

Abstract
Accumulating evidence suggests that p53 plays a suppressive role in cancer metastasis, yet the underlying mechanism remains largely unclear. Regulation of actin dynamics is essential for the control of cell migration, which is an important step in metastasis. The Arp2/3 complex is a major nucleation factor to initiate branched actin polymerization that drives cell migration. However, it is unknown whether p53 could suppress metastasis through modulating Arp2/3 function. Here, we report that WDR63 is transcriptionally upregulated by p53. We show with migration assays and mouse xenograft models that WDR63 negatively regulates cell migration, invasion, and metastasis downstream of p53. Mechanistically, WDR63 interacts with the Arp2/3 complex and inhibits Arp2/3‐mediated actin polymerization. Furthermore, WDR63 overexpression is sufficient to dampen the increase in cell migration, invasion, and metastasis induced by p53 depletion. Together, these findings suggest that WDR63 is an important player in the regulation of Arp2/3 function and also implicate WDR63 as a critical mediator of p53 in suppressing metastasis.
Keywords: Arp2/3, metastasis, p53, WDR63
Subject Categories: Cancer; Cell Adhesion, Polarity & Cytoskeleton
As a direct transcriptional target of p53, WDR63 interacts with the Arp2/3 complex and inhibits Arp2/3‐dependent actin polymerization, thereby mediating the inhibitory role of p53 on metastasis.

Introduction
p53 plays a pivotal role in tumor prevention 1, 2, 3, 4. The importance of p53 as a tumor suppressor is highlighted by the findings that p53 deficiency drives spontaneous tumorigenesis in mice and p53 is frequently inactivated in the development of human cancer 5, 6. The tumor‐suppressive function of p53 has been largely attributed to its intrinsic nature as a master transcription factor 7. By modulating target gene expression, p53 restrains cell proliferation in response to a variety of cellular stresses by inducing cell cycle arrest, apoptosis, and senescence 8. In addition, p53 has also been shown to regulate other cellular processes such as cell metabolism, autophagy, and ferroptosis 9, 10, 11, 12.
Increasing evidence suggests that p53 plays a critical role in suppressing metastasis 4, 13, 14, 15, 16. The suppressive function of p53 in metastasis involves the regulation of several canonical metastasis pathways, including cell adhesion, migration, and invasion. For instance, it has been shown that the loss of p53 activates Rho family GTPases Rac1, RhoA, and Cdc42, which are important regulators of actin cytoskeleton dynamics, thereafter promoting cell migration and invasion 17, 18, 19, 20, 21. In addition, p53 also indirectly reduces expression levels of Snail, Slug, and Zeb1, which are key transcription factors in regulating epithelial–mesenchymal transition (EMT), to suppress cell invasion 22, 23, 24. Moreover, a number of proteins have been linked to p53's function in inhibiting cell migration and invasion, including maspin, PAI‐1, MET, caldesmon, PCDH10, and GLS2 25, 26, 27, 28, 29, 30. However, the detailed mechanism of how p53 suppresses metastasis still remains largely unclear.
As an important step in metastasis, cell migration requires membrane protrusion at the leading edge of cells 31, 32. Membrane protrusion is powered by the actin‐related protein 2/3 (Arp2/3)‐mediated branched actin polymerization 33. The Arp2/3 complex is composed of seven evolutionarily conserved subunits (Arp2, Arp3, and ArpC1‐5), which serves as a nucleation core for de novo actin polymerization 34. On its own, the Arp2/3 complex displays low intrinsic actin nucleation activity and needs to be activated by nucleation‐promoting factors (NPFs), such as Wiskott–Aldrich syndrome protein (WASP) and WASP family verprolin‐homologous protein (WAVE). The NPFs bind to and activate Arp2/3 via their verprolin, cofilin, and acidic (VCA) domains 35, 36. Given the critical role of Arp2/3 in actin polymerization, it is not surprising that Arp2/3 function is subjected to intricate regulation in cells. For example, via the interaction with the Arp2/3 complex, PICK1 and arpin inhibit, whereas Exo70 stimulates, the function of Arp2/3 37, 38, 39. In addition, cortactin enhances Arp2/3‐mediated actin polymerization directly via activating Arp2/3 or indirectly via N‐WASP, while coronin 1B antagonizes cortactin and inhibits Arp2/3 function 40, 41, 42, 43. Moreover, cofilin affects Arp2/3‐mediated actin polymerization by promoting disassembly of actin filaments 44. These findings suggest the complexity of the regulation of Arp2/3‐mediated actin polymerization.
As mentioned earlier, p53 is able to inhibit cell migration and invasion via negatively regulating Rho family GTPase‐mediated modeling of the actin cytoskeleton. However, it is unknown whether p53 could regulate cell migration and invasion through modulating Arp2/3 function. In the present study, we report WD repeat‐containing protein 63 (WDR63) as a direct transcriptional target of p53. WDR63 functions as a negative regulator of cell migration, invasion, and metastasis through inhibiting Arp2/3‐mediated actin polymerization. Furthermore, WDR63 is shown to mediate p53's function in suppressing metastasis. Our study demonstrates WDR63 as an important regulator of Arp2/3 function and provides new insights into the mechanisms of how p53 suppresses metastasis.
Results
WDR63 inhibits cell migration, invasion, and metastasis
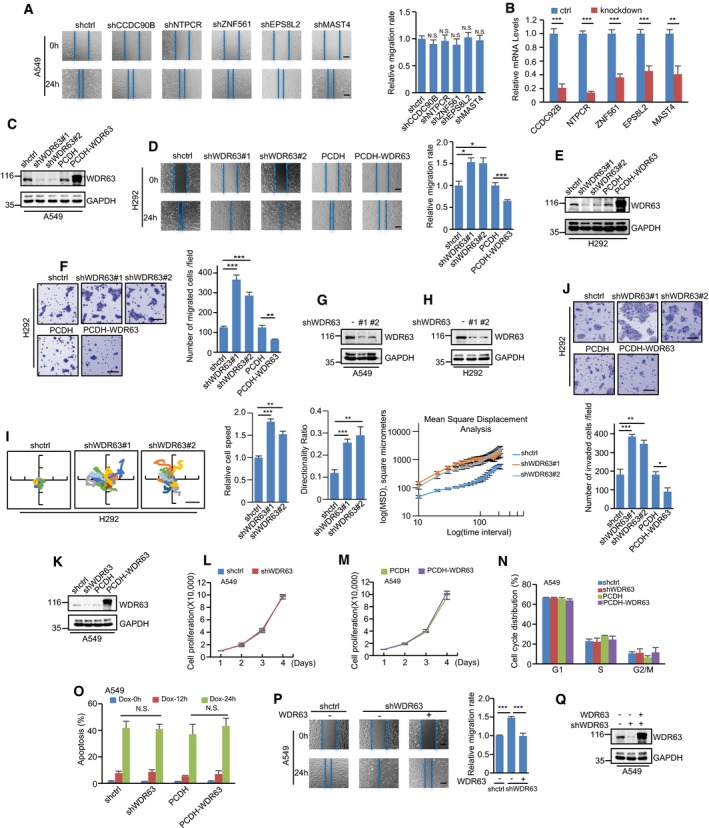
To generate new insights into the pivotal role of p53 in suppressing cancer metastasis, we sought to interrogate p53 target gene(s) with ability to regulate cell migration and invasion. The previous bioinformatic and experimental studies have documented a large amount of genuine and potential p53 target genes. By analyzing the top 116 p53‐activated target genes identified by different genome‐wide studies 45, we chose six potential p53‐activated targets (CCDC90B, NTPCR, WDR63, ZNF561, EPS8L2, and MAST4) with unknown function for further investigation. In the wound‐healing assay, knockdown of CCDC90B, NTPCR, ZNF561, EPS8L2, or MAST4 showed no obvious effect on the migration of A549 cells (Fig EV1A and B). Intriguingly, knockdown of WDR63 strongly enhanced directional cell migration toward a “wound”, whereas overexpression of WDR63 markedly inhibited it in both A549 and H292 cells (Figs 1A and EV1C–E). The inhibitory effect of WDR63 on the migration of A549 and H292 cells was further verified by transwell migration assay and single‐cell tracking analysis (Figs 1B and C, and EV1F–I). To examine whether WDR63 could also regulate cell invasion, we performed transwell invasion assay. Knockdown of WDR63 greatly increased, whereas overexpression of WDR63 significantly decreased, the invasive ability of A549 and H292 cells (Figs 1D and EV1J). Neither knockdown nor overexpression of WDR63 was shown to affect cell proliferation, cell cycle progression, and apoptosis (Fig EV1K–O). Collectively, these data indicate that WDR63 specifically inhibits cell migration and invasion. In support of this, WDR63 knockdown‐increased cell migration and invasion could be reversed by ectopic expression of shRNA‐resistant WDR63 (Figs 1E–G and EV1P and Q).
Figure EV1. WDR63 suppresses cell migration and invasion.
-
A, BA549 cells expressing control shRNA, CCDC90B shRNA, NTPCR shRNA, ZNF561 shRNA, EPS8L2 shRNA, or MAST4 shRNA were subjected to wound‐healing assay (A). The knockdown efficiency of the indicated proteins was analyzed by real‐time RT–PCR (B). The shown images are representative of three independent experiments. Data shown are mean ± SD (n = 3). **P < 0.01; ***P < 0.001; N.S., no significance; one‐way ANOVA. Scale bar: 200 μm.
-
CA549 cells were infected with lentiviruses expressing control shRNA, WDR63 shRNA#1, WDR63 shRNA#2, PCDH control, or PCDH‐Flag‐WDR63. Forty‐eight hours after infection, cell lysates were analyzed by Western blot. Data shown represent three independent experiments.
-
D–FH292 cells expressing control shRNA, WDR63 shRNA#1, WDR63 shRNA#2, PCDH control, or PCDH‐Flag‐WDR63 were subjected to wound‐healing (D) and transwell migration (F) assays. Lysates from these cells were also analyzed by Western blot (E). The shown images are representative of three independent experiments. Data shown are mean ± SD (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001; one‐way ANOVA. Scale bar in (D): 200 μm. Scale bar in (F): 100 μm.
-
GLysates from A549 cells expressing control shRNA, WDR63 shRNA#1, or WDR63 shRNA#2 were analyzed by Western blot.
-
H, IEffect of WDR63 knockdown on the migration of H292 cells was analyzed by single‐cell tracking with time‐lapse microscopy at 10‐min intervals for 4 h (I). Cell migration speed, persistence, and mean‐squared displacement (MSD) were also analyzed using an Excel macro described by Gorelik and Gautreau. Data shown are mean ± SD. n = 3 independent experiments. **P < 0.01; ***P < 0.001; one‐way ANOVA. The successful knockdown of WDR63 in H292 cells was also confirmed by Western blot analysis (H). Scale bar: 100 μm.
-
JH292 cells expressing control shRNA, WDR63 shRNA#1, WDR63 shRNA#2, PCDH control, or PCDH‐Flag‐WDR63 were subjected to transwell invasion assay. The shown images are representative of three independent experiments. Data shown are mean ± SD (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001; one‐way ANOVA. Scale bar: 100 μm.
-
K–NA549 cells were infected with lentiviruses expressing control shRNA, WDR63 shRNA#1, PCDH control, or PCDH‐Flag‐WDR63. Forty‐eight hours after infection, cell lysates were analyzed by Western blot (K). The cell growth curves were measured by counting cell numbers at the indicated time points (L and M). The cell cycle distribution was analyzed by flow cytometry (N). Data shown are mean ± SD; n = 3 independent experiments.
-
OA549 cells expressing control shRNA, WDR63 shRNA#1, PCDH control, or PCDH‐Flag‐WDR63 were treated with 1 μg/ml doxorubicin for the indicated periods of time. Cells were then stained with Hoechst 33342, and apoptotic cells were determined by the presence of nuclear fragmentation. Data shown are mean ± SD; n = 3 independent experiments. N.S., no significance; two‐tailed Student's t‐test.
-
P, QA549 cells expressing control shRNA, WDR63 shRNA#1, or WDR63 shRNA#1 plus shRNA‐resistant Flag‐WDR63 were subjected to wound‐healing assay (P). Lysates from these cells were also analyzed by Western blot (Q). The shown images are representative of three independent experiments. Data shown are mean ± SD (n = 3). ***P < 0.001; one‐way ANOVA. Scale bar: 200 μm.
Figure 1. WDR63 inhibits cell migration, invasion, and metastasis.

-
A, BA549 cells expressing control shRNA, WDR63 shRNA#1, WDR63 shRNA#2, PCDH control, or PCDH‐Flag‐WDR63 were subjected to wound‐healing (A) and transwell migration (B) assays. The shown images are representative of three independent experiments. Data shown are mean ± SD (n = 3). **P < 0.01; ***P < 0.001; one‐way ANOVA. Scale bar in (A): 200 μm. Scale bar in (B): 100 μm. The successful knockdown and overexpression of WDR63 are shown in Fig EV1C.
-
CEffect of WDR63 knockdown on the migration of A549 cells was analyzed by single‐cell tracking with time‐lapse microscopy at 10‐min intervals for 4 h. Scale bar: 200 μm. Cell migration speed, persistence, and mean‐squared displacement (MSD) were also analyzed using an Excel macro described by Gorelik and Gautreau. Data shown are mean ± SD. n = 3 independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001; one‐way ANOVA. The knockdown efficiency of WDR63 is shown in Fig EV1G.
-
DA549 cells expressing control shRNA, WDR63 shRNA#1, WDR63 shRNA#2, PCDH control, or PCDH‐Flag‐WDR63 were subjected to transwell invasion assay. The shown images are representative of three independent experiments. Data shown are mean ± SD (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001; one‐way ANOVA. Scale bar: 100 μm.
-
E–GA549 cells expressing control shRNA, WDR63 shRNA#1, or WDR63 shRNA#1 plus shRNA‐resistant Flag‐WDR63 were subjected to transwell migration (E), single‐cell tracking (F), and transwell invasion (G) assays. The shown images are representative of three independent experiments. Cell migration speed, persistence, and mean‐squared displacement (MSD) were also analyzed using an Excel macro described by Gorelik and Gautreau. Data shown are mean ± SD. n = 3 independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001; one‐way ANOVA. Scale bar in (E and G): 100 μm. Scale bar in (F): 200 μm. The successful knockdown and overexpression of WDR63 are shown in Fig EV1Q.
-
HA549 cells expressing control shRNA, WDR63 shRNA#1, or WDR63 shRNA#2 (each also expressing luciferase) were injected via tail vein into nude mice (n = 6 for each group). Three weeks after injection, tumor formation was monitored by bioluminescence imaging. Data shown are mean ± SD; *P < 0.05; one‐way ANOVA.
To further determine the role of WDR63 in cancer cell metastasis, we used a xenograft model of metastasis. A549 cells expressing control shRNA or WDR63 shRNA were injected via tail vein into nude mice. The tumor formation in whole animals and lungs was examined by D‐luciferin‐based bioluminescence. Mice that were injected with tumor cells expressing WDR63 shRNA exhibited enhanced tumor extravasation compared to mice injected with cells expressing control shRNA (Fig 1H). Analysis of tumor formation in lungs further verified the increased lung extravasation by WDR63 knockdown (Fig EV2A). Taken together, these data suggest that WDR63, as a potential p53‐activated target, inhibits cell migration, invasion, and metastasis. In support, WDR63 was shown to be down‐regulated in both lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) compared to their normal tissues (Fig EV2B and C). The expression levels of WR63 were also correlated with the malignancy of lung cancers, especially LUSC (Fig EV2D and E).
Figure EV2. WDR63 is upregulated by p53.

-
AA549 cells expressing control shRNA, WDR63 shRNA#1, or WDR63 shRNA#2 (each also expressing luciferase) were injected via tail vein into nude mice (n = 6 for each group). Six weeks after injection, tumor formation in lungs was monitored by bioluminescence imaging. Data shown are mean ± SD; *P < 0.05; one‐way ANOVA. Scale bar: 1 cm.
-
BWDR63 expression levels were analyzed in TCGA lung adenocarcinoma (LUAD) and their normal tissues. Data shown are mean ± SD; P‐value was calculated by two‐tailed Student's t‐test.
-
CWDR63 expression levels were analyzed in TCGA lung squamous cell carcinoma (LUSC) and their normal tissues. Data shown are mean ± SD; P‐value was calculated by two‐tailed Student's t‐test.
-
DWDR63 expression levels were analyzed in TCGA LUAD with different stages. Data shown are mean ± SD; P‐value was calculated by one‐way ANOVA.
-
EWDR63 expression levels were analyzed in TCGA LUSC with different stages. Data shown are mean ± SD; P‐value was calculated by one‐way ANOVA.
-
FA549 cells expressing control or p53 shRNA were treated with etoposide (50 μM) for the indicated periods of time. WDR63 mRNA and protein levels were then examined. Data shown are mean ± SD; n = 3 independent experiments. ***P < 0.001; two‐tailed Student's t‐test.
-
GA549 cells expressing control or p53 shRNA were treated with Nutlin (10 μM) for the indicated periods of time, followed by Western blot analysis. Data shown represent three independent experiments.
-
HHCT116 p53 +/+ and HCT116 p53 −/− cells were treated with Nutlin (10 μM) for the indicated periods of time, followed by Western blot analysis. Data shown represent three independent experiments.
-
IWDR63 expression levels were analyzed in TCGA LUAD harboring wild‐type or mutant TP53 gene using TCGA database. Data shown are mean ± SD; P‐value was calculated by two‐tailed Student's t‐test.
-
JWDR63 expression levels were analyzed in TCGA LUSC harboring wild‐type or mutant TP53 gene using TCGA database. Data shown are mean ± SD; P‐value was calculated by two‐tailed Student's t‐test.
-
KA549 cells expressing either control shRNA or p53 shRNA were transfected with the indicated reporter constructs plus Renilla luciferase plasmid. Twenty‐four hours later, reporter activity was measured. Data shown are mean ± SD; n = 3 independent experiments. **P < 0.01; ***P < 0.001; two‐tailed Student's t‐test.
WDR63 is a direct transcriptional target of p53
To verify whether WDR63 is a genuine p53 target, we first evaluated the effect of p53 on WDR63 expression. Ectopic expression of p53 increased, whereas knockdown of p53 decreased, both mRNA and protein levels of WDR63 in A549, H292, and HCT116 cells (Fig 2A and B). Treatment with the DNA‐damaging agents doxorubicin or etoposide in A549 cells led to a time‐dependent increase in WDR63 expression (Figs 2C and EV2F). However, this increased WDR63 expression was prevented when p53 was knocked down (Figs 2C and EV2F), indicating that WDR63 induction upon doxorubicin or etoposide treatment is p53‐dependent. Moreover, Nutlin‐3, a p53 activator 46, strongly induced WDR63 expression in A549 and HCT116 p53+/+ cells, but not p53 knockdown in A549 and HCT116 p53−/− cells (Fig EV2G and H). Together, these results demonstrate WDR63 as a p53‐inducible gene. In accordance with the relationship between p53 and WDR63 expression, analysis of TCGA database revealed that WDR63 was expressed at lower levels in both LUAD and LUSC harboring mutant TP53 gene than in TP53 wild‐type tumors (Fig EV2I and J).
Figure 2. WDR63 is a transcriptional target of p53.

-
AWDR63 mRNA and protein levels in A549, H292, and HCT116 cells transduced with empty vector (PCDH) or PCDH‐p53. Data shown are mean ± SD; n = 3 independent experiments. **P < 0.01; ***P < 0.001; two‐tailed Student's t‐test.
-
BWDR63 mRNA and protein levels in A549, H292, and HCT116 cells transduced with control (shctrl) or p53 (shp53) shRNA. Data shown are mean ± SD; n = 3 independent experiments. ***P < 0.001; two‐tailed Student's t‐test.
-
CA549 cells expressing control or p53 shRNA were treated with doxorubicin (Dox, 1 μg/ml) for the indicated periods of time. WDR63 mRNA and protein levels were then examined. Data shown are mean ± SD; n = 3 independent experiments. ***P < 0.001; two‐tailed Student's t‐test.
-
DWDR63 mRNA levels in H1299 cells transfected with empty vector (−), wild‐type p53, or the indicated p53 mutants. Data shown are mean ± SD; n = 3 independent experiments. ***P < 0.001; two‐tailed Student's t‐test.
-
ESchematic illustration of the first two exons of WDR63 gene. BS1 and BS2 represent 2 putative p53 binding sites predicted by the JASPAR database. Black bars represent p53 binding sites identified in the indicated p53 ChIP‐seq datasets. The pGL3‐based wild‐type and mutant reporter constructs used for luciferase assay are also shown.
-
FLysates from A549 cells were subjected to ChIP assay using control IgG or anti‐p53 antibody. ChIP products were amplified by real‐time PCR. Data shown are mean ± SD; n = 3 independent experiments. ***P < 0.001; two‐tailed Student's t‐test.
-
GA549 cells were transfected with empty vector (ctrl) or Flag‐p53 plus the indicated reporter constructs. Twenty‐four hours later, reporter activity was measured. Data shown are mean ± SD; n = 3 independent experiments. **P < 0.01; ***P < 0.001; two‐tailed Student's t‐test.
-
HA549 cells expressing control or p53 shRNA were transfected with pGL3‐BS1 and Renilla luciferase plasmids. Twenty‐four hours later, cells were treated with or without Nutlin (10 μM) for another 12 h. Reporter activity was then measured. Data shown are mean ± SD; n = 3 independent experiments. **P < 0.01; two‐tailed Student's t‐test.
To examine whether p53 transcriptionally upregulates WDR63 expression, we used three tumor‐associated p53 mutants (R175H, R273H, and G279E), which have lost the p53 transcriptional activity 5. Unlike wild‐type p53, all these three p53 mutants exhibited no inducible effects on WDR63 expression (Fig 2D), indicating that p53 could regulate WDR63 expression at the transcriptional level. We next sought to explore whether WDR63 is a direct target gene of p53. We used the JASPAR database to inspect the upstream and intronic regions of the WDR63 gene 47. Two putative p53 binding sites (BS1 and BS2) were identified within the first intron (Fig 2E). Analysis of published p53 ChIP‐seq datasets 48, 49, 50, 51, 52, 53 revealed the potential binding of p53 to the BS1 site (Fig 2E). The subsequent ChIP assay indeed verified the interaction of p53 with the chromatin fragment comprising the BS1 site (Fig 2F). To further determine whether the BS1 site confers p53‐dependent activity, luciferase reporter assay was performed. The transcriptional activity of luciferase reporter containing the wild‐type, but not the mutant BS1 site, was induced by ectopic expression of p53 and reduced by knockdown of p53 (Figs 2G and EV2K). In addition, Nutlin‐3 enhanced the transcriptional activity of reporter with the wild‐type BS1 site in control A549 cells, but not in p53 knockdown A549 cells (Fig 2H). Taken together, these data suggest that WDR63 is a bona fide transcriptional target of p53.
WDR63 interacts with the Arp2/3 complex
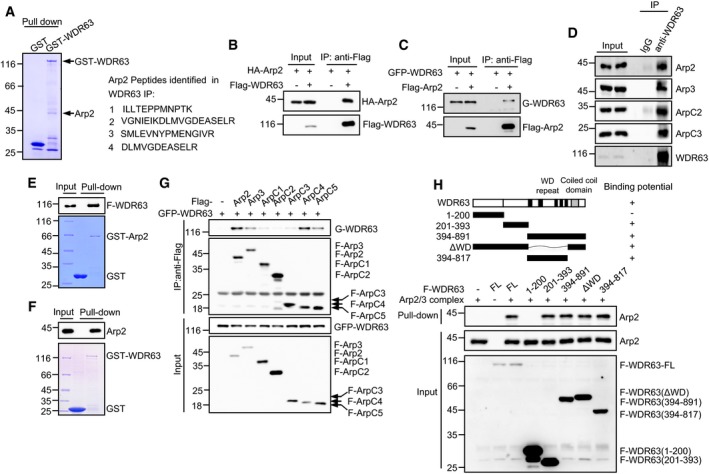
To investigate how WDR63 inhibits cell migration, invasion, and metastasis, we employed a GST pull‐down experiment to identify WDR63‐interacting proteins. The mass spectrometry analysis revealed Arp2 as a potential interacting partner for WDR63 (Fig 3A). Arp2 is a key component of the Arp2/3 complex, a seven‐subunit conserved complex that initiates branched actin polymerization 34. To further verify the interaction between WDR63 and Arp2, we performed immunoprecipitation assays. The results showed a strong interaction between exogenously expressed WDR63 and Arp2 (Fig 3B and C). Using a co‐immunoprecipitation assay with anti‐WDR63 antibody, the interaction between endogenous WDR63 and Arp2 was readily detected (Fig 3D). Furthermore, in vitro binding assays with purified WDR63 and Arp2 proteins showed that WDR63 is directly associated with Arp2 (Fig 3E and F). In addition to binding to the Arp2 subunit of the Arp2/3 complex, WDR63 was also shown to associate with Arp3, ArpC4, and ArpC5 when these proteins were exogenously expressed (Fig 3G). However, WDR63 did not interact with several well‐known Arp2/3‐regulating factors, such as coronin 1B, cortactin, N‐WASP, and cofilin (Fig EV3A and B). Collectively, these data indicate that WDR63 is a novel and specific binding partner for the Arp2/3 complex. In support of this, multiple endogenous subunits of Arp2/3, including Arp2, Arp3, ArpC2, and ArpC3, were successfully pulled down by WDR63 (Figs 3D and EV3C).
Figure 3. WDR63 interacts with the Arp2/3 complex.
-
AA ˜ 44 kD protein band specifically pulled down by GST‐WDR63 from A549 cell lysates was identified as Arp2 by mass spectrometry analysis. The Arp2 peptide sequences obtained by MS are shown.
-
BLysates from HEK293T cells expressing HA‐Arp2 alone or together with Flag‐WDR63 were immunoprecipitated by anti‐Flag antibody, followed by Western blot analysis. Data shown represent three independent experiments.
-
CLysates from HEK293T cells expressing GFP‐WDR63 alone or together with Flag‐Arp2 were immunoprecipitated by anti‐Flag antibody, followed by Western blot analysis. Data shown represent three independent experiments.
-
DLysates from A549 cells were immunoprecipitated with control IgG or anti‐WDR63 antibody, followed by Western blot analysis. Data shown represent three independent experiments.
-
EPurified GST or GST‐Arp2 proteins immobilized on glutathione beads were incubated with purified Flag‐WDR63. Input and bead‐bound proteins were analyzed by Western blot. Data shown represent three independent experiments.
-
FPurified GST or GST‐WDR63 proteins immobilized on glutathione beads were incubated with purified Arp2. Input and bead‐bound proteins were analyzed by Western blot. Data shown represent three independent experiments.
-
GLysates from HEK293T cells expressing GFP‐WDR63 alone or together with the indicated Flag‐tagged each single subunit of the Arp2/3 complex were immunoprecipitated with anti‐Flag antibody, followed by Western blot analysis. Data shown represent three independent experiments.
-
HPurified Flag‐tagged full‐length or the indicated truncated WDR63 proteins immobilized on anti‐Flag M2 beads were incubated with the purified Arp2/3 complex (Cytoskeleton, Inc.) in vitro. Input and bead‐bound proteins were analyzed by Western blot. Data shown represent three independent experiments.
Figure EV3. WDR63 interacts with Arp2/3 and inhibits FBS‐stimulated translocation of Arp2 to the leading edge of migrating cells.

-
ALysates from HEK293T cells expressing GFP‐WDR63 alone or GFP‐WDR63 plus Flag‐tagged Arp2, Arp3, ArpC2, and coronin 1B as indicated were immunoprecipitated by anti‐Flag antibody, followed by Western blot analysis. Data shown represent three independent experiments.
-
BLysates from HEK293T cells expressing GFP‐WDR63 alone or GFP‐WDR63 plus Flag‐tagged cortactin, N‐WASP, and cofilin as indicated were immunoprecipitated by anti‐Flag antibody, followed by Western blot analysis. Data shown represent three independent experiments.
-
CLysates from A549 cells were incubated with purified GST or GST‐WDR63 proteins immobilized on glutathione beads. Input and bead‐bound proteins were analyzed by Western blot. Data shown represent three independent experiments.
-
DThe cellular localization of exogenous GFP‐WDR63 was determined by immunofluorescence. Data shown represent three independent experiments. Scale bar: 20 μm.
-
EThe localization of endogenous WDR63 in A549 cells was evaluated by nuclear/cytosolic fractionation, followed by Western blot analysis. Data shown represent three independent experiments.
-
FA549 cells expressing Flag‐Arp2 alone or together with HA‐WDR63 were serum‐starved for 6 h. Cells were then treated with or without 10% FBS for 30 min, followed by immunofluorescence assay. Data shown represent three independent experiments. Scale bar: 20 μm.
-
GLysates from HEK293T cells expressing GFP‐WDR63 alone or together with Flag‐VCA were immunoprecipitated by anti‐Flag antibody, followed by Western blot analysis. Data shown represent three independent experiments.
To delineate the regions of WDR63 that are responsible for its interaction with the Arp2/3 complex, we generated a panel of WDR63 deletion mutants and performed an in vitro binding assay. Similar to full‐length WDR63, WDR63 deletion mutants (aa 394–891 and aa 394–817) that contain the central WD repeats region, WDR63 (aa 201–393), and WDR63 (ΔWD) are strongly associated with Arp2/3 (Fig 3H). In contrast, WDR63 (aa 1–200) exhibited no interaction with Arp2/3 (Fig 3H). These data suggest that multiple regions of WDR63 except the region of aa 1–200 mediate the interaction with the Arp2/3 complex.
WDR63 suppresses Arp2/3‐mediated actin polymerization
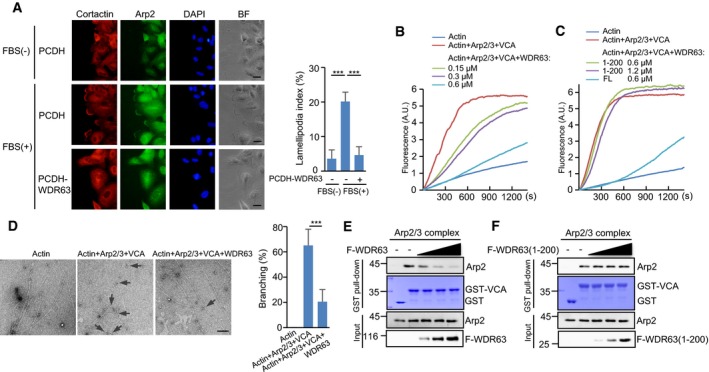
The Arp2/3 complex initiates the formation of the branched actin network at the leading edge of motile cells 31. Given the interaction of WDR63 with the Arp2/3 complex, we sought to investigate whether WDR63 could regulate Arp2/3 function. We first analyzed the cellular localization of WDR63. The results showed that WDR63 was predominantly and evenly distributed in the cytoplasm (Fig EV3D and E). The immunofluorescence assay revealed that ectopic expression of WDR63 was able to inhibit FBS (fetal bovine serum)‐stimulated lamellipodia formation by impairing the recruitment of Arp2 to the leading edge of migrating cells (Figs 4A and EV3F). The pyrene actin polymerization assay showed that WDR63 dose‐dependently reduced the rate of VCA‐stimulated Arp2/3‐mediated actin polymerization (Fig 4B). Unlike full‐length WDR63, the Arp2/3 binding‐defective mutant of WDR63 (aa 1–200) failed to show any effect on Arp2/3‐mediated actin polymerization (Fig 4C), indicating that the interaction with Arp2/3 is critical for WDR63 to inhibit actin polymerization. To further investigate the effect of WDR63 on Arp2/3‐dependent actin branching, we performed electron microscopy. The results showed that the fewer actin‐branched junctions were generated by Arp2/3 and VCA in the presence of WDR63 (Fig 4D). These data collectively indicate the inhibitory role of WDR63 in Arp2/3‐mediated actin polymerization.
Figure 4. WDR63 suppresses Arp2/3‐mediated actin polymerization.
-
AImmunolocalization of Arp2 in a wound‐healing assay. A549 cells expressing control or WDR63 were serum‐starved for 6 h before they were scratched. Cells were then treated with or without 10% FBS for 30 min, followed by immunofluorescence assay. The branched actin networks were shown by immunostaining with anti‐cortactin antibody. Data shown represent three independent experiments. Scale bar: 20 μm. The lamellipodia formation was quantified by measuring the length of the outer margin of lamellipodia in individual cell (n = 20 for each condition) and expressed as the proportion of the total length of the cell perimeter. Data shown are mean ± SD; n = 3 independent experiments. ***P < 0.001; one‐way ANOVA.
-
B, CEffects of full‐length WDR63 (B) and WDR63 (1–200) (C) on Arp2/3‐mediated actin polymerization were examined by pyrene actin polymerization assays. Various concentrations of WDR63 (B) or WDR63 (1–200) (C) were incubated with 2 μM actin (50% pyrene‐labeled), 10 nM Arp2/3, and 400 nM GST‐VCA as indicated. Data shown represent three independent experiments.
-
DEffect of WDR63 on Arp2/3‐mediated branched actin polymerization was analyzed by electron microscopy. 0.6 μM WDR63 was incubated with 2 μM actin, 10 nM Arp2/3, and 400 nM GST‐VCA as indicated. Actin structures were then stabilized with addition of 2 μM phalloidin, followed by electron microscopy analysis. The arrows indicate actin branch junctions. The percentage of branched actin filaments was expressed as the ratio of the number of actin filaments with branch junctions to the total numbers of actin filaments. For each condition, over 150 actin filaments were counted. Data shown are mean ± SD; n = 3 independent experiments. ***P < 0.001; two‐tailed Student's t‐test. Scale bar: 200 nm.
-
E, FIncreasing amounts of purified Flag‐WDR63 (0.1, 0.2, and 0.4 μM) (E) or Flag‐WDR63 (1–200) (0.25, 0.5, and 1 μM) (F) were incubated with the Arp2/3 complex (10 nM) and GST‐VCA as indicated in vitro, followed by GST pull‐down assay. Data shown represent three independent experiments.
We next sought to investigate how WDR63 exerts its function. The immunoprecipitation assay showed that WDR63 did not interact with VCA (Fig EV3G). We therefore asked whether WDR63 could compete with VCA for binding to the Arp2/3 complex. WDR63 indeed inhibited the association of VCA with Arp2/3 in a dose‐dependent manner (Fig 4E). However, the Arp2/3 binding‐defective mutant of WDR63 (aa 1–200) did not exhibit the similar effect (Fig 4F), which explained why this WDR63 mutant failed to regulate Arp2/3‐mediated actin polymerization (Fig 4C). Together, these data suggest that WDR63 suppresses Arp2/3‐mediated actin polymerization by disrupting the VCA‐Arp2/3 interaction.
WDR63 inhibits cell migration and invasion through the Arp2/3 complex
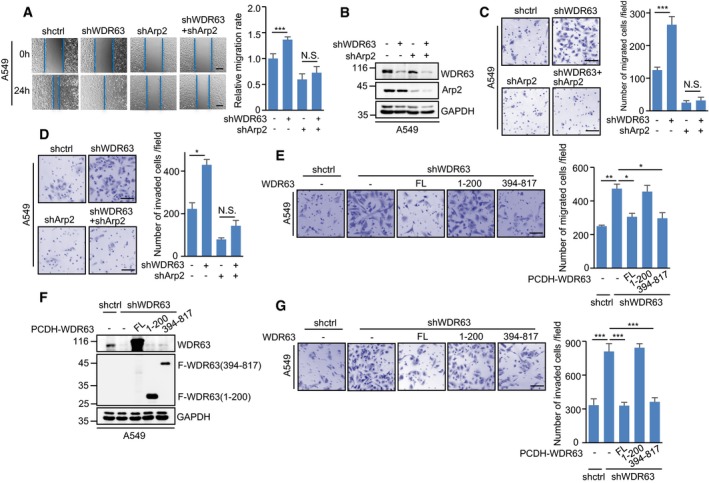
We next explored whether WDR63 regulates cell migration and invasion via the Arp2/3 complex. WDR63 and Arp2 were knocked down individually or combined in A549 and H292 cells. The migratory ability of these cells was then examined by wound‐healing, transwell migration, and single‐cell tracking assays. Knockdown of WDR63 consistently enhanced the migration of A549 and H292 cells (Figs 5A–C and EV4A–E). However, when Arp2 was knocked down in these cells, WDR63 did not exhibit any effect on cell migration (Figs 5A–C and EV4A–E). In addition, the transwell invasion assay showed that knockdown of WDR63 greatly increased the invasive ability of A549 and H292 cells; however, this promoting effect was minimized by Arp2 knockdown (Figs 5D and EV4F). Similar to knockdown of Arp2, inhibition of Arp2/3 by its specific inhibitor CK666 also greatly reversed the enhancing effect of WDR63 knockdown on cell migration and invasion (Fig EV4G and H). These findings indicate that WDR63 indeed regulates cell migration and invasion via the Arp2/3 complex.
Figure 5. WDR63 inhibits cell migration and invasion through the Arp2/3 complex.
-
A–DA549 cells expressing control shRNA, WDR63 shRNA#1, Arp2 shRNA, or both WDR63 and Arp2 shRNA were subjected to wound‐healing (A), transwell migration (C), and transwell invasion (D) assays. Lysates from these cells were also analyzed by Western blot (B). The shown images are representative of three independent experiments. Data shown are mean ± SD (n = 3). *P < 0.05; ***P < 0.001; N.S., no significance; one‐way ANOVA. Scale bar in (A): 200 μm. Scale bars in (C and D): 100 μm.
-
E–GA549 cells expressing either control shRNA or WDR63 shRNA#1 were infected with lentiviruses expressing Flag‐tagged full‐length or truncated WDR63 proteins as indicated. Cells were then subjected to transwell migration (E) and transwell invasion (G) assays. Lysates from these cells were also analyzed by Western blot (F). The shown images are representative of three independent experiments. Data shown are mean ± SD (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001; one‐way ANOVA. Scale bar: 100 μm.
Figure EV4. WDR63 suppresses cell migration and invasion via interacting with the Arp2/3 complex.

-
A–CH292 cells expressing control shRNA, WDR63 shRNA#1, Arp2 shRNA, or both WDR63 and Arp2 shRNA were subjected to wound‐healing (A) and transwell migration (C) assays. Lysates from these cells were also analyzed by Western blot (B). The shown images are representative of three independent experiments. Data shown are mean ± SD (n = 3). **P < 0.01; ***P < 0.001; N.S., no significance; one‐way ANOVA. Scale bar in (A): 200 μm. Scale bar in (C): 100 μm.
-
D, EA549 (D) and H292 (E) cells expressing control shRNA, WDR63 shRNA#1, or both WDR63 and Arp2 shRNA were analyzed by single‐cell tracking with time‐lapse microscopy at 10‐min intervals for 4 h. Scale bar in (D): 200 μm. Scale bar in (E): 100 μm. Cell migration speed, persistence, and mean‐squared displacement (MSD) were also analyzed using an Excel macro described by Gorelik and Gautreau. Data shown are mean ± SD. n = 3 independent experiments. **P < 0.01; ***P < 0.001; one‐way ANOVA.
-
FH292 cells expressing control shRNA, WDR63 shRNA#1, Arp2 shRNA, or both WDR63 and Arp2 shRNA were subjected to transwell invasion assay. The shown images are representative of three independent experiments. Data shown are mean ± SD (n = 3). *P < 0.05; ***P < 0.001; one‐way ANOVA. Scale bar: 100 μm.
-
G, HA549 cells expressing control shRNA and WDR63 shRNA#1 were treated with or without 100 μM CK666 for 24 h. The cells were then subjected to transwell migration (G) and transwell invasion (H) assays. The shown images are representative of three independent experiments. Data shown are mean ± SD (n = 3). *P < 0.05; ***P < 0.001; N.S., no significance; one‐way ANOVA. Scale bar: 100 μm.
-
IA549 cells expressing either control shRNA or WDR63 shRNA#1 were infected with lentiviruses expressing Flag‐tagged full‐length or truncated WDR63 proteins as indicated. Cells were then subjected to wound‐healing assay. The shown images are representative of three independent experiments. Data shown are mean ± SD (n = 3). **P < 0.01; ***P < 0.001; one‐way ANOVA. Scale bar: 200 μm.
To further determine whether the interaction with Arp2/3 is essential for WDR63 to regulate cell migration and invasion, we used two deletion mutants of WDR63 (aa 1–200 and aa 394–817) that lost and retained the Arp2/3‐binding ability, respectively (Fig 3H). Both full‐length WDR63 and WDR63 (394–817) could reverse WDR63 knockdown‐increased cell migration and invasion (Figs 5E–G and EV4I). However, WDR63 (1–200) failed to show the similar effect (Figs 5E–G and EV4I). Taken together, these data suggest that WDR63 inhibits cell migration and invasion through interacting with the Arp2/3 complex.
WDR63 mediates p53's function in suppressing cell migration, invasion, and metastasis
p53 plays a critical role in inhibiting cancer metastasis. Given the above findings that WDR63 is transcriptionally upregulated by p53 and WDR63 is able to inhibit cell migration, invasion, and metastasis, we asked whether WDR63 could mediate p53's function in suppressing cancer metastasis. Knockdown of p53 significantly enhanced the migration and invasion of both A549 and H292 cells, however, which was reversed by ectopic expression of WDR63 (Figs 6A–D and EV5A–D). We next evaluated the effects of WDR63 deletion mutants (aa 1–200 and aa 394–817) on p53‐regulated cell migration and invasion. Similar to full‐length WDR63, WDR63 (394–817), which retained the Arp2/3‐binding ability, was able to reduce p53 knockdown‐increased cell migration and invasion (Figs 6E and F, and EV5E and F). However, WDR63 (aa 1–200), which lost the Arp2/3‐binding ability, failed to show any effect on p53‐regulated cell migration and invasion (Figs 6E and F, and EV5E and F), indicating the critical role of the WDR63–Arp2/3 axis in mediating the suppressive effect of p53 on cell migration and invasion. By using a xenograft model of metastasis, we showed that knockdown of p53 dramatically increased lung extravasation of A549 cells (Figs 6G and EV5G). However, the promoting effect of p53 knockdown on lung extravasation was greatly abolished by overexpression of WDR63 (Figs 6G and EV5G). These results demonstrate that WDR63 is an important mediator of p53 in suppressing cell migration, invasion, and metastasis.
Figure 6. WDR63 mediates p53's function in suppressing cell migration, invasion, and metastasis.

-
A–DA549 cells expressing control shRNA, p53 shRNA, or p53 shRNA plus Flag‐WDR63 were subjected to wound‐healing (A), transwell migration (C), and transwell invasion (D) assays. Lysates from these cells were also analyzed by Western blot (B). The shown images are representative of three independent experiments. Data shown are mean ± SD (n = 3). **P < 0.01; ***P < 0.001; one‐way ANOVA. Scale bar in (A): 200 μm. Scale bars in (C and D): 100 μm.
-
E, FA549 cells expressing either control shRNA or p53 shRNA were infected with lentiviruses expressing Flag‐tagged full‐length or truncated WDR63 proteins as indicated. Cells were then subjected to transwell migration (E) and transwell invasion (F) assays. The shown images are representative of three independent experiments. Data shown are mean ± SD (n = 3). *P < 0.05; ***P < 0.001; one‐way ANOVA. Scale bar: 100 μm. The successful knockdown of p53 and overexpression of WDR63 proteins are shown in Fig EV5F.
-
GA549 cells expressing control shRNA, p53 shRNA, or p53 shRNA plus Flag‐WDR63 (each also expressing luciferase) were injected via tail vein into nude mice (n = 6 for each group). Three weeks after injection, tumor formation was monitored by bioluminescence imaging. Data shown are mean ± SD; *P < 0.05; one‐way ANOVA.
Figure EV5. WDR63 mediates the metastasis‐suppressive function of p53.

-
A–DH292 cells expressing control shRNA, p53 shRNA, or p53 shRNA plus Flag‐WDR63 were subjected to wound‐healing (A), transwell migration (C), and transwell invasion (D) assays. Lysates from these cells were also analyzed by Western blot (B). The shown images are representative of three independent experiments. Data shown are mean ± SD (n = 3). *P < 0.05; **P < 0.01; one‐way ANOVA. Scale bar in (A): 200 μm. Scale bars in (C and D): 100 μm.
-
E, FA549 cells expressing either control shRNA or p53 shRNA were infected with lentiviruses expressing Flag‐tagged full‐length or truncated WDR63 proteins as indicated. Cells were then subjected to wound‐healing assay (E). Lysates from these cells were also analyzed by Western blot (F). The shown images are representative of three independent experiments. Data shown are mean ± SD (n = 3). *P < 0.05; **P < 0.01; one‐way ANOVA. Scale bar: 200 μm.
-
GA549 cells expressing control shRNA, p53 shRNA, or p53 shRNA plus Flag‐WDR63 (each also expressing luciferase) were injected via tail vein into nude mice (n = 6 for each group). Six weeks after injection, tumor formation in lungs was monitored by bioluminescence imaging. Data shown are mean ± SD; *P < 0.05; one‐way ANOVA. Scale bar: 1 cm.
Discussion
The tumor suppressor p53 plays a prominent role in the protection against cancer 2, 54. Increasing evidence suggests a suppressive function of p53 in cancer metastasis 13, 55. The tumor‐derived p53 mutants also gain additional functions that promote metastasis 14, 56. It has been well recognized that as a master transcription factor, p53 transcribes its target genes to regulate a variety of cellular processes, which is central to its role as a tumor suppressor. While previous studies on the tumor‐suppressive function of p53 are mainly focused on its canonical anti‐proliferative effects, such as apoptosis, cell cycle arrest, and senescence, the mechanism of how p53 suppresses metastasis is much less understood. Therefore, identification of new p53‐responsive target with ability to regulate cell migration and invasion is of great importance to the comprehensive understanding of p53 biology. In this study, we report WDR63 as a bona fide transcriptional target of p53. The importance of p53‐induced WDR63 expression is also supported by the observation that WDR63 was expressed at lower levels in LUAD and LUSC harboring mutant TP53 gene. Functionally, WDR63 is able to mediate p53's function in suppressing metastasis through inhibiting Arp2/3‐mediated actin polymerization. Therefore, WDR63 is an important player in the regulation of p53 function.
WDR63 is the orthologue of the Chlamydomonas inner dynein arm intermediate chain (IC) gene IC140 57. Via the association with WDR78 (IC138 orthologue), WDR63 is potentially involved in the regulation of cilia function 58. WDR63 is also able to enhance osteogenic differentiation of dental tissue‐derived mesenchymal stem cells 59. The intragenic deletion of WDR63 has been implicated in human occipital encephalocele 60. However, the detailed function of WDR63 awaits further characterization.
Here, we clearly demonstrate that WDR63 is a negative regulator of cell migration and invasion. Further mechanistic investigation shows that WDR63 exerts its function through the Arp2/3 complex. WDR63 is able to interact with the Arp2/3 complex. As a result, WDR63 suppresses Arp2/3‐mediated actin polymerization by competing with VCA for binding to the Arp2/3 complex. The interaction with Arp2/3 appears to be essential for WDR63 to inhibit Arp2/3 function, as revealed by the findings that the Arp2/3 binding‐defective mutant of WDR63 (1–200) fails to show any regulatory effect on Arp2/3‐mediated actin polymerization, cell migration, and invasion. It has been shown that in agreement with the important role of Arp2/3‐mediated actin polymerization in cell migration 61, the deregulation of the Arp2/3 complex and its regulators has been linked to the progression of a variety of human cancers, indicating the important role of the Arp2/3 system in the regulation of cancer progression 62. Therefore, it is not surprising that accompanying with the inhibitory effect of WDR63 on cell migration and invasion, WDR63 is able to suppress lung extravasation of A549 cells.
As mentioned earlier, due to the low intrinsic actin nucleation activity of Arp2/3 on its own, Arp2/3 needs to be activated by nucleation‐promoting factors (NPFs). However, the NPFs normally adopt an autoinhibitory conformation 35. The VCA domains used to stimulate Arp2/3 have to be released from this inhibition by convergent signals involving activated Rho family GTPase 63. For example, cell migration and invasion involve activation of Rac1 and Cdc42, resulting in NPF‐stimulated Arp2/3‐mediated actin polymerization 64. It has been previously reported that p53 is capable of inhibiting the activity of Rac1 and Cdc42 19, 21. We here show that p53‐induced WDR63 is able to suppress Arp2/3‐mediated actin polymerization. Therefore, it is possible that p53 may inhibit cell migration and invasion through both Rho GTPase‐ and WDR63‐dependent regulation of Arp2/3‐mediated actin polymerization. Together, our study reveals that WDR63 is an important mediator of p53 in suppression of metastasis. Although we here clearly show that WDR63 mediates p53's function in suppressing cell migration and invasion, it is unlikely that WDR63 is the only determinant of the metastasis‐suppressive function for p53, because it has been previously reported that p53 is capable of inhibiting metastasis via several other mechanisms. For instance, p53 is able to inhibit tumor invasion and metastasis by down‐regulating IDO1 expression 65. In addition, the p53/p21 complex is shown to suppress cancer cell invasion by targeting Bcl‐2 family proteins 66. Moreover, a polymorphism in TP53 at amino acid 72 enhances migration and invasion of tumors through the ability to bind and regulate PGC‐1ɑ 67. These findings indicate that p53 may exert its function in suppressing metastasis through multiple different mechanisms.
Materials and Methods
Reagents and antibodies
The following reagents were used in this study: doxorubicin (Sigma, 1 μg/ml), doxycycline (Sigma, 1 μg/ml), etoposide (Sigma, 50 μM), CK666 (Sigma, 100 μM), Nutlin‐3 (Sigma, 10 μM), FITC‐labeled phalloidin (Sigma, P5282), complete EDTA‐free protease inhibitor cocktail (Roche Applied Science), glutathione beads (GE Healthcare), antibodies against Arp2 (Santa Cruz, sc‐166103, 1: 1:500), Arp3 (Proteintech, 13822‐1‐AP, 1:200), ArpC2 (Proteintech, 15058‐1‐AP, 1:200), ArpC3 (Proteintech, 14652‐1‐AP, 1:200), WDR63 (Abcam, ab216126, 1:200), p53 (Santa Cruz, sc‐126, 1:1,000), p21 (Sigma, P1484, 1:4,000), GAPDH (Santa Cruz, sc‐166545, 1:5,000), GFP (Santa Cruz, sc‐9996, 1:1,000), Flag for WB (Sigma, F3165, 1:4,000), Flag for immunofluorescence (Proteintech, 20543‐1‐AP, 1:200), HA for WB (Sigma, H9658, 1:4,000), HA for immunofluorescence (Sigma, H9658, 1:200), cortactin for immunofluorescence (Proteintech, 11381‐1‐AP, 1:200), HRP‐conjugated secondary antibodies against mouse (115‐035‐062, 1:10,000) and rabbit (111‐035‐144, 1:10,000) (Jackson ImmunoResearch), rhodamine‐conjugated secondary antibodies against mouse (115‐025‐146, 1:200) and rabbit (111‐025‐144, 1:200) (Jackson ImmunoResearch), FITC‐conjugated secondary antibody against mouse (115‐095‐146, 1:200) (Jackson ImmunoResearch), and normal mouse and rabbit IgG (Santa Cruz, sc‐2025 and sc‐2027).
Cell culture
A549, H292, and H1299 cells were maintained in RPMI 1640 medium (Sigma) supplemented with 10% FBS and 1% penicillin/streptomycin. HCT116 and HEK293T cells were cultured in DMEM (Invitrogen) supplemented with 10% FBS and 1% penicillin/streptomycin. All cell lines were routinely tested for mycoplasma contamination.
Generation of the lentiviral expression system
To generate lentiviruses expressing the indicated shRNAs, HEK293T cells were transfected with shRNAs (cloned in PLKO.1), pREV, pGag/Pol/PRE, and pVSVG with a ratio of 2:2:2:1. To generate lentiviruses expressing the indicated proteins, HEK293T cells were transfected with PCDH‐based construct, pmd2.g, and pspax2 with a ratio of 2:1:2. Twelve hours after transfection, cells were cultured in fresh medium for another 24 h. The culture medium containing lentivirus particles was filtered through a 0.45‐μm PVDF filter and used for infection. The shRNA target sequences are listed in Table EV1.
Real‐time RT–PCR
1 μg of total RNA isolated using TRIzol reagent (Invitrogen) was used to synthesize cDNA using the PrimeScriptTM RT Reagent Kit (TaKaRa) according to the manufacturer's instructions. Real‐time PCR was performed using SYBR Premix EX Taq (TaKaRa) and analyzed with the StepOnePlus Real‐Time PCR System (Thermo Fisher Scientific). The primer sequences are shown in Table EV1.
Protein expression and purification
The DNA sequence encoding WDR63 or Arp2 was cloned into pGEX‐4T‐1 vector, which contains a TEV protease cleavage site. The constructs were transformed into Escherichia coli Rosetta 2 (DE3). After induction with isopropyl D‐thiogalactoside for 20 h at 16°C, GST‐fusion proteins were purified by glutathione affinity chromatography. To obtain GST tag‐free Arp2, the glutathione bead‐bound GST‐Arp2 was incubated with TEV protease.
To overexpress Flag‐WDR63 or Flag‐WDR63 (1–200) proteins, Flag‐WDR63 or Flag‐WDR63 (1–200)‐expressing construct was transfected into HEK293T cells. Cell lysates were immunoprecipitated with anti‐Flag M2 beads (Sigma). To remove non‐specific binding proteins, the beads were sequentially washed with lysis buffer containing 0.25, 0.5, and 1 M KCl as we previously described 68. The bead‐bound Flag‐tagged proteins were eluted with 3× Flag peptide (Sigma).
GST pull‐down and immunoprecipitation
For the GST pull‐down assay, GST‐fusion proteins immobilized on glutathione beads were incubated with the indicated proteins in the pull‐down buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, and 0.5% Triton X‐100) for 4 h at 4°C. Input and bead‐bound proteins were analyzed by Western blot. For the immunoprecipitation assay, cells were lysed in IP lysis buffer (50 mM Tris–HCl, pH 7.4, 0.5% NP‐40, 0.5% Triton X‐100, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, and 10% glycerol) supplemented with 1× protease inhibitor cocktail. Cell lysates were pre‐cleared with protein A/G‐coupled Sepharose beads for 2 h before they were immunoprecipitated with the indicated antibodies. The input and immunoprecipitates were then analyzed by Western blot.
Identification of Arp2 as a WDR63‐interacting protein
To identify WDR63‐interacting proteins, we employed a GST pull‐down experiment. 1 × 107 A549 cells were lysed in IP lysis buffer (50 mM Tris–HCl, pH 7.4, 0.5% NP‐40, 0.5% Triton X‐100, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, and 10% glycerol) supplemented with 1× protease inhibitor cocktail. Cell lysates were then incubated with purified GST or GST‐WDR63 immobilized on glutathione beads. After extensive washing, bead‐bound proteins were separated by SDS–PAGE and visualized by Coomassie blue staining. The strong protein bands specifically present in GST‐WDR63 pull‐down complex were analyzed by mass spectrometry, and a ~ 44 kD protein band was identified as Arp2.
Chromosome immunoprecipitation assay
The chromosome immunoprecipitation (ChIP) assay was performed by using the ChIP Assay Kit (Beyotime Biotechnology, Shanghai, China) as we previously described 69. Normal mouse IgG and anti‐p53 antibody were used for immunoprecipitation. The bound DNA fragments were analyzed by real‐time PCR using the specific primers (Table EV1).
Luciferase reporter assay
To determine whether WDR63 is transcriptionally regulated by p53, A549 cells expressing control shRNA or p53 shRNA, or A549 cells expressing control or Flag‐p53 were transfected with the indicated pGL3‐based reporter constructs plus Renilla luciferase plasmid. Twenty‐four hours later, firefly and Renilla luciferase activities were measured by the Dual‐Luciferase Reporter Assay System (Promega), and Renilla luciferase activity was used to normalize firefly luciferase activity.
Pyrene actin polymerization assay
The pyrene actin polymerization assay was performed using the Actin Polymerization Biochem Kit (Cytoskeleton, Inc.) according to the manufacturer's instructions. The Arp2/3 protein complex and GST‐tagged VCA domain of WASP were also purchased from Cytoskeleton, Inc. Reaction conditions were: 2 μM actin (50% pyrene‐labeled), 10 nM Arp2/3, and 400 nM VCA plus full‐length WDR63 or WDR63 (1–200) at the indicated concentrations. Pyrene fluorescence was recorded using a CLARIOstar microplate reader (BMG LABTECH) at excitation and emission wavelengths of 365 and 407 nm, respectively.
Electron microscopy
To examine the effect of WDR63 on Arp2/3‐stimulated branched actin polymerization, 0.6 μM WDR63 was incubated with 2 μM actin, 10 nM Arp2/3, and 400 nM GST‐VCA for 20 min at 25°C in the polymerization buffer (5 mM Tris–HCl, pH 8.0, 50 mM KCl, 2 mM MgCl2, 0.2 mM CaCl2, 5 mM guanidine carbonate, 1 mM ATP, and 1 mM DTT). Actin structures were then stabilized with addition of 2 μM phalloidin. Negative staining was used to visualize F‐actin under electron microscopy. The samples were loaded onto copper grids coated with formvar and carbon for 90 s, and excess liquid was removed with filter paper. Actin bound on the grids was washed twice and stained with 2% uranyl acetate (Sigma) for 90 s. Excess liquid was removed, and grids were air‐dried. Electron microscopy observation was performed using a transmission electron microscope (Tecnai Spirit, FEI) operated at 120 kV. The percentage of branched actin filaments was expressed as the ratio of the number of actin filaments with branch junctions to the total numbers of actin filaments. For each condition, over 150 actin filaments were counted.
Cell migration and invasion assays
The wound‐healing and transwell migration assays were used to measure cell migratory ability. The transwell invasion assay was used to examine cell invasive ability. For the wound‐healing assay, cells seeded in six‐well plate were serum‐starved for 6 h, and then, a straight scratch was created using a pipette tip. Wound healing was observed at 24 h after wounding. The transwell system (Corning) was used for cell migration and invasion studies according to the manufacturer's instructions. Briefly, cells in serum‐free medium were seeded into the upper uncoated (migration assay) or Matrigel‐coated chamber (invasion assay). Medium containing 10% FBS was placed in the lower chamber as a chemoattractant. Twenty‐four hours later, cells migrated/invaded to the lower surface were fixed and stained with 0.1% crystal violet. The stained cells were then photographed, and the numbers of cells counted in three randomly chosen fields (200×) were averaged.
Single‐cell tracking analysis
Cells were cultured in glass‐bottomed dish with Leibovitz's L‐15 medium (Gibco) and placed in a temperature‐controlled chamber. The movement of cells was tracked with time‐lapse microscopy at 10‐min intervals for 4 h. Cell migration paths were then analyzed using ImageJ software (NIH) and Microsoft Excel program. Cell migration speed, persistence, and mean‐squared displacement (MSD) were also analyzed using an Excel macro described by Gorelik and Gautreau 70.
In vivo lung metastasis assay
A549 stable cell lines (2 × 106) were injected into 6‐week‐old Balb/c nude mice via tail vein (n = 6 in each group were from three independent experiments). Tumor formation in whole animals was analyzed 3 weeks later. The mice were injected with D‐luciferin and imaged for 5 min using the IVIS Spectrum Imaging System (PerkinElmer). To evaluate tumor extravasation into lungs, mice were sacrificed 6 weeks later. The lungs were then dissected and imaged. Quantification was performed by Living Image software (PerkinElmer). Studies on mice were conducted with approval from the Animal Research Ethics Committee of the University of Science and Technology of China.
Reproducibility
Each experiment was repeated independently at least three times with similar results.
Statistical analysis
Statistical analysis was carried out using Microsoft Excel software and GraphPad Prism to assess differences between experimental groups. Statistical significance was analyzed by two‐tailed Student's t‐test or one‐way ANOVA and expressed as a P‐value. P < 0.05 was considered to be statistically significant. *, **, and *** indicate P < 0.05, P < 0.01, and P < 0.001, respectively. N.S. indicates no significance.
Author contributions
KZ and YM conceived and designed the project. KZ, DW, XZ, CW, YG, and KL performed all the experiments and analyzed the data. FW, XWu, XWa, LS, and JZ provided the reagents and technical help. YM wrote the manuscript. All authors discussed the results and commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Review Process File
Acknowledgements
This work was supported by grants from Ministry of Science and Technology of China (2019YFA0802600), National Natural Science Foundation of China (31871440, 31671487 and 91957108), the Fundamental Research Funds for Central Universities (WK9110000007 and YD2070002007), and Collaborative Innovation Programs of Hefei Science Center, CAS (2019HSC‐CIP010).
EMBO Reports (2020) 21: e49269
References
- 1. Vousden KH, Prives C (2009) Blinded by the light: the growing complexity of p53. Cell 137: 413–431 [DOI] [PubMed] [Google Scholar]
- 2. Kastenhuber ER, Lowe SW (2017) Putting p53 in context. Cell 170: 1062–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aylon Y, Oren M (2016) The paradox of p53: what, how, and why? Cold Spring Harb Perspect Med 6: a026328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mello SS, Attardi LD (2018) Deciphering p53 signaling in tumor suppression. Curr Opin Cell Biol 51: 65–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Freed‐Pastor WA, Prives C (2012) Mutant p53: one name, many proteins. Genes Dev 26: 1268–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, Bradley A (1992) Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356: 215–221 [DOI] [PubMed] [Google Scholar]
- 7. Pfister NT, Prives C (2017) Transcriptional regulation by wild‐type and cancer‐related mutant forms of p53. Cold Spring Harb Perspect Med 7: a026054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hafner A, Bulyk ML, Jambhekar A, Lahav G (2019) The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol 20: 199–210 [DOI] [PubMed] [Google Scholar]
- 9. Labuschagne CF, Zani F, Vousden KH (2018) Control of metabolism by p53 – cancer and beyond. Biochim Biophys Acta Rev Cancer 1870: 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. White E (2016) Autophagy and p53. Cold Spring Harb Perspect Med 6: a026120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W (2015) Ferroptosis as a p53‐mediated activity during tumour suppression. Nature 520: 57–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri‐Mergny M, D'Amelio M, Criollo A, Morselli E, Zhu C, Harper F et al (2008) Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 10: 676–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Powell E, Piwnica‐Worms D, Piwnica‐Worms H (2014) Contribution of p53 to metastasis. Cancer Discov 4: 405–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Muller PA, Vousden KH, Norman JC (2011) p53 and its mutants in tumor cell migration and invasion. J Cell Biol 192: 209–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thompson TC, Park SH, Timme TL, Ren C, Eastham JA, Donehower LA, Bradley A, Kadmon D, Yang G (1995) Loss of p53 function leads to metastasis in ras+myc‐initiated mouse prostate cancer. Oncogene 10: 869–879 [PubMed] [Google Scholar]
- 16. Lewis BC, Klimstra DS, Socci ND, Xu S, Koutcher JA, Varmus HE (2005) The absence of p53 promotes metastasis in a novel somatic mouse model for hepatocellular carcinoma. Mol Cell Biol 25: 1228–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xia M, Land H (2007) Tumor suppressor p53 restricts Ras stimulation of RhoA and cancer cell motility. Nat Struct Mol Biol 14: 215–223 [DOI] [PubMed] [Google Scholar]
- 18. Guo F, Zheng Y (2004) Rho family GTPases cooperate with p53 deletion to promote primary mouse embryonic fibroblast cell invasion. Oncogene 23: 5577–5585 [DOI] [PubMed] [Google Scholar]
- 19. Guo F, Gao Y, Wang L, Zheng Y (2003) p19Arf‐p53 tumor suppressor pathway regulates cell motility by suppression of phosphoinositide 3‐kinase and Rac1 GTPase activities. J Biol Chem 278: 14414–14419 [DOI] [PubMed] [Google Scholar]
- 20. Gadea G, de Toledo M, Anguille C, Roux P (2007) Loss of p53 promotes RhoA‐ROCK‐dependent cell migration and invasion in 3D matrices. J Cell Biol 178: 23–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gadea G, Roger L, Anguille C, de Toledo M, Gire V, Roux P (2004) TNFalpha induces sequential activation of Cdc42‐ and p38/p53‐dependent pathways that antagonistically regulate filopodia formation. J Cell Sci 117: 6355–6364 [DOI] [PubMed] [Google Scholar]
- 22. Kim NH, Kim HS, Li XY, Lee I, Choi HS, Kang SE, Cha SY, Ryu JK, Yoon D, Fearon ER et al (2011) A p53/miRNA‐34 axis regulates Snail1‐dependent cancer cell epithelial‐mesenchymal transition. J Cell Biol 195: 417–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang SP, Wang WL, Chang YL, Wu CT, Chao YC, Kao SH, Yuan A, Lin CW, Yang SC, Chan WK et al (2009) p53 controls cancer cell invasion by inducing the MDM2‐mediated degradation of Slug. Nat Cell Biol 11: 694–704 [DOI] [PubMed] [Google Scholar]
- 24. Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW, Yu WH, Rehman SK, Hsu JL, Lee HH et al (2011) p53 regulates epithelial‐mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol 13: 317–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kunz C, Pebler S, Otte J, von der Ahe D (1995) Differential regulation of plasminogen activator and inhibitor gene transcription by the tumor suppressor p53. Nucleic Acids Res 23: 3710–3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zou Z, Gao C, Nagaich AK, Connell T, Saito S, Moul JW, Seth P, Appella E, Srivastava S (2000) p53 regulates the expression of the tumor suppressor gene maspin. J Biol Chem 275: 6051–6054 [DOI] [PubMed] [Google Scholar]
- 27. Hwang CI, Matoso A, Corney DC, Flesken‐Nikitin A, Korner S, Wang W, Boccaccio C, Thorgeirsson SS, Comoglio PM, Hermeking H et al (2011) Wild‐type p53 controls cell motility and invasion by dual regulation of MET expression. Proc Natl Acad Sci USA 108: 14240–14245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shi D, Murty VV, Gu W (2015) PCDH10, a novel p53 transcriptional target in regulating cell migration. Cell Cycle 14: 857–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang C, Liu J, Zhao Y, Yue X, Zhu Y, Wang X, Wu H, Blanco F, Li S, Bhanot G et al (2016) Glutaminase 2 is a novel negative regulator of small GTPase Rac1 and mediates p53 function in suppressing metastasis. Elife 5: e10727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mukhopadhyay UK, Eves R, Jia L, Mooney P, Mak AS (2009) p53 suppresses Src‐induced podosome and rosette formation and cellular invasiveness through the upregulation of caldesmon. Mol Cell Biol 29: 3088–3098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Insall RH, Machesky LM (2009) Actin dynamics at the leading edge: from simple machinery to complex networks. Dev Cell 17: 310–322 [DOI] [PubMed] [Google Scholar]
- 32. Krause M, Gautreau A (2014) Steering cell migration: lamellipodium dynamics and the regulation of directional persistence. Nat Rev Mol Cell Biol 15: 577–590 [DOI] [PubMed] [Google Scholar]
- 33. Swaney KF, Li R (2016) Function and regulation of the Arp2/3 complex during cell migration in diverse environments. Curr Opin Cell Biol 42: 63–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goley ED, Welch MD (2006) The ARP2/3 complex: an actin nucleator comes of age. Nat Rev Mol Cell Biol 7: 713–726 [DOI] [PubMed] [Google Scholar]
- 35. Campellone KG, Welch MD (2010) A nucleator arms race: cellular control of actin assembly. Nat Rev Mol Cell Biol 11: 237–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rotty JD, Wu C, Bear JE (2013) New insights into the regulation and cellular functions of the ARP2/3 complex. Nat Rev Mol Cell Biol 14: 7–12 [DOI] [PubMed] [Google Scholar]
- 37. Dang I, Gorelik R, Sousa‐Blin C, Derivery E, Guerin C, Linkner J, Nemethova M, Dumortier JG, Giger FA, Chipysheva TA et al (2013) Inhibitory signalling to the Arp2/3 complex steers cell migration. Nature 503: 281–284 [DOI] [PubMed] [Google Scholar]
- 38. Rocca DL, Martin S, Jenkins EL, Hanley JG (2008) Inhibition of Arp2/3‐mediated actin polymerization by PICK1 regulates neuronal morphology and AMPA receptor endocytosis. Nat Cell Biol 10: 259–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zuo X, Zhang J, Zhang Y, Hsu SC, Zhou D, Guo W (2006) Exo70 interacts with the Arp2/3 complex and regulates cell migration. Nat Cell Biol 8: 1383–1388 [DOI] [PubMed] [Google Scholar]
- 40. Chan KT, Creed SJ, Bear JE (2011) Unraveling the enigma: progress towards understanding the coronin family of actin regulators. Trends Cell Biol 21: 481–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cai L, Makhov AM, Schafer DA, Bear JE (2008) Coronin 1B antagonizes cortactin and remodels Arp2/3‐containing actin branches in lamellipodia. Cell 134: 828–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Helgeson LA, Nolen BJ (2013) Mechanism of synergistic activation of Arp2/3 complex by cortactin and N‐WASP. Elife 2: e00884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schnoor M, Stradal TE, Rottner K (2018) Cortactin: cell functions of a multifaceted actin‐binding protein. Trends Cell Biol 28: 79–98 [DOI] [PubMed] [Google Scholar]
- 44. Coumans JVF, Davey RJ, Moens PDJ (2018) Cofilin and profilin: partners in cancer aggressiveness. Biophys Rev 10: 1323–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fischer M (2017) Census and evaluation of p53 target genes. Oncogene 36: 3943–3956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C et al (2004) In vivo activation of the p53 pathway by small‐molecule antagonists of MDM2. Science 303: 844–848 [DOI] [PubMed] [Google Scholar]
- 47. Mathelier A, Fornes O, Arenillas DJ, Chen CY, Denay G, Lee J, Shi W, Shyr C, Tan G, Worsley‐Hunt R et al (2016) JASPAR 2016: a major expansion and update of the open‐access database of transcription factor binding profiles. Nucleic Acids Res 44: D110–D115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zeron‐Medina J, Wang X, Repapi E, Campbell MR, Su D, Castro‐Giner F, Davies B, Peterse EF, Sacilotto N, Walker GJ et al (2013) A polymorphic p53 response element in KIT ligand influences cancer risk and has undergone natural selection. Cell 155: 410–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hafner A, Stewart‐Ornstein J, Purvis JE, Forrester WC, Bulyk ML, Lahav G (2017) p53 pulses lead to distinct patterns of gene expression albeit similar DNA‐binding dynamics. Nat Struct Mol Biol 24: 840–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Andrysik Z, Galbraith MD, Guarnieri AL, Zaccara S, Sullivan KD, Pandey A, MacBeth M, Inga A, Espinosa JM (2017) Identification of a core TP53 transcriptional program with highly distributed tumor suppressive activity. Genome Res 27: 1645–1657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Younger ST, Kenzelmann‐Broz D, Jung H, Attardi LD, Rinn JL (2015) Integrative genomic analysis reveals widespread enhancer regulation by p53 in response to DNA damage. Nucleic Acids Res 43: 4447–4462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Menietti E, Xu X, Ostano P, Joseph JM, Lefort K, Dotto GP (2016) Negative control of CSL gene transcription by stress/DNA damage response and p53. Cell Cycle 15: 1767–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McDade SS, Patel D, Moran M, Campbell J, Fenwick K, Kozarewa I, Orr NJ, Lord CJ, Ashworth AA, McCance DJ (2014) Genome‐wide characterization reveals complex interplay between TP53 and TP63 in response to genotoxic stress. Nucleic Acids Res 42: 6270–6285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kaiser AM, Attardi LD (2018) Deconstructing networks of p53‐mediated tumor suppression in vivo . Cell Death Differ 25: 93–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mak AS (2014) p53 in cell invasion, podosomes, and invadopodia. Cell Adh Migr 8: 205–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, Lukashchuk N, Gillespie DA, Ludwig RL, Gosselin P et al (2009) Mutant p53 drives invasion by promoting integrin recycling. Cell 139: 1327–1341 [DOI] [PubMed] [Google Scholar]
- 57. Pazour GJ, Agrin N, Walker BL, Witman GB (2006) Identification of predicted human outer dynein arm genes: candidates for primary ciliary dyskinesia genes. J Med Genet 43: 62–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang Y, Chen Y, Zheng J, Wang J, Duan S, Zhang W, Yan X, Zhu X (2019) Vertebrate Dynein‐f depends on Wdr78 for axonemal localization and is essential for ciliary beat. J Mol Cell Biol 11: 383–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Diao S, Yang DM, Dong R, Wang LP, Wang JS, Du J, Wang SL, Fan Z (2015) Enriched trimethylation of lysine 4 of histone H3 of WDR63 enhanced osteogenic differentiation potentials of stem cells from apical papilla. J Endod 41: 205–211 [DOI] [PubMed] [Google Scholar]
- 60. Hofmeister W, Pettersson M, Kurtoglu D, Armenio M, Eisfeldt J, Papadogiannakis N, Gustavsson P, Lindstrand A (2018) Targeted copy number screening highlights an intragenic deletion of WDR63 as the likely cause of human occipital encephalocele and abnormal CNS development in zebrafish. Hum Mutat 39: 495–505 [DOI] [PubMed] [Google Scholar]
- 61. Suraneni P, Rubinstein B, Unruh JR, Durnin M, Hanein D, Li R (2012) The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J Cell Biol 197: 239–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Molinie N, Gautreau A (2018) The Arp2/3 regulatory system and its deregulation in cancer. Physiol Rev 98: 215–238 [DOI] [PubMed] [Google Scholar]
- 63. Pollitt AY, Insall RH (2009) WASP and SCAR/WAVE proteins: the drivers of actin assembly. J Cell Sci 122: 2575–2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yamazaki D, Kurisu S, Takenawa T (2005) Regulation of cancer cell motility through actin reorganization. Cancer Sci 96: 379–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tang D, Yue L, Yao R, Zhou L, Yang Y, Lu L, Gao W (2017) P53 prevent tumor invasion and metastasis by down‐regulating IDO in lung cancer. Oncotarget 8: 54548–54557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kim EM, Jung CH, Kim J, Hwang SG, Park JK, Um HD (2017) The p53/p21 complex regulates cancer cell invasion and apoptosis by targeting Bcl‐2 family proteins. Cancer Res 77: 3092–3100 [DOI] [PubMed] [Google Scholar]
- 67. Basu S, Gnanapradeepan K, Barnoud T, Kung CP, Tavecchio M, Scott J, Watters A, Chen Q, Kossenkov AV, Murphy ME (2018) Mutant p53 controls tumor metabolism and metastasis by regulating PGC‐1alpha. Genes Dev 32: 230–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhao K, Yang Y, Zhang G, Wang C, Wang D, Wu M, Mei Y (2018) Regulation of the Mdm2‐p53 pathway by the ubiquitin E3 ligase MARCH7. EMBO Rep 19: 305–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang C, Yang Y, Zhang G, Li J, Wu X, Ma X, Shan G, Mei Y (2019) Long noncoding RNA EMS connects c‐Myc to cell cycle control and tumorigenesis. Proc Natl Acad Sci USA 116: 14620–14629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gorelik R, Gautreau A (2014) Quantitative and unbiased analysis of directional persistence in cell migration. Nat Protoc 9: 1931–1943 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Review Process File