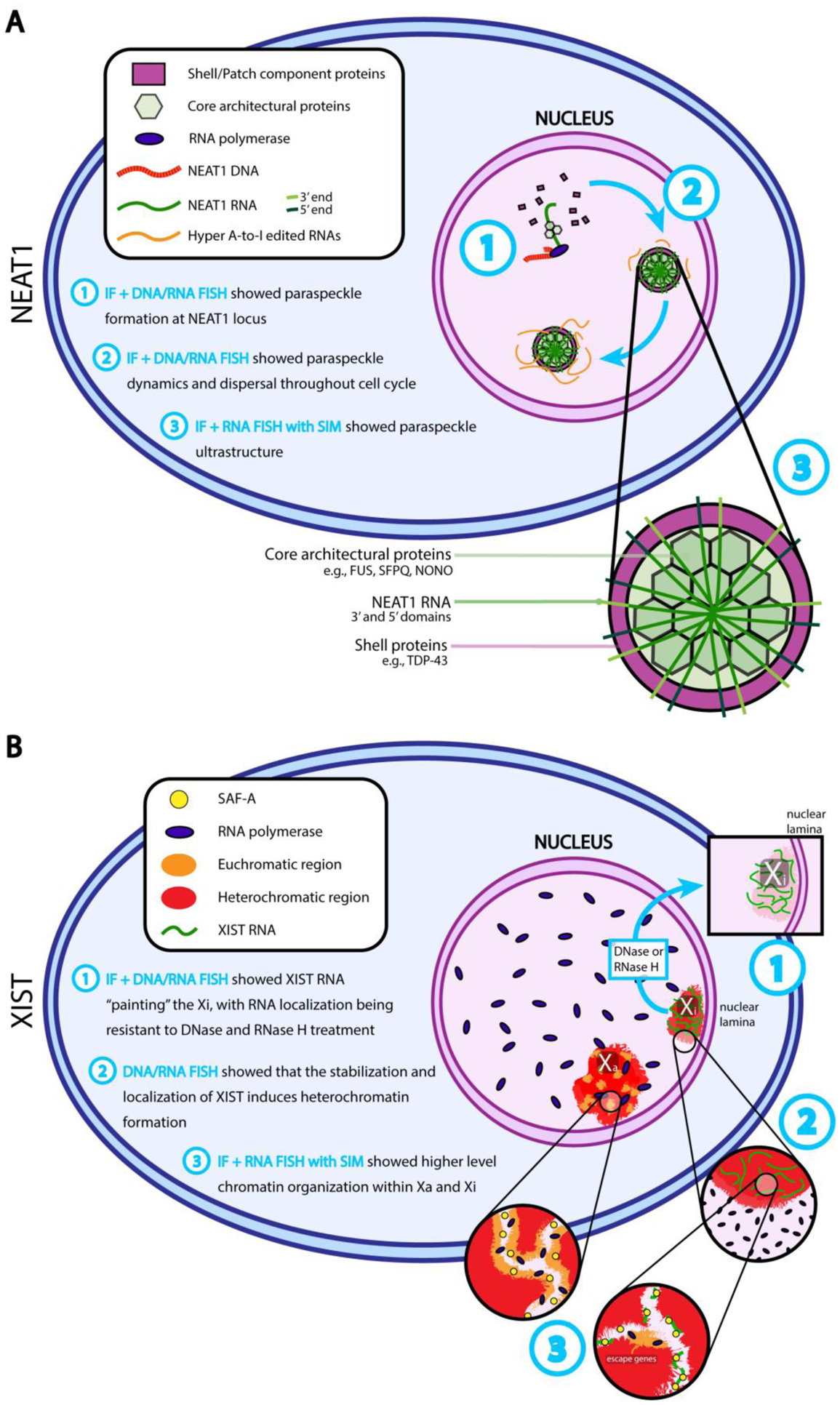
Key insights gained from molecular cytology on the nuclear function of NEAT1 and XIST lncRNAs. A) NEAT1 is transcribed and paraspeckle proteins are recruited to accumulated transcripts in the nucleus (Step 1). Paraspeckle components then organize (Step 2) into a core-shell structure, where NEAT-1 RNA transcripts are oriented with their 3’ and 5’ ends bundled along the shell while key paraspeckle proteins and the middle of NEAT-1 transcripts localize to the center (Step 3). B) XIST RNA localizes to, and “paints”, the inactive X chromosome in the nucleus, in a manner that is resistant to DNase or RNaseH treatment (Step 1). XIST coating of chromatin induces a suite of epigenetic effects ultimately resulting in heterochromatin formation (Step 2) and gene silencing within condensed folds of the chromosome, except for a few “escape genes” in transcriptionally permissive pockets (Step 3). Matrix-associating protein SAF-A is required for proper localization of Xist to the inactivated X chromosome (Hasegawa et al., 2010; Sunwoo et al., 2017), yet XIST’s localization dependency on SAF-A may vary in different cellular contexts (Kolpa et al., 2016).
There are many in vitro methods to examine fine-scale RNA-protein interactions and sequence targets, but they do not speak to any structural or organizational changes of cells in response to binding changes between RNA and protein partners within their native cellular environments or at the single cell level (Choi et al., 2017; Murthy and Rangarajan, 2010; Niranjanakumari et al., 2002; Popova et al., 2015; Poria and Ray, 2017). While molecular cytology may not provide quantitative binding affinity data, it does, in combination with genomic tools, allow for in situ observation of the effects of RNA sequence and expression changes on specific RNA-protein interactions. For example, co-immunoFISH for key paraspeckle proteins and NEAT1 truncated transcripts revealed interactions between specific regions of the NEAT1 transcript and paraspeckle proteins where the final 10kb of the longer NEAT1 isoform, but not the shorter isoform, is involved in recruitment and retention of paraspeckle proteins (Li et al., 2017; Naganuma et al., 2012; Nakagawa et al., 2011; Sasaki et al., 2009).
The use of super-resolution microscopy has allowed for a more detailed understanding of the organization of these paraspeckles with increasing resolution. For example, the use of SIM along with single molecule FISH (smFISH) illuminated a core-shell architecture in paraspeckles, in which the 3’ and 5’ ends of the long NEAT1 isoform transcript are bundled along the shell, while the middle of the transcript and structural paraspeckle proteins reside within the paraspeckle core (West et al., 2016; Yamazaki et al., 2018). Shell component proteins are distinct from structurally crucial core proteins, and so may interact with other nucleoplasmic elements to facilitate paraspeckle function (West et al., 2016), thus generating a refined candidate list of NEAT1 interacting proteins for further study.
XIST
The 17kb XIST lncRNA was first discovered and mapped to the X Inactivation Center (XIC/Xic), a locus on the X chromosome known to be required in cis for X chromosome inactivation (Brown et al., 1991b; Maxfield Boumil, 2001). It is transcribed exclusively from the inactivated X chromosome of cells with active dosage compensation (Brown et al., 1991a; Brown et al., 1991b; Kay et al., 1993; McCarrey and Dilworth, 1992; Rastan and Robertson, 1985; Richler et al., 1992). A high degree of conservation of the gene location and X chromosome-delimited expression within mouse and human cells pointed to an involvement of the gene in the X inactivation process (Ballabio and Willard, 1992; Borsani et al., 1991; Brockdorff et al., 1991; Brown et al., 1991a; McCarrey and Dilworth, 1992).
Through the use of RNA FISH, Brown and colleagues discovered that the XIST transcript itself was localized specifically to the inactive X chromosome (Brown et al., 1992), despite its being spliced, capped, and poly-adenylated (Cerase et al., 2015). Given this distinct nuclear localization and a lack of conserved open reading frames between mouse and human Xist/XIST genes (Migeon, 1994), this argued for a role of the RNA product of Xist/XIST itself (Brown et al., 1992; Migeon, 1994), which was later found to be required for X inactivation (Penny et al., 1996). The first glimpse of the molecular role of XIST RNA in X inactivation was demonstrated by DNA and RNA co-FISH for X chromosomal DNA and XIST RNA (Clemson, 1996). This study revealed a physical association between XIST RNA and the X chromosome territory, in which mature XIST transcripts “painted” the inactivated X chromosome in a transcription-independent manner (Clemson, 1996). Furthermore, XIST RNA retained its distinctive pattern of localization even after RNase H and DNase treatment which directly led to the idea that its localization might be facilitated by close interactions with the nuclear matrix, or scaffold (Clemson, 1996).
A number of cytological studies suggested that the characteristic in cis localization of XIST/Xist to the inactive X chromosome was key to X inactivation (Clemson, 1996; Duthie et al., 1999; Lee et al., 1996). Further, more specific RNA FISH for XIST using Xist transgene truncations allowed for determination of the regions responsible for the silencing and localization abilities of Xist RNA, wherein the 5’ A-repeat region of the transcript was found to be essential for silencing but not for its association with the X chromosome (Clemson et al., 1998; Hall and Lawrence, 2003; Wutz et al., 2002). XIST/Xist RNA has also been found to cause extensive remodeling of the inactivated X chromosome (Dyer et al., 1989; Escamilla-Del-Arenal et al., 2011; Galupa and Heard, 2018; Teller et al., 2011). These structural changes to the chromosomal territory were initially, and continue to be, observable only through molecular cytology-based methods. The simultaneous detection of multiple labels identified the repeat-rich and transcription-deficient nuclear compartment as forming the heart of the inactivated X territory, coincident with the Barr body (Calabrese et al., 2012; Chaumeil et al., 2006; Chow et al., 2010; Clemson et al., 2006; Escamilla-Del-Arenal et al., 2011; Jégu et al., 2017; Teller et al., 2011). Electron microscopy and light microscopy with DNA FISH showed highly condensed folds of heterochromatin in the inactivated X chromosome territory, distinct from both the active X chromosome and other territories of condensed heterochromatin in the nucleus (Rego et al., 2008). More recent work using 3D SIM adds even greater detail to our picture of the architecture of the inactivated X chromosome, suggesting that the inactive X is comprised of pockets of heterochromatin scattered with less compact transcriptionally permissive pockets (Smeets et al., 2014). Xist RNA localizes in foci throughout the inactivated chromosome territory in these permissive pockets, potentially associating stochastically with competent sites along the pocket (Smeets et al., 2014) (Fig. 1B).
Cytological methods also uniquely have the capacity to provide great insight into the cell cycle dynamics of any given RNA species in situ. XIST/Xist RNAs coat the inactive X chromosome in both human and mouse cells, but previous studies of its localization throughout the cell cycle hinted at potential species-specific differences in the dynamics of XIST/Xist retention on X chromosomes throughout the cell cycle. (Clemson, 1996; Duthie et al., 1999; Escamilla-Del-Arenal et al., 2011; Hall and Lawrence, 2003; Jonkers et al., 2008). However, the key limitation of the fixation-based methods used in these prior studies was that any one preparation offered but a single snapshot into a cell line prepared a specific way at a certain time. Live-cell imaging, however, has the capacity to circumvent some of these issues inherent in fixation-based cytological methods. Through the use of a MS2-tagged autosomal Xist transgene in male mouse cells, Ng et al. were able to present compelling evidence for the dissociation of transcripts from the X chromosome during mitosis followed by dynamic binding and displacement of Xist transcripts upon resynthesis (Ng et al., 2011).
More detailed live-imaging based techniques are likely to continue to reveal the dynamic nature of the transcription and interactions between other lncRNA species and their binding partners. The study of XIST RNA has not only served as a potent “gold standard” model to inform our understanding of the global mechanisms of nuclear lncRNAs, but it has also fueled the development of the molecular cytology-based tools to study the function of lncRNAs, which continue to be applied to their study today.
HSATII
A substantial percentage of eukaryotic genomes consists of repetitive elements, including satellites, DNA transposons, LINE retrotransposons, and others – together, they comprise up to half or more of the whole human genome (Garrido-Ramos, 2017; Levine et al., 2016; Levy et al., 2007). Tandemly repeating satellite sequences themselves make up a substantial percent of the human genome, with the most abundant species being alpha satellite (aSAT) and satellite 2/3 (HSATII/III) (Levy et al., 2007; Miga, 2015). Because assembly algorithms cannot uniquely map repetitive satellite reads, satellite DNA comprises a substantial proportion of unmapped reads of the human genome (Altemose et al., 2014). As a result, satellites are not as easily studied through standard genetic and genomic tools, and biochemical studies using extraction-based methods cannot easily be mapped to individual sites of repeats residing in the genome, thus necessitating the application of cytological analysis to both refine satellite repeat genomic mapping and to discern their functional roles.
Co-immunoFISH on extended chromatin fibers can further reveal satellite DNA composition with much higher spatial resolution than whole-cell staining (Sullivan, 2010), demonstrating a chromatin environment at centromeres distinct from surrounding euchromatin and heterochromatin (Sullivan and Karpen, 2004). Misregulation of this chromatin environment may result in the aberrant transcription of satellite sequences, particularly within the pericentric region (Biscotti et al., 2015; Brückmann et al., 2018; Carone and Lawrence, 2013; Hall et al., 2012; McNulty and Sullivan, 2018; Smurova and De Wulf, 2018). HSATII, characterized by a family of tandemly arranged sequence variants of a ~26 base-pair consensus sequence in the pericentric regions of a subset of human chromosomes (Tagarro et al., 1994), was initially shown to be overexpressed in a global expression survey of epithelial and pancreatic cancers (Bersani et al., 2015; Ting et al., 2011).
Though this initial observation was discovered using extraction-based methods, progress to reveal the localization and potential functions of HSATII lncRNA was revealed by a later study employing molecular cytology (Hall et al., 2017). HSATII transcripts were first visualized within tumor cell lines with abnormally large focal RNA accumulations in the nucleus (Hall et al., 2017). Because FISH probes may be designed and hybridized with high specificity to a target sequence, accumulated HSATII RNA nuclear foci could be traced to originate from individual HSATII loci (Hall et al., 2017). RNA FISH for HSATII sequence variants led to two key discoveries: 1) HSATII RNA remains in cis relative to its site of transcription, and 2) is not transcribed from the mega-arrays on chromosomes 1 and 16, but instead from smaller loci, such as chromosome 7 (Hall et al., 2017). Co-immunoFISH for HSATII DNA/RNA and Polycomb group proteins led to the development of a model for the activity of HSATII in cancer cells such that when the mega-arrays of HSATII on chromosomes 1 and 16 become demethylated, they nucleate aggregations of PRC1, which is concomitant with in cis expression of HSATII from other loci. HSATII RNA nuclear accumulations then recruit, and may sequester, DNA methyl-binding protein MeCP2 within the nucleus (Hall et al., 2017)(Fig. 2A).
Fig 2.
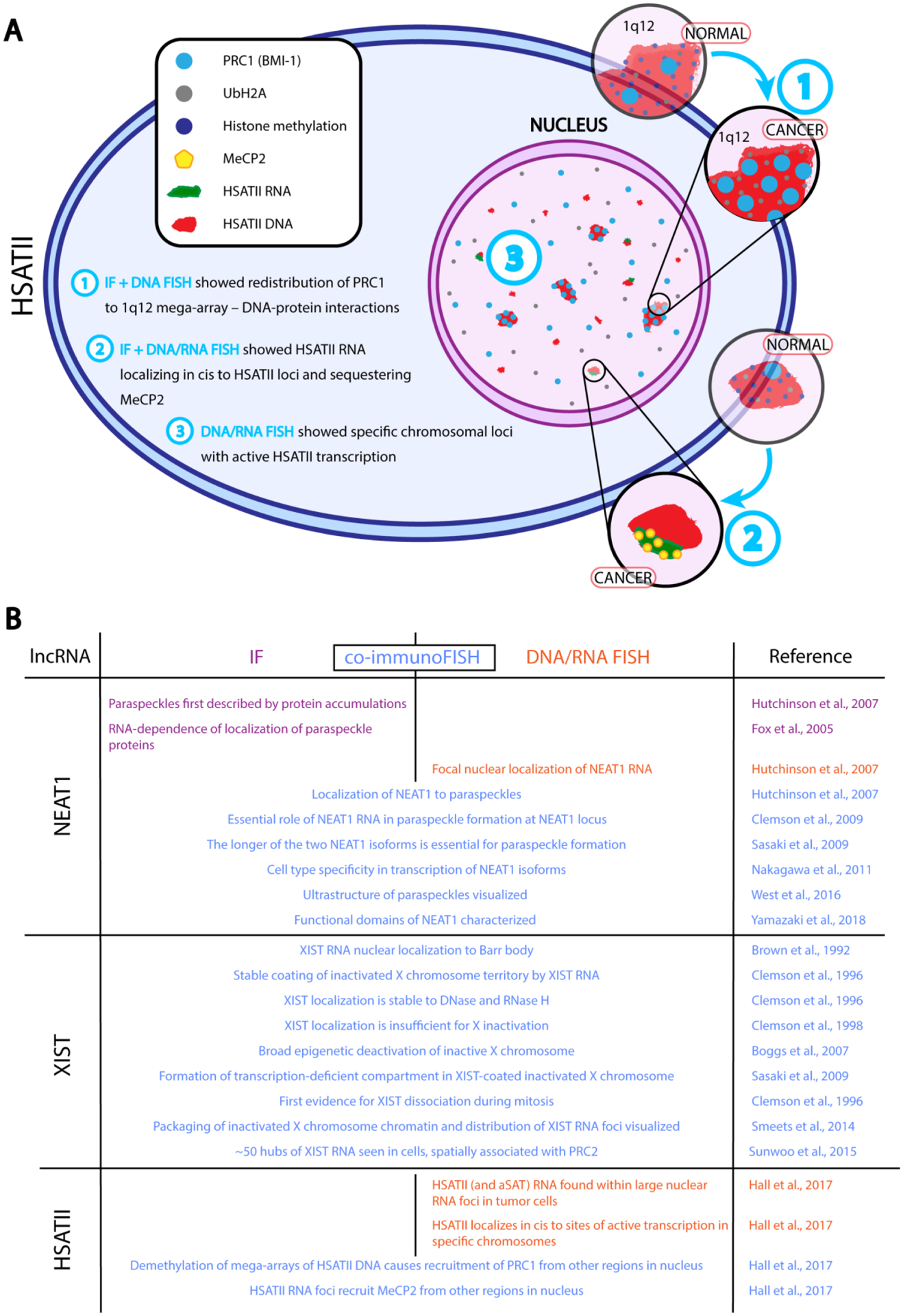
A) Key insights gained from molecular cytology on the nuclear function of HSATII lncRNA. Larger HSATII genomic locations (Chr1q12) accumulate PRC1 polycomb marks (BMI-1 and UbH2A) in cancer cells leading to an abnormal distribution of these nuclear proteins in cancer cells (Step 1). Smaller HSATII loci do not recruit polycomb marks and instead are transcribed, where HSATII lncRNA accumulates in cis and recruits MeCP2 proteins in cancer cells (Steps 2 and 3), leading to sequestration of key nuclear regulatory proteins. B) Summary of findings from cytological studies of NEAT1, XIST, and HSATII.
There is a wealth of information available on the biological functions of the other large families of human satellites. aSAT, HSATI, and HSATIII transcripts in particular have been found to be functionally relevant to centromere activity and chromosome segregation (Chan et al., 2012; Ideue et al., 2014; McNulty et al., 2017; Rošić et al., 2014; Talbert and Henikoff, 2018). Because satellite regions generally reside within facultative heterochromatin regions, their transcription may be affected by changes within the chromatin environment throughout the cell cycle. In accordance with this idea, mouse minor satellite transcription increases throughout the cell cycle from G0/G1 to S phase, to a peak in G2 (Ferri et al., 2009; Talbert and Henikoff, 2018). However, neither aSAT nor HSATIII show any such cell cycle dependent expression pattern, despite their requirement in proper kinetochore formation and chromosome segregation (McNulty et al., 2017; Rošić et al., 2014; Talbert and Henikoff, 2018). Overexpression of satellites such as aSAT can result in segregation errors and chromosomal instability, in the form of aneuploidy, chromosomal bridges, abnormal segregation, or micronuclei (Bouzinba-Segard et al., 2006; Ichida et al., 2018; Ting et al., 2011). HSATIII has also been found to be involved in the heat-shock response pathway and the formation of phase separated droplets within nuclei of cells under heat shock (nuclear stress bodies), nucleated by HSATIII transcripts (Biamonti and Vourc’h, 2010; Drino and Schaefer, 2018; Goenka et al., 2016; Ideue et al., 2014; Jolly et al., 2004; Rošić et al., 2014; Valgardsdottir et al., 2008). The RNA accumulations of HSATII that can be visualized through RNA FISH known as “CAST” (Cancer-Associated Satellite Transcript) bodies, are quite reminiscent of nuclear stress bodies, though little is known, to date, about their biochemical properties and stability (Hall et al., 2017).
Given their potentially pathological role in recruiting nuclear proteins in cis within cancer cell nuclei and the difficulty inherent in mapping HSATII transcripts, molecular cytology provides a promising means to begin to evaluate and understand their function. As illustrated here with HSATII and the other example lncRNAs (NEAT1 and XIST), molecular cytological methods provide a powerful set of tools to study the in situ transcription, activity, and dynamics of lncRNAs within single cells (Fig. 2B), while preserving the key information held in their spatiotemporal occurrence, which is more difficult with extraction-based and population-averaged methods, which we next discuss.
APPLYING EXTRACTION-BASED METHODS TO THE STUDY OF NUCLEAR lncRNA
Visualization of the spatial distribution and localization of a lncRNA of interest within the nucleus can be an excellent starting place to gather information about the potential function of the lncRNA of interest, particularly if candidate binding partners can be identified. One of the key features that has emerged as a more general role for nuclear lncRNAs is their ability to complex with chromatin, other RNA molecules, and proteins. This implies that each cell type would have specific ribonuclear protein complexes (RNPs), consisting of a distinctive combination of lncRNAs and RNA binding proteins (RBPs), which form and carry out their unique functions within the nucleus. While using molecular cytology to identify candidate RBPs of a lncRNA of interest is a sound strategy for lncRNA that are already known to colocalize with proteins (e.g. NEAT1, XIST, HSATII), there are also clear limitations to taking a candidate protein approach. Foremost, in order to study the proteins binding to a specific nuclear lncRNA, one first needs a list of candidate nuclear RBPs thought to interact with the RNA of interest. Thus, a candidate list of proteins that have the more general ability to bind RNA can be a good first step toward identifying potential binding partners of a lncRNA of interest.
Systematic Identification of RNA Binding Proteins in Diverse Cell Types
The systematic identification of RNA binding proteins has greatly expanded the number of RNA binding proteins, elucidated new functions, and led to a shift in our understanding of the function of RNA in RNPs (Baltz et al., 2012; Beckmann et al., 2015; Castello et al., 2012; Gerstberger et al., 2014). Central to these methods is a cross-linking step that covalently links bound proteins to RNA, ensuring that legitimate binding relationships are not lost as RNA-protein complexes are extracted from the cell (Baltz et al., 2012). UV crosslinking is more effective in identifying RNA-protein interactions, as crosslinking using formaldehyde or other methods can lead to isolation of indirect RNA-protein interactions and high background levels (Friedersdorf and Keene, 2014), even after exposure to denaturing conditions meant to disrupt such interactions (McHugh and Guttman, 2018; Ule et al., 2005). Labeling techniques further enhance the specificity of UV crosslinking through the use of nucleoside mimics that are incorporated into newly synthesized RNA, but not DNA (Favre et al., 1998), thus reducing background detection of DNA-protein interactions. Further advances have been made in reducing background levels and false positives through the use of stable isotope labeling by amino acids in cell culture (SILAC) in which heavy and light amino acids are labeled in separate samples, allowing for the comparison of relative amounts of peptides present in “treated” and “untreated” samples. This has proved to be an effective method for both determining proteins that are specifically enriched in crosslinked precipitates and eliminating background proteins (Mann, 2006).
These techniques have been successfully applied to identify the complete set of RBPs in a variety of cell types (Table 1A), setting the stage for further investigation into candidate RBPs (Baltz et al., 2012; Beckmann et al., 2015; Castello et al., 2012), and the possibility to investigate these identified RBPs for their ability to bind lncRNAs. In one study, the authors found the mRNA-bound proteome to contain nearly 800 proteins in a human embryonic kidney cell line, a third of which were not previously annotated as RNA binding and another 15 percent that were not even predicted to interact with RNA based on computational algorithms (Baltz et al., 2012). In another similar study, the authors used conventional UV-crosslinking and photoactivatable-ribonucleoside-enhanced-crosslinking (PAR-CL), with 4SU incorporation in parallel, as well as oligo(dT) purification to identify 860 proteins that directly bind to all mRNAs in a HeLa cell line (Castello et al., 2012). In addition to identifying more than 300 novel RBPs, this systematic identification revealed new roles for RNA-binding enzymes of intermediary metabolism and identified that many of the mRNA interacting proteins in HeLa cells consist of regions of short repetitive amino acids motifs and intrinsically disordered domains of RBPs (Castello et al., 2012).
Table 1.
Overview of biochemical approaches for probing lncRNA structure and function, including techniques that focus on A) RBP identification, B) techniques that have not previously focused on lncRNAs, but have the potential to rapidly expand our understanding of lncRNA structure, RBP binding and chromatin interactions, and C) techniques that have been successfully applied to specific lncRNAs. Rows shaded purple indicate techniques classified as RBP-centric in Fig. 3. Rows shaded blue mark techniques classified as RNP-centric and orange corresponds to those techniques that are lncRNA-centric in Fig. 3.
| A. Global Identification of RNA Binding Proteins in Various Cell Types | ||
|---|---|---|
| Cell Line | Overview | Reference |
| ESCs | Incorporation of 4SU and 6-SG nucleosides into RNA using UV crosslinking. Oligo(dT) purification used to isolate mRNA. | (Baltz et al., 2012) |
| HeLa | Standard UV-crosslinking and photoactivatable-ribonucleoside-enhanced-crosslinking (PAR-CL), using 4SU incorporation, were used to determine proteins that bind mRNAs. | (Castello et al., 2012) |
| Saccharomyces cerevisiae and Huh-7 | Applied both conventional (cCL) and PAR-CL to yeast and Huh-7 cells, to capture and compare the mRNA interactome across diverse cell types. | (Beckmann et al., 2015) |
| B. Techniques for studying RNA binding and overall RNA structure | |||
|---|---|---|---|
| Goal of Technique | Technique Name | Overview | Reference |
| Identify RBDs/RBRs within RBPs | RBDMap | Used UV crosslinking and two rounds of oligo d(T) capture, as well as protease and RNase treatment to identify the RBDs within RBPs. | (Castello et al., 2016) |
| Identify RBDs/RBRs within RBPs | RBRID | Determined optimal set of crosslinking chemistries in ESCs to identify all RBRs, rather than just those that bind polyadenylated mRNAs. | (He et al., 2016a) |
| Establish Transcripts Bound by RBPs | RBNS (RNA Bind-n-Seq) | Tagged RBPs with streptavidin binding tag and mixed with pool of RNA to pulldown and sequence bound RNAs. | (Lambert et al., 2014) |
| Establish Transcripts Bound by RBPs | RNAcompeteS | Incorporation of a competitive binding reaction along with a computational pipeline to interrogate RNP sequence and structure. | (Cook et al., 2017) |
| Establish Transcripts Bound by RBPs | HyperTRIBE | Fusion of hyperactivate A to I editing enzyme to RBP of interest allows for determination of transcripts bound by an RBP of interest. | (Nguyen et al., 2018) |
| Establish Transcripts Bound by RBPs | Improved CLIP | FLAG tagging of protein of interest allows for selective purification of RNAs bound to a single protein. | (Liu et al., 2019) |
| Establish Transcripts Bound by RBPs | fRIP-seq | Formaldehyde crosslinking, followed by RNA-immunoprecipitation of chromatin regulators to reveal RNA binding capabilities. | (Hendrickson et al., 2016) |
| Determine DNA Binding Sites of RBPs | ChIP-seq | Performed RBP-specific ChIP-seq to identify RBP- chromatin associations. | (Van Nostrand et al., 2018)(pre-print) |
| Determine DNA Binding Sites of RBPs | large-scale RBP ChIP-seq analysis | Applied RBP-ChIP-seq to two cell lines in order to map RBP occupancy to specific gene promoters and elucidate the role of RBPs in regulating transcription. | (Xiao et al., 2019) |
| Determine RNA Structure | DMS (dimethyl sulfate) foot printing | Use of distinct reactions that probe tertiary interactions, adenosine stacking and base-paring, to determine RNA structure. | (Peattie and Gilbert, 1980) |
| Determine RNA Structure | Structure-seq | DMS footprinting-based RNA structural analysis of chemically modified residues at large scale, yielding genome-wide data. | (Ding et al., 2015b) |
| Determine RNA Structure | SHAPE (selective 2′-hydroxyl acylation analyzed by primer extension) | Differential reactivities of each base with NMIA is used to determine RNA structure. | (Merino et al., 2005) |
| Determine RNA Structure | SHAPE-MaP (selective 2′- hydroxyl acylation analyzed by primer extension and mutational profiling) | Combines RNA structure identification with deep sequencing to more accurately predict in vivo folding. | (Siegfried et al., 2014) |
| Identify RNA-RNA Interactions | LIGR-seq | Utilizes incorporation of RNA intercalator 4’- aminome thyltrioxalen and UV irradiation to cause inter-strand adducts to form | (Sharma et. al., 2016) |
| Identify RNA-RNA Interactions | PARIS | Four main steps include UV crosslinking, 2D gel purification, proximity ligation and high- throughput sequencing | (Lu et. al., 2018) |
| C. Techniques for probing function that have been applied to lncRNAs of interest | ||||
|---|---|---|---|---|
| Goal of Technique | Technique | lncRNA(s) of Interest | Overview | Reference |
| Establish Protein Interactomes for lncRNAs of Interest | CHIRP-MS (Comprehensive Identification of RNA-Binding Proteins by Mass Spectrometry) | XIST | Short complementary tiling oligos and formaldehyde cross-linking are used to identify a broader set of protein interactors | (Chu et al., 2015a) |
| Establish Protein Interactomes for lncRNAs of Interest | RAP-MS (RNA Antisense Purification-Mass Spectrometry | XIST | Utilizes longer complementary tiling oligos, UV-crosslinking and identifies a smaller number of protein interactors | (McHugh et al., 2015) |
| Establish Protein Interactomes for lncRNAs of Interest | HyperMS (Hybridization purification of RNA-protein complexes followed by Mass Spectrometry) | MALAT1, NEAT1, NORAD | Multiplexing allows for identification of RBPs that interact with multiple lncRNAs at once | (Spiniello et al., 2018) |
| Map DNA Binding Sites of lncRNAs | CHART (Capture Hybridization Analysis of RNA Targets) | roX2 | Use of short, affinity-tagged oligonucleotides (C-oligos) to capture lncRNA of interest and map to genomic binding loci | (Simon et al., 2011) |
| Map DNA Binding Sites of lncRNAs | CHART | NEAT1 and MALAT1 | Application of CHART to multiple RNAs to compare binding patterns | (West et al., 2014) |
| Map DNA Binding Sites of lncRNAs | LongTarget | ANRIL, H19, Igf2-AS, Airn, Gnas-AS1, Kcnq1ot1 | First computational analysis for determining lncRNA DNA- binding motifs and binding | (He et al., 2015) |
| Map DNA Binding Sites of lncRNAs | TDF (Triplex Domain Finder) | Fendrr, HOTAIR, MEG3, GATA6- AS | Computational method that detects triplexes and thus characterizes DNA-binding domains and DNA targets statistically | (Kuo et al., 2019) |
| lncRNA Knockdown | RNAi | NEAT1 | Knocked down NEAT1 transcription using a pool of siRNAs to determine NEAT1 is required for paraspeckle formation. | (Clemson et al., 2009) |
| lncRNA Knockdown | LNA | Xist | Used LNAs to target XIST for release from inactive X Chr. | (Sarma et al., 2010) |
| lncRNA Knockdown | gapmeR | MALAT-1 | ASO-mediated targeting of MALAT-1 revealed role as molecular scaffold and therapeutic potential | (Sun et al., 2018), (Amodio et al., 2018b) |
An alternative study took a comparative approach to identify RBPs that are conserved between yeast and human Huh-7 cells. Intriguingly, this comparative analysis found that highly conserved RBPs also contain intrinsically disordered regions (Beckmann et al., 2015), highlighting our need to understanding these protein domains in relation to their ability to bind RNA (Jarvelin et al., 2016), especially in light of recent evidence that these regions can interact promiscuously with other proteins and contribute to phase separation and granule formation (Lin et al., 2017; Protter et al., 2018). While these studies have expanded our understanding of the types of proteins that bind RNA, we highlight there is also potential to expand the number of cell types to more fully characterize the cell-type specificity of the RNA interactome (Fig. 3). Another limitation is that, to date, these studies have been focused on identifying proteins that bind to polyadenylated mRNA, meaning it is possible that a unique set of yet-to-be-identified proteins bind to lncRNAs populations that are not polyadenylated (Zhang et al., 2014).
Fig 3.
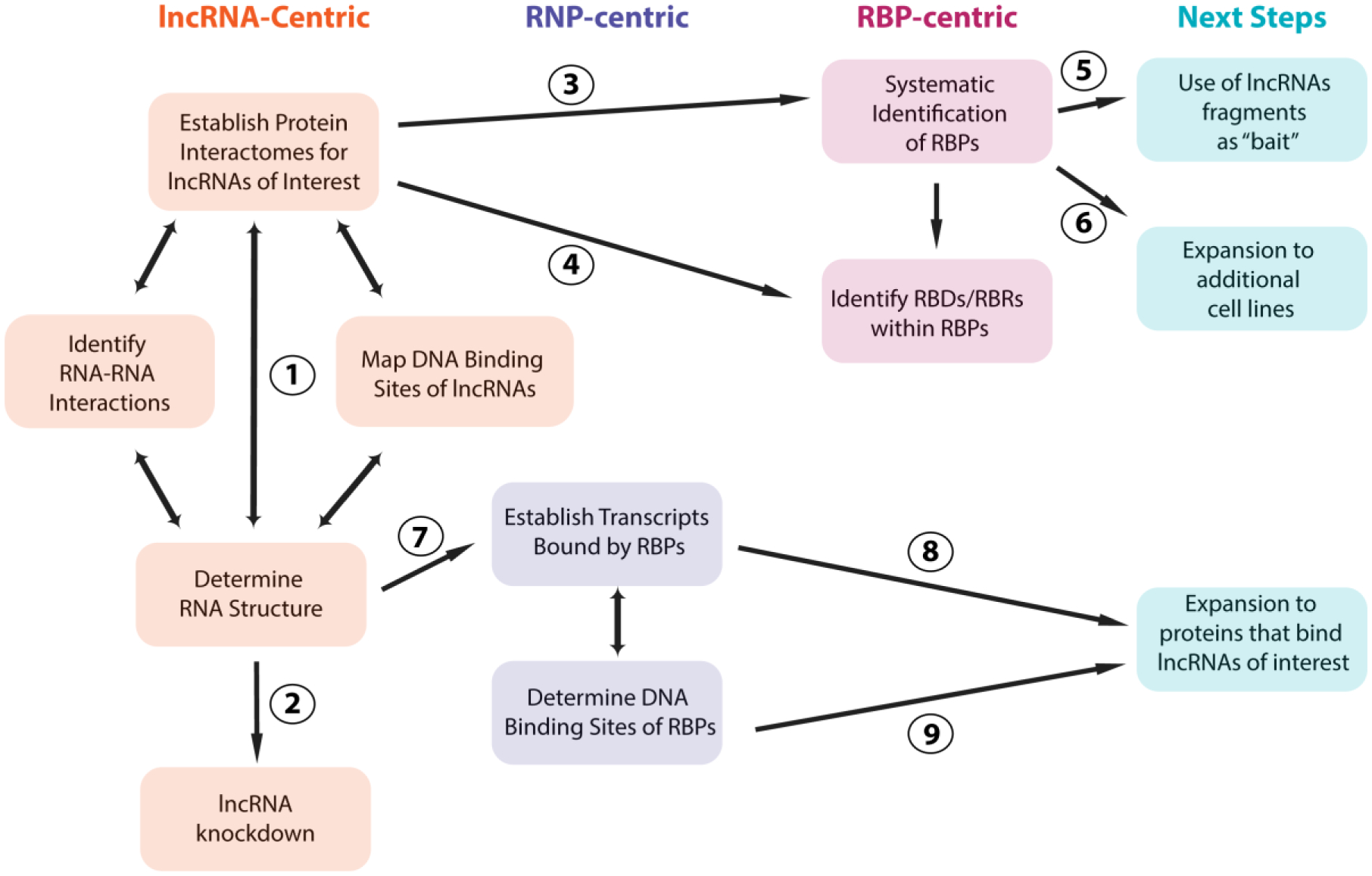
Potential pipeline for investigating lncRNAs of interest. Initial steps include determining protein interactomes, RNA structure, DNA binding sites and RNA-RNA interactions for lncRNAs of interest. This is likely to be an iterative process (1). While difficult for lncRNAs that either form densely packed RNPs or primarily associate with DNA and/or RNA, lncRNA knockdown is a valuable tool for determining the function of lncRNAs of interest (2). Identified proteins that bind a lncRNA of interest are then cross referenced against an established database of identified RBPs in the same cell line to either confirm that the proteins are known to bind RNA (3) or, if not, the protein may be a novel RBP with the capability to bind a specific lncRNA of interest. The RBDs and RBRs within RBPs should be analyzed for known lncRNA interactions and regions of RBPs containing RBDs and RBRs should be established for newly identified proteins (4). Previous databases of all RBPs should be expanded by using lncRNAs to pulldown and identify additional RBPs (5), and by application to additional cell lines (6). After establishing RNA structure, the transcripts bound by RBPs and their DNA binding sites should be determined (7). Both of these techniques have the potential to be applied to proteins that specifically bind lncRNAs of interest (8,9). Individual steps are shaded in colored boxes to indicate their overall classification (lncRNA-centric, RNP-centric, RBP-centric, Next Steps). Refer to Table 1 for a summary of individual techniques designed to achieve each step within colored boxes.
Investigation of lncRNA Interactomes and lncRNA-DNA Interactions
It is clear that identifying proteins with the ability to bind lncRNAs in various cell types is an important first step, yet in order to get a better understanding of how RBPs functionally interact with unique lncRNAs, it is essential to determine the specific nuclear proteins that bind to a lncRNA of interest. The mechanisms driving lncRNA-dependent changes in gene regulation and nuclear organization still remain undetermined for most lncRNAs (Rinn et al., 2012). However, techniques aimed toward identifying proteins that directly interact with lncRNAs have been developed and applied to some well-studied lncRNAs (Table 1C). For example, several studies have been conducted to identify the proteins that associate with XIST RNA. One such study developed CHIRP-MS (Comprehensive Identification of RNA-Binding Proteins by Mass Spectrometry) in which biotinylated tiling oligos complementary to Xist RNA were hybridized to fomaldehyde crosslinked cells, to ultimately identify 81 proteins that interact with Xist (Chu et al., 2015b). From this, nuclear RNA binding protein hnRNPK was found to directly bind to Xist and play a role in initiating gene silencing and the establishment of repressive histone modifications. Further, the Drosophila Split end homolog, Spen, was found to specifically interact with the A-repeat domain of Xist (Chu et al., 2015b). RNA antisense purification mass spectrometry (RAP-MS), an alternate technique for enriching for lncRNA, utilizes longer biotinylated probes coupled with UV-crosslinked cells and SILAC labeling (McHugh and Guttman, 2018). RAP-MS was also applied to identify Xist-binding proteins and identified a smaller set of ten proteins that directly bind to Xist RNA (McHugh et al., 2015). By knocking down each one of these ten proteins using siRNAs and assaying for inability to effectively silence gene expression on the X chromosome, three proteins that are required for XIST-mediated transcriptional repression were discovered (McHugh et al., 2015). One of these proteins, SHARP, is essential for both silencing the inactive X chromosome and excluding RNA polymerase II (Pol II) from the chromosome, through recruitment of HDAC3. While CHIRP-MS and RAP-MS present excellent tools to identify the proteins that specifically bind to a single RNA of interest and establish a list of proteins with functions to further investigate, multiplexing experiments have the potential to more rapidly identify the proteins that bind to multiple lncRNAs of interest. HyPR-MS (hybridization purification of RNA-protein complexes followed by mass spectrometry), a technique developed to identify the proteins that interact with multiple lncRNAs at once, was recently demonstrated for MALAT1, NEAT1 and NORAD lncRNAs. This technique has the potential to be applied to various lncRNAs in a multiplexed fashion to determine both the proteins that bind diverse lncRNAs and those that more specifically interact with individual lncRNAs (Spiniello et al., 2018).
Investigating the genomic binding sites of lncRNAs of interest can provide further insight into the nuclear function of lncRNAs and can confirm or inform cytology-based localization studies. The genomic binding sites of roX2, a ncRNA from Drosophila responsible for facilitating dosage compensation, were determined using capture hybridization analysis of RNA targets (CHART), revealing that this lncRNA binds to chromatin sites to which the MSL chromatin regulating complex reside (Simon et al., 2011). RAP has been used to study the binding sites of Xist transcripts across the X chromosome during the initiation of mammalian X inactivation, where it was found that Xist transcripts utilize 3D spatial proximity to spread from their site of transcription (Engreitz et al., 2013). This is entirely consistent with a cytological study utilizing the super resolution technique STORM, that proposed a “hit-and-run” model of methylation, whereby the limited number of Xist-PRC2 complexes in cells transiently associate with and methylate sites as they spread along the chromosome (Sunwoo et al., 2015). Similar applications of CHART has shown that NEAT1 and MALAT1 bind to sites of active transcription, consistent with their role in post-transcriptional regulation (West et al., 2014). Of note, a range of chromatin binding proteins have the capability to bind RNA (He et al., 2016b; Hendrickson et al., 2016), and RBPs and transcription factors often bind the same open-chromatin domains, suggesting a more widespread regulatory role in transcription (Xiao et al., 2019). While this remains an active area of research, it is emerging that in addition to binding genomic loci of 11interest, lncRNAs also interact with key chromatin binding proteins and regulate their function.
Determination of RNA Binding Domains (RBDs) and RNA Binding Regions (RBRs)
A more detailed understanding of the participating residues and structures within RBPs that bind RNA is critical for understanding how RNPs form and how protein and RNA function might be affected by binding. Prior to recent studies that have systematically identified RBDs, the focus of most attention was on the so-called “classical RNA binding domains”, which contain well defined RNA binding domains, such as the RNA recognition motif (RRM) (Maris et al., 2005), K homology (KH) domain, DEAD motif, double stranded RNA-binding motif (DSRM) or zinc-finger domain. This was primarily due to the fact that structural information on RNPs was obtained by X-ray crystallography, which requires the types of rigid folds found in globular protein domains, but not in intrinsically unfolded proteins (Beckmann et al., 2016). A variety of new techniques have recently been developed to identify the full set of RBDs in an unbiased fashion. RBPmap allows for the identification of RNA-binding regions from hundreds of RNA binding proteins in a single approach (Castello et al., 2016). RBR-ID, an alternative to RBD map, has been used to identify RNA-binding domains in embryonic stem cells (ESCs) (He et al., 2016b). Surprisingly, Castello et. al. found that more than half of RBPs do not contain conventional RBDs, but instead proteins with intrinsically disordered regions (IDRs) comprise half of the nearly 1,200 identified binding sites, adding to evidence citing the importance of unstructured protein domains as components of many RNPs (Jarvelin et al., 2016). Further, these RBDs are well conserved, suggesting important functional roles for these protein domains (Castello et. al., 2016, Beckman et. al., 2015). Disordered protein regions have been shown to be used by transcription factors to bind DNA (Vuzman and Levy, 2012) and roles in phase transitions and granule formation through RNA interactions with YGG repeats have been suggested (Zhang et al., 2015). For example, half of the identified paraspeckle proteins associated with NEAT1 (discussed above) contain an IDR (Nakagawa et al., 2018).. The IDR of one of these proteins, FUS, is required for phase-separated paraspeckle formation in vivo (Hennig et al., 2015). Abundant evidence has indicated the critical role that IDRs of paraspeckle proteins play in associating with NEAT1 and promoting paraspeckle formation under specific cellular conditions (Maharana et al., 2018; Yamazaki et al., 2018). The presence and conservation of distinct motifs within disordered regions across nonhomologous RBPs suggest a range of biological functions and interactions that have yet to be fully explored but represent an exciting area of ongoing research.
Methods to Predict RNA Structure and Next Generation Techniques
Techniques such as RBDmap and RBR-ID focus on protein-lncRNA interactions, yet the structure(s) that RNA molecules themselves may fold into within the nucleus can also dictate which proteins they associate with. Technological advancements in the development and adaptation of chemistry techniques have made it possible to study both the in vitro and in vivo folded structure of RNAs. The development of chemical reagents to probe RNA secondary and tertiary structure relies on the common principle that alkylating agents have more access to react with RNA bases that are available (not base-paired) as opposed to protected (by intramolecular base-pairing). Two of these alkylating chemical reagents used to probe RNA structure, DMS (dimethlysulfoxide) and NMIA (N-methylisotoic anhydride), differ in their substrate preference, with DMS reacting with only adenosine and cytosine residues while NMIA reacts with the 2’OH in any RNA base. DMS footprinting has long been used to probe DNA and RNA structure in vitro (Bevilacqua et al., 2016; Peattie and Gilbert, 1980; Tijerina et al., 2007) and has more recently been adapted for the in vivo study of lncRNA structure at single-nucleotide resolution by combining DMS footprinting with deep sequencing (Ding et al., 2015a). Similarly, the NMIA-based SHAPE (Selective 2’-Hydroxyl Acylation and Primer Extension) (Merino et al., 2005) technique can also be combined with deep sequencing to identify RNA bases involved in intramolecular base pairing to create predictions for RNA folding in vivo (SHAPE-MaP) (Siegfried et al., 2014).
Both of these techniques have been applied to the study of full-length nuclear-restricted lncRNAs including XIST (Fang et al., 2015; Smola et al., 2015), with each technique revealing slightly different structural predictions for Xist RNA folding, likely due to the experimental conditions and computational parameters (Pintacuda et al., 2017). SHAPE mapping of Neat1 has revealed structural differences between mouse and human transcripts suggesting that there may not be a singular, conserved lncRNA structure within paraspeckles, but highlight that long-range interactions within both human and mouse Neat1 transcripts may play an integral architectural role (Lin et al., 2018). Future work, largely dependent on obtaining larger datasets with functional validation of RNA structural predictions, holds much promise to more tightly connect lncRNA structure with functional interactions within the nucleus. Toward this aim, a recently developed technique, PARIS (Psoralen Analysis of RNA Structures and Interactions), has been applied in combination with other structural determination techniques to probe the connection between lncRNA structure and function (Lu, 2018; McCown et al., 2019). The four main steps of PARIS are in vivo crosslinking, 2D gel purification, proximity ligation and high throughput sequencing. Critically, the crosslinking step uses psoralen, which causes pyrimidine bases on opposite RNA strands to react which allows for RNA structure to be determined (Lu, 2018). As PARIS captures both intramolecular and intermolecular reactions between pyrimidine bases, PARIS allows for the determination of the structure of individual RNAs and the entire RNA interactome in a single experiment. Given the variety of techniques available for determining RNA structure, there is great combinatorial power in utilizing multiple techniques to determine the structure of lncRNAs for which structure may be difficult to determine. For example, a hypothetical secondary structure for MALAT1 was recently determined through RNA structure information derived from DMS-seq, PARIS and PARS (a technique in which RNA is extracted and then folded in vitro) datasets. Further analysis revealed that structural rearrangements may be induced by specific RNA modifications or mutations, and that these rearrangements may reveal or hide microRNA binding sites (McCown et al., 2019). Combining analysis of RNA structure with changes upon mutation, loss of interacting partners, and in specific cell types and developmental stages is an exciting potential application of these techniques. In the future, it will be possible to combine information obtained from RNA structural analyses and predictions from techniques such as RPBmap and RBR-ID to more accurately map RNA-protein interactions within RNP complexes (Fig. 3).
A variety of next-generation methods are currently being developed and modified to refine the relationships between RNA and protein (Table 1B). CLIP (crosslinking and immunoprecipitation) has remained a “gold-standard” approach for identifying transcripts bound to specific RBPs by using antibodies to specific proteins to pull down crosslinked RNAs bound to RBPs (Ule et al., 2005), and has resulted in the identification of many RNA-protein interactions in a variety of cell types and tissues, especially following the development of modifications to the technique to increase sensitivity and resolution (reviewed in (Lee and Ule, 2018)). In one such example, a recent improvement on the CLIP technique has been used to determine the set of transcripts (inclusive of lncRNAs) bound by IDH1, a recently identified RBP, revealing that this RBP binds RNA involved in diverse cellular processes including chromatin regulation and RNA processing (Liu et al., 2019). This technique, called FBioClip-Seq (Crosslinking and Immunoprecipitation via a FLAG- and Biotin- double tags followed by sequencing), utilizes a rigorous, two-step purification of RNA-protein complexes and yields greater sensitivity and an improved signal-to-noise ratio compared to previously developed CLIP-seq techniques (Liu et al., 2019). FBioClip-Seq has the potential to uncover the complete set of transcripts bound by newly identified RBPs, as well as previously unidentified lower abundance transcripts bound by more well-known RBPs for which traditional CLIP-based methods has previously been performed. However, one limitation to the CLIP-based techniques is the reliance on a large number of cells, thus limiting the application of these techniques to samples in which cells are in high abundance (i.e. cell culture and large tissues). To circumvent the need for large sample size, TRIBE (targets of RNA-binding proteins identified by editing) was recently developed, which utilizes the in vivo expression of the fusion of an RBP of interest to the catalytic domain of ADAR, an RNA-editing enzyme, allowing for the RNA nucleotide targets of a specific RBP to be identified by sequencing the RNA (McMahon et al., 2016). The double-stranded RNA binding domain (dsRBD) regions of the ADAR protein are not present in the fusion protein, meaning that the A to I edited sites read out by sequencing are determined solely by the features of the fused RBP of interest that bind RNA. Importantly, TRIBE is effective in determining the targets of RBDs in small cell populations, allowing for the determination of RBD targets from as few as 150 fly neurons (McMahon et al., 2016). A previously characterized hyperactive mutant of the catalytic domain of ADAR, E488Q, is used in HyperTRIBE (Xu et al., 2018). HyperTribe was used to determine the differential activity between cell types of the RNA binding protein Musashi-2 (MSI2), an RBD that has been shown to be regulate differentiation, as well as self-renewal capacity, in both normal hematopoietic stem and progenitor cells (HSPCs) and leukemic stem cells (LSCs) (Kharas et al., 2010; Park, 2014; Park, 2015) . MSI2 was shown to have decreased RNA binding activity in HSPCs compared to LSCs (Nguyen et al., 2018). Thus HyperTribe has the potential to determine differences in activity, as well as potential differences in binding sites, for RBPs that have important functions in rare cell types and/or are present in a small number of cells In order to expand our understanding of nuclear RNP formation and function in future studies, the application of improved CLIP technology, in more abundant cells populations, and TRIBE-based techniques, in more limited cell populations, to an increased number of RBPs has the potential to reveal the molecular interactions between lncRNAs and RBPs in high resolution (Fig. 3).
In addition to probing protein-RNA interactions, a range of new techniques have been developed to map RNA-RNA interactions, thus expanding our understanding of the molecular interactions of specific lncRNAs beyond RNA-protein and RNA-DNA interactions (Engreitz et al., 2014; Lu et al., 2016; Nguyen et al., 2016; Sharma et al., 2016). RAP-RNA, a technique that is based on RNA antisense purification (RAP), can be applied to a range of RNAs of interest in order to determine the full set of RNAs that interact. Importantly, this technique can be applied to RNAs of various size and is sensitive enough to determine the RNA interactome of low abundance RNAs (Engreitz et al., 2014). Another technique, ligation of interacting RNA and high-throughput sequencing (LIGR-seq), utilizes the RNA intercalator 4’-aminome-thyltrioxalen, which, following 365nm UV irradiation, causes the formation of inter-strand adducts between pyrimidine bases (Sharma et al., 2016). This technique was applied to uncover a highly complex network of RNA-RNA interactions that includes mRNA and a variety of ncRNAs in human HEK 293T cells, highlighting previously unknown interactions between small nucleolar (sno) RNAs and mRNAs (Sharma et al., 2016). Downstream functional analysis, such as the knockdown of newly identified ncRNAs that target mRNAs, can reveal the effect these interactions have on the stability and expression of target mRNAs, improving our understanding of the function of newly identified regulatory ncRNAs (Sharma et al., 2016). Coupling RNA-RNA interaction data with 3D chromatin conformation information obtained from techniques such as HiC (Lieberman-Aiden et al., 2009) and SPRITE (Quinodoz et al., 2018) poses the enticing possibility to more fully examine the intricate relationship between RNA-protein, RNA-DNA and RNA-RNA in a context-dependent manner. As the majority of these biochemical techniques have only been applied to a few cell types, lncRNAs, or RBPs of interest, there is great potential for application of these techniques to a broad range of lncRNAs and the RBPs that associate with them, resulting in a much more complete picture of lncRNP formation and function in the nucleus (Fig. 3).
Lnc-ing nuclear RNAs to function
It is important to recognize that an understanding of the specific RNA-protein interactions, especially when coupled with localization information obtained from molecular cytology-based studies, provides a strong foothold into the study of the molecular function of lncRNAs, but does not prove function without further study. Within the molecular toolkit aimed to examine function, a common genetic approach is to examine what occurs upon loss of function by knockout or knockdowns. Knockdown of nuclear lncRNAs has historically been more difficult than knockdown of mRNAs, likely due to the low abundance, cell-type and tissue specificity, and redundancy of many lncRNA transcripts (for more comprehensive review see (Ponting et al., 2009) and (Kopp and Mendell, 2018)). Further challenges lie in attempting to target lncRNAs embedded within nuclear architecture in that the RNA may not be available for targeting if it is tightly packed within nuclear compartments and complexed with DNA, protein, and other RNAs. Additionally, while RNAi is very effective for targeting mRNAs that are exported to the cytoplasm, targeting of nuclear lncRNAs has been much less effective. This is likely due to the localization of the components of the RNAi pathway within the cytoplasmic compartment (Behlke, 2016) and additional off-target effects. However, successful knockdown of NEAT1 was achieved by the use of an siRNA pool, ultimately demonstrating that NEAT1 was required for the formation of paraspeckles and indicating that some lncRNAs can be efficiently knocked down using conventional siRNA mechanisms (Clemson et al., 2009). To achieve higher potency in knocking down even highly abundant lncRNAs, techniques involving targeting by antisense oligonucleotides (ASOs) have been most successful. These ASOs often have modified bases (phosphothioate (PS), locked nucleic acids (LNA) or 2’O-Methyl RNA bases) to enhance specificity and increase stability (Zong et al., 2015). Many different types of ASOs have been used to target lncRNAs, whose ability to elicit lncRNA knockdown relies on recruiting endogenous RNAseH1, which is active within the nuclear compartment, to target the RNA-DNA hybrid formed upon base-pairing of the ASO to the target lncRNA. For example, targeting of LNA oligonucleotides to regions of Xist RNA revealed three key insights into its function: 1) that release of Xist RNA from the chromosome did not affect Xist transcription; 2) the A repeat region was required for chromosome coating; 3) components of the PRC2 complex were released along with Xist RNA (Sarma et al., 2010). ASO technology has also been successfully applied to target the lncRNA MALAT-1, which is overexpressed and responsible for pathological effects at the onset of multiple myeloma (MM), among other diseases (reviewed in (Amodio et al., 2018a)). ASO-mediated knockdown studies of MALAT-1 contributed key functional information leading to the current model of MALAT-1 acting as a molecular scaffold to enhance molecular interactions between protein-protein, protein-RNA, and protein-DNA at nuclear speckles (Sun et al., 2018). Further, targeted knockdown of MALAT-1 using an LNA-gapmeR resulted in decreased cell proliferation and triggered apoptosis in a mouse xenograft model of human MM (Amodio et al., 2018c). In addition to the use of ASO-mediated knockdown to reveal key functional effects of lncRNAs, this technology also holds much promise for therapeutic use. In one example, ASOs targeting expressed CTG repeat expansions within the DMPK gene in myotonic dystrophy type 1 (DM1) led to a 70% reduction in nuclear repeat foci and improved body weight and muscle strength in a mouse model of DM1 (Jauvin et al., 2017). Several other ASO-driven therapies are currently in clinical development (Bennett, 2019), highlighting this as a potent and effective mechanism to study the function of a lncRNA of interest in addition to potential uses as a therapeutic technology in a subset of diseases caused by misregulation of lncRNAs.
PERSPECTIVE
Significant progress has been made in studying the properties and functions of lncRNAs. We highlight here the diversity of information that can be gleaned from molecular cytology approaches, especially in the power to examine a specific lncRNA within its native context in a single nucleus. Further insight into a lncRNA’s abundance, cell cycle dynamics, mode of action (i.e. cis vs. trans), and candidate interacting partners have solidified cytology-based methods as a benchmark for the study of lncRNAs. These techniques have been instrumental in the study of many individual nuclear lncRNAs including model examples such as XIST and NEAT1 RNAs and have revealed key insights into their molecular function, yet drawbacks related to the low throughput of such techniques and correlative information that is often obtained (i.e. colocalization) can plague cytology-based studies. Biochemical approaches hold advantages in their high throughput nature, where information can be obtained from hundreds to millions of cells, and their power to reveal the nature of molecular interactions between lncRNA and nuclear proteins, chromatin, and other nuclear RNAs, yet this also has the distinct disadvantage that this information is averaged over a population of cells. Additionally, for lncRNAs that are in low abundance or only expressed in specific stages of the cell cycle, extraction-based techniques may miss their mark. In order to understand the mechanisms and functions of nuclear long noncoding RNAs in a more holistic manner, lncRNA researchers must utilize a vast array of these technologies available in their toolkit, with care taken to combine molecular cytology and extraction-based approaches to mitigate these inherent biases. Highlighting this, combinatorial use of these two broad approaches have revealed the most significant advances in our understanding of the functions of nuclear lncRNAs (e.g. XIST, NEAT1, HSATII, MALAT1, etc), as we highlight here.
From these technologies, emerging roles for lncRNA now suggest that lncRNA plays an active role in the organization of the nucleus into compartments with distinct functions in transcription and post-transcriptional regulation. One potentially exciting way in which lncRNA may mediate this organizational role is via its ability to recruit specific cofactors (chromatin, RNA, proteins) into phase-separated, liquid-liquid domains within the nucleus (reviewed in (Mir et al., 2019)). We note that this concept is congruous with early biochemical data suggesting that treatment of nuclei with RNase caused collapse of chromatin in the nucleus, suggesting that the bulk of RNA is required to maintain proper nuclear structure (Nickerson et al., 1989). We are poised on the precipice of understanding the myriad roles of lncRNAs in nuclear organization and genome regulation and the rapid development of techniques described here have been responsible for our arrival. Key to continued momentum and furthering our understanding of lncRNA functions will be the application and continued development of a variety of the technologies discussed here, with strategic combination of both cytological and biochemical-based information, to uncover the functions of nuclear long noncoding RNAs.
Acknowledgements:
This work was supported by funding to DMC from the Charles E. Kaufman Foundation.
List of Abbreviations
- lncRNA
long non-coding RNA
- miRNA
microRNA
- piRNA
piwi-interacting RNA
- crasiRNA
centromere repeat-associated small interacting RNA
- FISH
fluorescence in situ hybridization
- SIM
structured illumination microscopy
- STED
stimulated emission depletion
- STORM
stochastic optical reconstruction microscopy
- PALM
photo-activated localization microscopy
- NEAT1
nuclear-enriched abundant transcript 1
- XIST
X-inactive specific transcript
- HSAT
human satellite
- LINE
long interspersed nuclear element
- CAST
cancer-associated satellite transcript
- RNP
ribonuclear protein
- RBP
RNA binding protein
- SILAC
stable isotope labeling by amino acids in cell culture
- CHIRP
comprehensive identification of RNA-binding proteins
- RAP
RNA antisense purification
- MS
mass spectrometry
- CHART
capture hybridization analysis of RNA targets
- RRM
RNA recognition motif
- RBD
RNA binding domain
- RBR
RNA binding region
- IDR
intrinsically disordered region
- SHAPE
selective 2’-hydroxyl acylation and primer extension
- PARIS
psoralen analysis of RNA structures and interactions
- CLIP
crosslinking and immunoprecipitation
- TRIBE
targets of RNA-binding proteins identified by editing
- LIGR
ligation of interacting RNA
- ASO
antisense oligonucleotide
- LNA
locked nucleic acid
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
REFERENCES
- Altemose N, Miga KH, Maggioni M and Willard HF (2014). Genomic characterization of large heterochromatic gaps in the human genome assembly. PLoS Comput Biol 10, e1003628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amodio N, Raimondi L, Juli G, Stamato MA, Caracciolo D, Tagliaferri P and Tassone P (2018a). MALAT1: a druggable long non-coding RNA for targeted anti-cancer approaches. In J Hematol Oncol, vol. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amodio N, Stamato MA, Juli G, Morelli E, Fulciniti M, Manzoni M, Taiana E, Agnelli L, Cantafio MEG, Romeo E et al. (2018b). Drugging the lncRNA MALAT1 via LNA gapmeR ASO inhibits gene expression of proteasome subunits and triggers anti-multiple myeloma activity. Leukemia 32, 1948–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amodio N, Stamato MA, Juli G, Morelli E, Fulciniti M, Manzoni M, Taiana E, Agnelli L, Cantafio MEG, Romeo E et al. (2018c). Drugging the lncRNA MALAT1 via LNA gapmeR ASO inhibits gene expression of proteasome subunits and triggers anti-multiple myeloma activity. Leukemia 32, 1948–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballabio A and Willard HF (1992). Mammalian X-chromosome inactivation and the XIST gene. Curr Opin Genet Dev 2, 439–47. [DOI] [PubMed] [Google Scholar]
- Baltz A, Munschauer M, Schwanhausser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M et al. (2012). The mRNA-Bound Proteome and Its Global Occupancy Profile on Protein-Coding Transcripts. Molecular Cell 46, 674–690. [DOI] [PubMed] [Google Scholar]
- Beckmann B, Castello A and Medenbach J (2016). The expanding universe of ribonucleoproteins: of novel RNA-binding proteins and unconventional interactions. Pflugers Archiv-European Journal of Physiology 468, 1029–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann B, Horos R, Fischer B, Castello A, Eichelbaum K, Alleaume A, Schwarzl T, Curk T, Foehr S, Huber W et al. (2015). The RNA-binding proteomes from yeast to man harbour conserved enigmRBPs. Nature Communications 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behlke M (2016). Mini-review on current strategies to knockdown long non-coding RNAs. Journal of Rare Diseases Research & Treatment 1, 66–70. [Google Scholar]
- Beliveau BJ, Joyce EF, Apostolopoulos N, Yilmaz F, Fonseka CY, McCole RB, Chang Y, Li JB, Senaratne TN, Williams BR et al. (2012). Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proc Natl Acad Sci U S A 109, 21301–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett CF (2019). Therapeutic Antisense Oligonucleotides Are Coming of Age. Annu Rev Med 70, 307–321. [DOI] [PubMed] [Google Scholar]
- Bersani F, Lee E, Kharchenko PV, Xu AW, Liu M, Xega K, MacKenzie OC, Brannigan BW, Wittner BS, Jung H et al. (2015). Pericentromeric satellite repeat expansions through RNA-derived DNA intermediates in cancer. Proc Natl Acad Sci U S A 112, 15148–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevilacqua PC, Ritchey LE, Su Z and Assmann SM (2016). Genome-Wide Analysis of RNA Secondary Structure. Annu Rev Genet 50, 235–266. [DOI] [PubMed] [Google Scholar]
- Biamonti G and Vourc’h C (2010). Nuclear stress bodies. Cold Spring Harb Perspect Biol 2, a000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biscotti MA, Canapa A, Forconi M, Olmo E and Barucca M (2015). Transcription of tandemly repetitive DNA: functional roles. Chromosome Res 23, 463–77. [DOI] [PubMed] [Google Scholar]
- Bond CS and Fox AH (2009). Paraspeckles: nuclear bodies built on long noncoding RNA. The Journal of Cell Biology 186, 637–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev B and Cavalli G (2016). Organization and function of the 3D genome. Nature Reviews Genetics 17, 661–678. [DOI] [PubMed] [Google Scholar]
- Borsani G, Tonlorenzi R, Simmler MC, Dandolo L, Arnaud D, Capra V, Grompe M, Pizzuti A, Muzny D, Lawrence C et al. (1991). Characterization of a murine gene expressed from the inactive X chromosome. Nature 351, 325–9. [DOI] [PubMed] [Google Scholar]
- Bouzinba-Segard H, Guais A and Francastel C (2006). Accumulation of small murine minor satellite transcripts leads to impaired centromeric architecture and function. Proc Natl Acad Sci U S A 103, 8709–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockdorff N, Ashworth A, Kay GF, Cooper P, Smith S, McCabe VM, Norris DP, Penny GD, Patel D and Rastan S (1991). Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature 351, 329–31. [DOI] [PubMed] [Google Scholar]
- Brown CJ, Ballabio A, Rupert JL, Lafreniere RG, Grompe M, Tonlorenzi R and Willard HF (1991a). A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 349, 38–44. [DOI] [PubMed] [Google Scholar]
- Brown CJ, Lafreniere RG, Powers VE, Sebastio G, Ballabio A, Pettigrew AL, Ledbetter DH, Levy E, Craig IW and Willard HF (1991b). Localization of the X inactivation centre on the human X chromosome in Xq13. Nature 349, 82–4. [DOI] [PubMed] [Google Scholar]
- Brown J, Hendrich BD and Rupert JL (1992). The Human XIST Gene: Analysis of a 17 kb Inactive X-Specific RNA That Contains Conserved Repeats and Is Highly Localized within the Nucleus. Cell 71, 16. [DOI] [PubMed] [Google Scholar]
- Brückmann NH, Pedersen CB, Ditzel HJ and Gjerstorff MF (2018). Epigenetic Reprogramming of Pericentromeric Satellite DNA in Premalignant and Malignant Lesions. Mol Cancer Res 16, 417–427. [DOI] [PubMed] [Google Scholar]
- Buchwalow I, Samoilova V, Boecker W and Tiemann M (2018). Multiple immunolabeling with antibodies from the same host species in combination with tyramide signal amplification. Acta Histochem 120, 405–411. [DOI] [PubMed] [Google Scholar]
- Byron M, Hall LL and Lawrence JB (2013). A multifaceted FISH approach to study endogenous RNAs and DNAs in native nuclear and cell structures. Curr Protoc Hum Genet Chapter 4, Unit 4.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese JM, Sun W, Song L, Mugford JW, Williams L, Yee D, Starmer J, Mieczkowski P, Crawford GE and Magnuson T (2012). Site-specific silencing of regulatory elements as a mechanism of X inactivation. Cell 151, 951–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carone DM and Lawrence JB (2013). Heterochromatin instability in cancer: from the Barr body to satellites and the nuclear periphery. Semin Cancer Biol 23, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carone DM, Zhang C, Hall LE, Obergfell C, Carone BR, O’Neill MJ and O’Neill RJ (2013). Hypermorphic expression of centromeric retroelement-encoded small RNAs impairs CENP-A loading. Chromosome Res 21, 49–62. [DOI] [PubMed] [Google Scholar]
- Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann B, Strein C, Davey N, Humphreys D, Preiss T, Steinmetz L et al. (2012). Insights into RNA Biology from an Atlas of Mammalian mRNA-Binding Proteins. Cell 149, 1393–1406. [DOI] [PubMed] [Google Scholar]
- Castello A, Fischer B, Frese C, Horos R, Alleaume A, Foehr S, Curk T, Krijgsveld J and Hentze M (2016). Comprehensive Identification of RNA-Binding Domains in Human Cells. Molecular Cell 63, 696–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerase A, Pintacuda G, Tattermusch A and Avner P (2015). Xist localization and function: new insights from multiple levels. Genome Biology 16, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FL, Marshall OJ, Saffery R, Kim BW, Earle E, Choo KH and Wong LH (2012). Active transcription and essential role of RNA polymerase II at the centromere during mitosis. Proc Natl Acad Sci U S A 109, 1979–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaumeil J, Le Baccon P, Wutz A and Heard E (2006). A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes Dev 20, 2223–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LL and Carmichael GG (2009). Altered nuclear retention of mRNAs containing inverted repeats in human embryonic stem cells: functional role of a nuclear noncoding RNA. Mol Cell 35, 467–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Park C, Kim KE and Kim KK (2017). An in vitro technique to identify the RNA binding-site sequences for RNA-binding proteins. Biotechniques 63, 28–33. [DOI] [PubMed] [Google Scholar]
- Chow JC, Ciaudo C, Fazzari MJ, Mise N, Servant N, Glass JL, Attreed M, Avner P, Wutz A, Barillot E et al. (2010). LINE-1 activity in facultative heterochromatin formation during X chromosome inactivation. Cell 141, 956–69. [DOI] [PubMed] [Google Scholar]
- Chu C, Zhang Q, da Rocha S, Flynn R, Bharadwaj M, Calabrese J, Magnuson T, Heard E and Chang H (2015a). Systematic Discovery of Xist RNA Binding Proteins. Cell 161, 404–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C, Zhang Qiangfeng C., da Rocha Simão T., Flynn Ryan A., Bharadwaj M, Calabrese JM, Magnuson T, Heard E and Chang Howard Y. (2015b). Systematic Discovery of Xist RNA Binding Proteins. Cell 161, 404–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemson CM (1996). XIST RNA paints the inactive X chromosome at interphase: evidence for a novel RNA involved in nuclear/chromosome structure. The Journal of Cell Biology 132, 259–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemson CM, Chow JC, Brown CJ and Lawrence JB (1998). Stabilization and Localization of Xist RNA are Controlled by Separate Mechanisms and are Not Sufficient for X Inactivation. The Journal of Cell Biology 142, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemson CM, Hall LL, Byron M, McNeil J and Lawrence JB (2006). The X chromosome is organized into a gene-rich outer rim and an internal core containing silenced nongenic sequences. Proc Natl Acad Sci U S A 103, 7688–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemson CM, Hutchinson JN, Sara SA, Ensminger AW, Fox AH, Chess A and Lawrence JB (2009). An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol Cell 33, 717–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook KB, Vembu S, Ha KCH, Zheng H, Laverty KU, Hughes TR, Ray D and Morris QD (2017). RNAcompete-S: Combined RNA sequence/structure preferences for RNA binding proteins derived from a single-step in vitro selection. Methods 126, 18–28. [DOI] [PubMed] [Google Scholar]
- Darzynkiewicz Z, Traganos F, Zhao H, Halicka HD and Li J (2011). Cytometry of DNA replication and RNA synthesis: Historical perspective and recent advances based on “click chemistry”. Cytometry A 79, 328–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Kwok CK, Tang Y, Bevilacqua PC and Assmann SM (2015a). Genome-wide profiling of. Nature Protocols 10, 1050–1066. [DOI] [PubMed] [Google Scholar]
- Ding Y, Kwok CK, Tang Y, Bevilacqua PC and Assmann SM (2015b). Genome-wide profiling of in vivo RNA structure at single-nucleotide resolution using structure-seq. Nature Protocols 10, 1050–1066. [DOI] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F et al. (2012). Landscape of transcription in human cells. Nature 489, 101–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drino A and Schaefer MR (2018). RNAs, Phase Separation, and Membrane-Less Organelles: Are Post-Transcriptional Modifications Modulating Organelle Dynamics? Bioessays 40, e1800085. [DOI] [PubMed] [Google Scholar]
- Duthie SM, Nesterova TB, Formstone EJ, Keohane AM, Turner BM, Zakian SM and Brockdorff N (1999). Xist RNA exhibits a banded localization on the inactive X chromosome and is excluded from autosomal material in cis. Hum Mol Genet 8, 195–204. [DOI] [PubMed] [Google Scholar]
- Dyer KA, Canfield TK and Gartler SM (1989). Molecular cytological differentiation of active from inactive X domains in interphase: implications for X chromosome inactivation. Cytogenet Cell Genet 50, 116–20. [DOI] [PubMed] [Google Scholar]
- Eminaga S, Teekakirikul P, Seidman CE and Seidman JG (2016). Detection of Cell Proliferation Markers by Immunofluorescence Staining and Microscopy Imaging in Paraffin-Embedded Tissue Sections. Curr Protoc Mol Biol 115, 14.25.1–14.25.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Pandya-Jones A, McDonel P, Shishkin A, Sirokman K, Surka C, Kadri S, Xing J, Goren A, Lander ES et al. (2013). The Xist lncRNA Exploits Three-Dimensional Genome Architecture to Spread Across the X Chromosome. Science 341, 1237973–1237973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Sirokman K, McDonel P, Shishkin AA, Surka C, Russell P, Grossman SR, Chow AY, Guttman M and Lander ES (2014). RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent Pre-mRNAs and chromatin sites. Cell 159, 188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escamilla-Del-Arenal M, da Rocha ST and Heard E (2011). Evolutionary diversity and developmental regulation of X-chromosome inactivation. Hum Genet 130, 307–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Moss WN, Rutenberg-Schoenberg M and Simon MD (2015). Probing Xist RNA Structure in Cells Using Targeted Structure-Seq. PLOS Genetics 11, e1005668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favre A, Saintomé C, Fourrey JL, Clivio P and Laugâa P (1998). Thionucleobases as intrinsic photoaffinity probes of nucleic acid structure and nucleic acid-protein interactions. J Photochem Photobiol B 42, 109–24. [DOI] [PubMed] [Google Scholar]
- Ferri F, Bouzinba-Segard H, Velasco G, Hubé F and Francastel C (2009). Non-coding murine centromeric transcripts associate with and potentiate Aurora B kinase. Nucleic Acids Res 37, 5071–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox AH, Lam YW, Leung AK, Lyon CE, Andersen J, Mann M and Lamond AI (2002). Paraspeckles: a novel nuclear domain. Curr Biol 12, 13–25. [DOI] [PubMed] [Google Scholar]
- Fox AH and Lamond AI (2010). Paraspeckles. Cold Spring Harbor Perspectives in Biology 2, a000687–a000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox AH, Nakagawa S, Hirose T and Bond CS (2018). Paraspeckles: Where Long Noncoding RNA Meets Phase Separation. Trends Biochem Sci 43, 124–135. [DOI] [PubMed] [Google Scholar]
- Friedersdorf M and Keene J (2014). Advancing the functional utility of PAR-CLIP by quantifying background binding to mRNAs and lncRNAs. Genome Biology 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith CG and Galbraith JA (2011). Super-resolution microscopy at a glance. J Cell Sci 124, 1607–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall JG and Pardue ML (1969). Formation and detection of RNA-DNA hybrid molecules in cytological preparations. Proc Natl Acad Sci U S A 63, 378–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galupa R and Heard E (2018). X-Chromosome Inactivation: A Crossroads Between Chromosome Architecture and Gene Regulation. Annu Rev Genet 52, 535–566. [DOI] [PubMed] [Google Scholar]
- Garrido-Ramos MA (2017). Satellite DNA: An Evolving Topic. Genes (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstberger S, Hafner M and Tuschl T (2014). A census of human RNA-binding proteins. Nature Reviews Genetics 15, 829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goenka A, Sengupta S, Pandey R, Parihar R, Mohanta GC, Mukerji M and Ganesh S (2016). Human satellite-III non-coding RNAs modulate heat-shock-induced transcriptional repression. J Cell Sci 129, 3541–3552. [DOI] [PubMed] [Google Scholar]
- Gratzner HG (1982). Monoclonal antibody to 5-bromo- and 5-iododeoxyuridine: A new reagent for detection of DNA replication. Science 218, 474–5. [DOI] [PubMed] [Google Scholar]
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP et al. (2009). Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemeijer MC, Vonk AM, Monastyrska I, Rottier PJ and de Haan CA (2012). Visualizing coronavirus RNA synthesis in time by using click chemistry. J Virol 86, 5808–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall LE, Mitchell SE and O’Neill RJ (2012). Pericentric and centromeric transcription: a perfect balance required. Chromosome Res 20, 535–46. [DOI] [PubMed] [Google Scholar]
- Hall LL, Byron M, Carone DM, Whitfield TW, Pouliot GP, Fischer A, Jones P and Lawrence JB (2017). Demethylated HSATII DNA and HSATII RNA Foci Sequester PRC1 and MeCP2 into Cancer-Specific Nuclear Bodies. Cell Rep 18, 2943–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall LL and Lawrence JB (2003). The cell biology of a novel chromosomal RNA: chromosome painting by XIST/Xist RNA initiates a remodeling cascade. Semin Cell Dev Biol 14, 369–78. [DOI] [PubMed] [Google Scholar]
- Hasegawa Y, Brockdorff N, Kawano S, Tsutui K and Nakagawa S (2010). The matrix protein hnRNP U is required for chromosomal localization of Xist RNA. Dev Cell 19, 469–76. [DOI] [PubMed] [Google Scholar]
- Haukenes G, Szilvay AM, Brokstad KA, Kanestrøm A and Kalland KH (1997). Labeling of RNA transcripts of eukaryotic cells in culture with BrUTP using a liposome transfection reagent (DOTAP). Biotechniques 22, 308–12. [DOI] [PubMed] [Google Scholar]
- He C, Sidoli S, Warneford-Thomson R, Tatomer D, Wilusz J, Garcia B and Bonasio R (2016a). High-Resolution Mapping of RNA-Binding Regions in the Nuclear Proteome of Embryonic Stem Cells. Molecular Cell 64, 416–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Sidoli S, Warneford-Thomson R, Tatomer Deirdre C., Wilusz Jeremy E., Garcia Benjamin A. and Bonasio R (2016b). High-Resolution Mapping of RNA-Binding Regions in the Nuclear Proteome of Embryonic Stem Cells. Molecular Cell 64, 416–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L and Hannon GJ (2019). MicroRNAs: small RNAs with a big role in gene regulation. Nature Reviews Genetics 5, 522–531. [DOI] [PubMed] [Google Scholar]
- He S, Zhang H, Liu H and Zhu H (2015). LongTarget: a tool to predict lncRNA DNA-binding motifs and binding sites via Hoogsteen base-pairing analysis. Bioinformatics 31, 178–86. [DOI] [PubMed] [Google Scholar]
- Hendrickson D, Kelley D, Tenen D, Bernstein B and Rinn J (2016). Widespread RNA binding by chromatin-associated proteins. Genome Biology 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig S, Kong G, Mannen T, Sadowska A, Kobelke S, Blythe A, Knote G, Iyer K, Ho D, Newcombe E et al. (2015). Prion-like domains in RNA binding proteins are essential for building subnuclear paraspeckles. Journal of Cell Biology 210, 529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horisawa K (2014). Specific and quantitative labeling of biomolecules using click chemistry. Front Physiol 5, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson JN, Ensminger AW, Clemson CM, Lynch CR, Lawrence JB and Chess A (2007). A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 8, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichida K, Suzuki K, Fukui T, Takayama Y, Kakizawa N, Watanabe F, Ishikawa H, Muto Y, Kato T, Saito M et al. (2018). Overexpression of satellite alpha transcripts leads to chromosomal instability via segregation errors at specific chromosomes. Int J Oncol. [DOI] [PubMed] [Google Scholar]
- Ideue T, Cho Y, Nishimura K and Tani T (2014). Involvement of satellite I noncoding RNA in regulation of chromosome segregation. Genes Cells 19, 528–38. [DOI] [PubMed] [Google Scholar]
- Jao CY and Salic A (2008). Exploring RNA transcription and turnover in vivo by using click chemistry. Proc Natl Acad Sci U S A 105, 15779–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvelin A, Noerenberg M, Davis I and Castello A (2016). The new (dis)order in RNA regulation. Cell Communication and Signaling 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jauvin D, Chrétien J, Pandey SK, Martineau L, Revillod L, Bassez G, Lachon A, McLeod AR, Gourdon G, Wheeler TM et al. (2017). Targeting DMPK with Antisense Oligonucleotide Improves Muscle Strength in Myotonic Dystrophy Type 1 Mice. In Mol Ther Nucleic Acids, vol. 7, pp. 465–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsson P, Lipovich L, Grandér D and Morris KV (2014). Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim Biophys Acta 1840, 1063–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly C, Metz A, Govin J, Vigneron M, Turner BM, Khochbin S and Vourc’h C (2004). Stress-induced transcription of satellite III repeats. J Cell Biol 164, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers I, Monkhorst K, Rentmeester E, Grootegoed JA, Grosveld F and Gribnau J (2008). Xist RNA Is Confined to the Nuclear Territory of the Silenced X Chromosome throughout the Cell Cycle. Molecular and Cellular Biology 28, 5583–5594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jégu T, Aeby E and Lee JT (2017). The X chromosome in space. Nat Rev Genet 18, 377–389. [DOI] [PubMed] [Google Scholar]
- Kay F, Penny D, Ashworth A and Drockdorff N (1993). Expression of XIST During Mouse Development Suggests a Role in the Initiation of X Chromosome Inactivation. Cell 72, 12. [DOI] [PubMed] [Google Scholar]
- Kharas M, Lengner C, Al-Shahrour F, Bullinger L, Ball B, Zaidi S, Morgan K, Tam W, Paktinat M, Okabe R et al. (2010). Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nature Medicine 16, 903–U101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klec C, Prinz F and Pichler M (2019). Involvement of the long noncoding RNA NEAT1 in carcinogenesis. Molecular Oncology 13, 46–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolpa HJ, Fackelmayer FO and Lawrence JB (2016). SAF-A Requirement in Anchoring XIST RNA to Chromatin Varies in Transformed and Primary Cells. Dev Cell 39, 9–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp F and Mendell JT (2018). Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 172, 393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CC, Hänzelmann S, Sentürk Cetin N, Frank S, Zajzon B, Derks JP, Akhade VS, Ahuja G, Kanduri C, Grummt I et al. (2019). Detection of RNA-DNA binding sites in long noncoding RNAs. Nucleic Acids Res 47, e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert N, Robertson A, Jangi M, McGeary S, Sharp PA and Burge CB (2014). RNA Bind-n-Seq: quantitative assessment of the sequence and structural binding specificity of RNA binding proteins. Mol Cell 54, 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JB, Singer RH and Marselle LM (1989). Highly localized tracks of specific transcripts within interphase nuclei visualized by in situ hybridization. Cell 57, 493–502. [DOI] [PubMed] [Google Scholar]
- Lee FCY and Ule J (2018). Advances in CLIP Technologies for Studies of Protein-RNA Interactions. Mol Cell 69, 354–369. [DOI] [PubMed] [Google Scholar]
- Lee JT, Strauss WM, Dausman JA and Jaenisch R (1996). A 450 kb Transgene Displays Properties of the Mammalian X-Inactivation Center. Cell 86, 83–94. [DOI] [PubMed] [Google Scholar]
- Lee YS, Shibata Y, Malhotra A and Dutta A (2009). A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev 23, 2639–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengronne A, Pasero P, Bensimon A and Schwob E (2001). Monitoring S phase progression globally and locally using BrdU incorporation in TK(+) yeast strains. Nucleic Acids Res 29, 1433–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Ting DT and Greenbaum BD (2016). P53 and the defenses against genome instability caused by transposons and repetitive elements. Bioessays 38, 508–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G et al. (2007). The diploid genome sequence of an individual human. PLoS Biol 5, e254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Harvey AR, Hodgetts SI and Fox AH (2017). Functional dissection of NEAT1 using genome editing reveals substantial localization of the NEAT1_1 isoform outside paraspeckles. RNA 23, 872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO et al. (2009). Comprehensive mapping of long range interactions reveals folding principles of the human genome. Science 326, 289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Currie SL and Rosen MK (2017). Intrinsically disordered sequences enable modulation of protein phase separation through distributed tyrosine motifs. J Biol Chem 292, 19110–19120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Schmidt BF, Bruchez MP and McManus CJ (2018). Structural analyses of NEAT1 lncRNAs suggest long-range RNA interactions that may contribute to paraspeckle architecture. Nucleic Acids Res 46, 3742–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Li T, Song G, He Q, Yin Y, Lu J, Bi X, Wang K, Luo S, Chen Y et al. (2019). Insight into novel RNA-binding activities via large-scale analysis of lncRNA-bound proteome and IDH1-bound transcriptome. Nucleic Acids Research 47, 2244–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z (2018). PARIS: Psoralen Analysis of RNA Interactions and Structures with High Throughput and Resolution, vol. 1649 Methods in Molecular Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Zhang QC, Lee B, Flynn RA, Smith MA, Robinson JT, Davidovich C, Gooding AR, Goodrich KJ, Mattick JS et al. (2016). RNA Duplex Map in Living Cells Reveals Higher-Order Transcriptome Structure. Cell 165, 1267–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharana S, Wang J, Papadopoulos D, Richter D, Pozniakovsky A, Poser I, Bickle M, Rizk S, Guillen-Boixet J, Franzmann T et al. (2018). RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 360, 918–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann M (2006). Functional and quantitative proteomics using SILAC. Nature Reviews Molecular Cell Biology 7, 952–958. [DOI] [PubMed] [Google Scholar]
- Maris C, Dominguez C and Allain F (2005). The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. Febs Journal 272, 2118–2131. [DOI] [PubMed] [Google Scholar]
- Mattout A, Cabianca DS and Gasser SM (2015). Chromatin states and nuclear organization in development — a view from the nuclear lamina. Genome Biology 16, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield Boumil R (2001). Forty years of decoding the silence in X-chromosome inactivation. Human Molecular Genetics 10, 2225–2232. [DOI] [PubMed] [Google Scholar]
- McCarrey JR and Dilworth DD (1992). Expression of Xist in mouse germ cells correlates with X-chromosome inactivation. Nat Genet 2, 200–3. [DOI] [PubMed] [Google Scholar]
- McCown PJ, Wang MC, Jaeger L and Brown JA (2019). Secondary Structural Model of Human MALAT1 Reveals Multiple Structure-Function Relationships. Int J Mol Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh C, Chen C, Chow A, Surka C, Tran C, McDonel P, Pandya-Jones A, Blanco M, Burghard C, Moradian A et al. (2015). The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 521, 232-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh CA and Guttman M (2018). RAP-MS: A Method to Identify Proteins that Interact Directly with a Specific RNA Molecule in Cells In RNA Detection: Methods and Protocols, (ed. Gaspar I), pp. 473–488. New York, NY: Springer New York. [DOI] [PubMed] [Google Scholar]
- McMahon A, Rahman R, Jin H, Shen J, Fieldsend A, Luo W and Rosbash M (2016). TRIBE: Hijacking an RNA-Editing Enzyme to Identify Cell-Specific Targets of RNA-Binding Proteins. Cell 165, 742–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNulty SM and Sullivan BA (2018). Alpha satellite DNA biology: finding function in the recesses of the genome. Chromosome Res 26, 115–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNulty SM, Sullivan LL and Sullivan BA (2017). Human Centromeres Produce Chromosome-Specific and Array-Specific Alpha Satellite Transcripts that Are Complexed with CENP-A and CENP-C. Dev Cell 42, 226–240.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino E, Wilkinson K, Coughlan J and Weeks K (2005). RNA structure analysis at single nucleotide resolution by selective 2 ‘-hydroxyl acylation and primer extension (SHAPE). Journal of the American Chemical Society 127, 4223–4231. [DOI] [PubMed] [Google Scholar]
- Miga KH (2015). Completing the human genome: the progress and challenge of satellite DNA assembly. Chromosome Res 23, 421–6. [DOI] [PubMed] [Google Scholar]
- Migeon BR (1994). X-chromosome inactivation: molecular mechanisms and genetic consequences. Trends Genet 10, 230–5. [DOI] [PubMed] [Google Scholar]
- Mir M, Bickmore W, Furlong EEM and Narlikar G (2019). Chromatin topology, condensates and gene regulation: shifting paradigms or just a phase? [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moazed D (2009). Small RNAs in transcriptional gene silencing and genome defence. Nature 457, 413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy UM and Rangarajan PN (2010). Identification of protein interaction regions of VINC/NEAT1/Men epsilon RNA. FEBS Lett 584, 1531–5. [DOI] [PubMed] [Google Scholar]
- Naganuma T, Nakagawa S, Tanigawa A, Sasaki YF, Goshima N and Hirose T (2012). Alternative 3′-end processing of long noncoding RNA initiates construction of nuclear paraspeckles: LncRNA processing for nuclear body architecture. The EMBO Journal 31, 4020–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Naganuma T, Shioi G and Hirose T (2011). Paraspeckles are subpopulation-specific nuclear bodies that are not essential in mice. The Journal of Cell Biology 193, 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Yamazaki T and Hirose T (2018). Molecular dissection of nuclear paraspeckles: towards understanding the emerging world of the RNP milieu. Open Biology 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng K, Daigle N, Bancaud A, Ohhata T, Humphreys P, Walker R, Ellenberg J and Wutz A (2011). A system for imaging the regulatory noncoding Xist RNA in living mouse embryonic stem cells. Mol Biol Cell 22, 2634–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D, Lu Y, Choo Z, Chin C, Prieto C, Gourkanti S, Leslie C and Kharas M (2018). The RNA Binding Protein MSI2 Has Increased RNA Binding Activity in Leukemic Stem Cells Compared to Normal Hematopoietic Stem Cells. Blood 132. [Google Scholar]
- Nguyen T (2016). Mapping RNA-RNA interactome and RNA structure in vivo by MARIO. Nature Communications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TC, Cao X, Yu P, Xiao S, Lu J, Biase FH, Sridhar B, Huang N, Zhang K and Zhong S (2016). Mapping RNA–RNA interactome and RNA structure in vivo by MARIO. Nature Communications 7, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson JA, Krochmalnic G, Wan KM and Penman S (1989). Chromatin architecture and nuclear RNA. Proc Natl Acad Sci U S A 86, 177–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niranjanakumari S, Lasda E, Brazas R and Garcia-Blanco MA (2002). Reversible cross-linking combined with immunoprecipitation to study RNA-protein interactions in vivo. Methods 26, 182–90. [DOI] [PubMed] [Google Scholar]
- Nozawa RS and Gilbert N (2019). RNA: Nuclear Glue for Folding the Genome. Trends Cell Biol 29, 201–211. [DOI] [PubMed] [Google Scholar]
- Pardue ML and Gall JG (1969). Molecular hybridization of radioactive DNA to the DNA of cytological preparations. Proc Natl Acad Sci U S A 64, 600–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S-M (2014). Musashi-2 controls cell fate, lineage bias, and TGF-β signaling in HSCs, vol. 211, pp. 71–87. Journal of Experimental Medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SM (2015). Musashi2 sustains the mixed-lineage leukemia–driven stem cell regulatory program, vol. 125, pp. 1286–1298. The Journal of Clinical Investigation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peattie DA and Gilbert W (1980). Chemical probes for higher-order structure in RNA. Proc Natl Acad Sci U S A 77, 4679–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson T (2011). The nucleus introduced. Cold Spring Harb Perspect Biol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penny GD, Kay GF, Sheardown SA, Rastan S and Brockdorff N (1996). Requirement for Xist in X chromosome inactivation. Nature 379, 131–7. [DOI] [PubMed] [Google Scholar]
- Pintacuda G, Young AN and Cerase A (2017). Function by Structure: Spotlights on Xist Long Non-coding RNA. Front Mol Biosci 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting CP, Oliver PL and Reik W (2009). Evolution and functions of long noncoding RNAs. Cell 136, 629–41. [DOI] [PubMed] [Google Scholar]
- Popova VV, Kurshakova MM and Kopytova DV (2015). [Methods to study the RNA-protein interactions]. Mol Biol (Mosk) 49, 472–81. [DOI] [PubMed] [Google Scholar]
- Poria DK and Ray PS (2017). RNA-protein UV-crosslinking Assay. Bio Protoc 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protter DSW, Rao BS, Van Treeck B, Lin Y, Mizoue L, Rosen MK and Parker R (2018). Intrinsically Disordered Regions Can Contribute Promiscuous Interactions to RNP Granule Assembly. Cell Rep 22, 1401–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinodoz SA, Ollikainen N, Tabak B, Palla A, Schmidt JM, Detmar E, Lai MM, Shishkin AA, Bhat P, Takei Y et al. (2018). Higher-Order Inter-chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell 174, 744–757.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A and Tyagi S (2008). Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5, 877–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastan S and Robertson EJ (1985). X-chromosome deletions in embryo-derived (EK) cell lines associated with lack of X-chromosome inactivation. J Embryol Exp Morphol 90, 379–88. [PubMed] [Google Scholar]
- Rego A, Sinclair PB, Tao W, Kireev I and Belmont AS (2008). The facultative heterochromatin of the inactive X chromosome has a distinctive condensed ultrastructure. J Cell Sci 121, 1119–27. [DOI] [PubMed] [Google Scholar]
- Richler C, Soreq H and Wahrman J (1992). X inactivation in mammalian testis is correlated with inactive X-specific transcription. Nat Genet 2, 192–5. [DOI] [PubMed] [Google Scholar]
- Rinn J, Chang H and Kornberg R (2012). Genome Regulation by Long Noncoding RNAs. Annual Review of Biochemistry , Vol 81 81, 145–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rošić S, Köhler F and Erhardt S (2014). Repetitive centromeric satellite RNA is essential for kinetochore formation and cell division. J Cell Biol 207, 335–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudkin GT and Stollar BD (1977). High resolution detection of DNA-RNA hybrids in situ by indirect immunofluorescence. Nature 265, 472–3. [DOI] [PubMed] [Google Scholar]
- Sadoni N and Zink D (2004). Nascent RNA synthesis in the context of chromatin architecture. Chromosome Res 12, 439–51. [DOI] [PubMed] [Google Scholar]
- Salic A and Mitchison TJ (2008). A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A 105, 2415–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma K, Levasseur P, Aristarkhov A and Lee JT (2010). Locked nucleic acids (LNAs) reveal sequence requirements and kinetics of Xist RNA localization to the X chromosome. Proc Natl Acad Sci U S A 107, 22196–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki YT, Ideue T, Sano M, Mituyama T and Hirose T (2009). MENepsilon/beta noncoding RNAs are essential for structural integrity of nuclear paraspeckles. Proc Natl Acad Sci U S A 106, 2525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma E, Sterne-Weiler T, O’Hanlon D and Blencowe BJ (2016). Global Mapping of Human RNA-RNA Interactions. Mol Cell 62, 618–26. [DOI] [PubMed] [Google Scholar]
- Siegfried N, Busan S, Rice G, Nelson J and Weeks K (2014). RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nature Methods 11, 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal YM, Zhou R and Zhuang X (2018). Visualizing and discovering cellular structures with super-resolution microscopy. Science 361, 880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon M, Wang C, Kharchenko P, West J, Chapman B, Alekseyenko A, Borowsky M, Kuroda M and Kingston R (2011). The genomic binding sites of a noncoding RNA. Proceedings of the National Academy of Sciences of the United States of America 108, 20497–20502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer RH and Ward DC (1982). Actin gene expression visualized in chicken muscle tissue culture by using in situ hybridization with a biotinated nucleotide analog. Proc Natl Acad Sci U S A 79, 7331–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeets D, Markaki Y, Schmid VJ, Kraus F, Tattermusch A, Cerase A, Sterr M, Fiedler S, Demmerle J, Popken J et al. (2014). Three-dimensional super-resolution microscopy of the inactive X chromosome territory reveals a collapse of its active nuclear compartment harboring distinct Xist RNA foci. Epigenetics Chromatin 7, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smola M, Calabrese J and Weeks K (2015). Detection of RNA-Protein Interactions in Living Cells with SHAPE. Biochemistry 54, 6867–6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smurova K and De Wulf P (2018). Centromere and Pericentromere Transcription: Roles and Regulation … in Sickness and in Health. Front Genet 9, 674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiniello M, Knoener RA, Steinbrink MI, Yang B, Cesnik AJ, Buxton KE, Scalf M, Jarrard DF and Smith LM (2018). HyPR-MS for Multiplexed Discovery of MALAT1, NEAT1, and NORAD lncRNA Protein Interactomes. J Proteome Res 17, 3022–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan BA (2010). Optical mapping of protein-DNA complexes on chromatin fibers. Methods in molecular biology 659, 99–115. [DOI] [PubMed] [Google Scholar]
- Sullivan BA and Karpen GH (2004). Centromeric chromatin exhibits a histone modification pattern that is distinct from both euchromatin and heterochromatin. Nat Struct Mol Biol 11, 1076–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Hao Q and Prasanth KV (2018). Nuclear Long Noncoding RNAs: Key Regulators of Gene Expression. Trends Genet 34, 142–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunwoo H, Colognori D, Froberg JE, Jeon Y and Lee JT (2017). Repeat E anchors Xist RNA to the inactive X chromosomal compartment through CDKN1A-interacting protein (CIZ1). Proceedings of the National Academy of Sciences 114, 10654–10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunwoo H, Dinger ME, Wilusz JE, Amaral PP, Mattick JS and Spector DL (2009). MEN epsilon/beta nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res 19, 347–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunwoo H, Wu JY and Lee JT (2015). The Xist RNA-PRC2 complex at 20-nm resolution reveals a low Xist stoichiometry and suggests a hit-and-run mechanism in mouse cells. Proceedings of the National Academy of Sciences 112, E4216–E4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagarro I, Fernández-Peralta AM and González-Aguilera JJ (1994). Chromosomal localization of human satellites 2 and 3 by a FISH method using oligonucleotides as probes. Hum Genet 93, 383–8. [DOI] [PubMed] [Google Scholar]
- Talbert PB and Henikoff S (2018). Transcribing Centromeres: Noncoding RNAs and Kinetochore Assembly. Trends Genet 34, 587–599. [DOI] [PubMed] [Google Scholar]
- Tam J and Merino D (2015). Stochastic optical reconstruction microscopy (STORM) in comparison with stimulated emission depletion (STED) and other imaging methods. J Neurochem 135, 643–58. [DOI] [PubMed] [Google Scholar]
- Teller K, Illner D, Thamm S, Casas-Delucchi CS, Versteeg R, Indemans M, Cremer T and Cremer M (2011). A top-down analysis of Xa- and Xi-territories reveals differences of higher order structure at ≥ 20 Mb genomic length scales. Nucleus 2, 465–77. [DOI] [PubMed] [Google Scholar]
- Thorn K (2016). A quick guide to light microscopy in cell biology. Mol Biol Cell 27, 219–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tijerina P, Mohr S and Russell R (2007). DMS footprinting of structured RNAs and RNA-protein complexes. Nat Protoc 2, 2608–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting DT, Lipson D, Paul S, Brannigan BW, Akhavanfard S, Coffman EJ, Contino G, Deshpande V, Iafrate AJ, Letovsky S et al. (2011). Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science 331, 593–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J, Jensen K, Mele A and Darnell R (2005). CLIP: A method for identifying protein-RNA interaction sites in living cells. Methods 37, 376–386. [DOI] [PubMed] [Google Scholar]
- Valgardsdottir R, Chiodi I, Giordano M, Rossi A, Bazzini S, Ghigna C, Riva S and Biamonti G (2008). Transcription of Satellite III non-coding RNAs is a general stress response in human cells. Nucleic Acids Res 36, 423–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Freese P, Pratt GA, Wang X, Wei X, Xiao R, Blue SM, Chen J-Y, Cody NAL, Dominguez D et al. (2018). A Large-Scale Binding and Functional Map of Human RNA Binding Proteins. bioRxiv, 179648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuzman D and Levy Y (2012). Intrinsically disordered regions as affinity tuners in protein-DNA interactions. Mol Biosyst 8, 47–57. [DOI] [PubMed] [Google Scholar]
- Wansink DG, Schul W, van der Kraan I, van Steensel B, van Driel R and de Jong L (1993). Fluorescent labeling of nascent RNA reveals transcription by RNA polymerase II in domains scattered throughout the nucleus. J Cell Biol 122, 283–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Somanathan S, Samarabandu J and Berezney R (1999). Three-dimensional visualization of transcription sites and their association with splicing factor-rich nuclear speckles. J Cell Biol 146, 543–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weick E-M and Miska EA (2014). piRNAs: from biogenesis to function. [DOI] [PubMed] [Google Scholar]
- West Jason A., Davis Christopher P., Sunwoo H, Simon Matthew D., Sadreyev Ruslan I., Wang Peggy I., Tolstorukov Michael Y. and Kingston Robert E. (2014). The Long Noncoding RNAs NEAT1 and MALAT1 Bind Active Chromatin Sites. Molecular Cell 55, 791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JA, Mito M, Kurosaka S, Takumi T, Tanegashima C, Chujo T, Yanaka K, Kingston RE, Hirose T, Bond C et al. (2016). Structural, super-resolution microscopy analysis of paraspeckle nuclear body organization. The Journal of Cell Biology 214, 817–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wutz A, Rasmussen TP and Jaenisch R (2002). Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat Genet 30, 167–74. [DOI] [PubMed] [Google Scholar]
- Xiao R, Chen J, Liang Z, Luo D, Chen G, Lu Z, Chen Y, Zhou B, Li H, Du X et al. (2019). Pervasive Chromatin-RNA Binding Protein Interactions Enable RNA-Based Regulation of Transcription. Cell 178, 107-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Rahman R and Rosbash M (2018). Mechanistic implications of enhanced editing by a HyperTRIBE RNA-binding protein. Rna 24, 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T and Hirose T (2015). The building process of the functional paraspeckle with long non-coding RNAs. Front Biosci (Elite Ed) 7, 1–41. [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Souquere S, Chujo T, Kobelke S, Chong YS, Fox AH, Bond CS, Nakagawa S, Pierron G and Hirose T (2018). Functional Domains of NEAT1 Architectural lncRNA Induce Paraspeckle Assembly through Phase Separation. Molecular Cell 70, 1038–1053.e7. [DOI] [PubMed] [Google Scholar]
- Yu K, Liu C, Kim BG and Lee DY (2015). Synthetic fusion protein design and applications. Biotechnol Adv 33, 155–164. [DOI] [PubMed] [Google Scholar]
- Zhang H, Elbaum-Garfinkle S, Langdon E, Taylor N, Occhipinti P, Bridges A, Brangwynne C and Gladfelter A (2015). RNA Controls PolyQ Protein Phase Transitions. Molecular Cell 60, 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XO, Yin QF, Chen LL and Yang L (2014). Gene expression profiling of non-polyadenylated RNA-seq across species. Genom Data 2, 237–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z and Carmichael GG (2001). The fate of dsRNA in the nucleus: a p54(nrb)-containing complex mediates the nuclear retention of promiscuously A-to-I edited RNAs. Cell 106, 465–75. [DOI] [PubMed] [Google Scholar]
- Zong X, Huang L, Tripathi V, Peralta R, Freier SM, Guo S and Prasanth KV (2015). Knockdown of nuclear-retained long noncoding RNAs using modified DNA antisense oligonucleotides. Methods Mol Biol 1262, 321–31. [DOI] [PubMed] [Google Scholar]