

Abstract
An elevated intracellular NADH/NAD+ ratio, or “reductive stress”, has been associated with multiple diseases, including disorders of the mitochondrial electron transport chain (ETC). As the intracellular NADH/NAD+ ratio can be in near equilibrium with the circulating lactate/pyruvate ratio, we hypothesized that reductive stress could be alleviated by oxidizing extracellular lactate to pyruvate. We engineered LOXCAT, a fusion of bacterial lactate oxidase (LOX) and catalase (CAT), which irreversibly converts lactate and oxygen to pyruvate and water. Addition of purified LOXCAT to the media of cultured human cells with a defective ETC decreased the extracellular lactate/pyruvate ratio, normalized the intracellular NADH/NAD+ ratio, upregulated glycolytic ATP production, and restored cellular proliferation. In mice, tail-vein-injected LOXCAT reduced the circulating lactate/pyruvate ratio, blunted a metformin-induced rise in blood lactate/pyruvate ratio, and improved NADH/NAD+ balance in the heart and brain. Our study lays the groundwork for a new class of injectable therapeutic enzymes that alleviates intracellular redox imbalances by directly targeting circulating redox-coupled metabolites.
Maintenance of a proper NADH/NAD+ ratio is crucial for numerous fundamental cellular processes, including energy metabolism, calcium homeostasis, cell death and proliferation. The mitochondrial electron transport chain (ETC) complex I and cytoplasmic enzyme lactate dehydrogenase (LDH) are major determinants of this ratio by oxidizing NADH to NAD+. Although the lack of mitochondria-derived ATP has been classically described as the major driver of pathology in ETC dysfunction, emerging data suggest that the accompanying elevation in the NADH/NAD+ ratio can also contribute to pathogenesis1–3. An elevated NADH/NAD+ ratio is one form of “reductive stress”, which more broadly refers to the buildup of reducing equivalents (e.g. elevated NADH/NAD+, NADPH/NADP+, GSH/GSSG, or cysteine/cystine ratio)4,5. Here we focus on NADH reductive stress, which can stall scores of NAD+-dependent pathways and generate toxic reactive oxygen species (ROS)5. Excessive NADH reductive stress can also impede glycolysis, presumably by inhibiting the activity of the NAD+-dependent enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH)6,7, further lowering the cell’s ability to buffer ATP concentrations in ETC dysfunction. Our recent work utilizing LbNOX, a water-forming NADH oxidase that directly oxidizes intracellular NADH to NAD+, demonstrates that reductive stress can cause cellular pathologies such as impaired proliferation in the face of ETC dysfunction3.
The extracellular lactate/pyruvate ratio has classically been used as a marker of intracellular NADH/NAD+ status8 due to the high LDH reaction flux and robust transport of lactate and pyruvate across the plasma membrane by monocarboxylate transporters9. LDH catalyzes the oxidation of NADH to NAD+ while transferring two electrons from NADH to pyruvate to make lactate. Blood lactate/pyruvate levels are often elevated in a variety of diseases, including mitochondrial ETC dysfunction2,10, in response to an increased intracellular NADH/NAD+ ratio. Previously, it was shown that decreasing the extracellular lactate/pyruvate ratios by adding exogenous pyruvate in the media lowers the intracellular NADH/NAD+ ratios in cultured cells.11,12 Sodium pyruvate has also been tested as a therapy for mitochondrial dysfunction13,14. However, this approach has been ineffective clinically in part because it requires administering stoichiometric amounts of the metabolite.
Here, we investigate the possibility of directly targeting the circulating milieu to alleviate intracellular redox imbalance by taking advantage of redox metabolite pairs that rapidly exchange between the inside and outside of cells. Such a strategy would allow us to indirectly modulate tissue redox homeostasis by direct targeting of cell-permeable blood metabolite pairs that are coupled in a redox reaction in the cell. We focused on the extracellular lactate and pyruvate couple, as these two metabolites are interconverted in the lactate dehydrogenase (LDH) reaction in the cytosol. We envisioned irreversibly converting glycolysis-derived extracellular lactate back to pyruvate, which can then enter the cell to participate in the LDH reaction and facilitate further tissue oxidation of NADH (Fig. 1a). The end result of such catalytic regeneration of pyruvate is oxidation of one mole of intracellular NADH per mole of extracellular lactate oxidized. Our approach is based on lactate oxidase (LOX), a bacterial enzyme that converts lactate and oxygen to pyruvate and hydrogen peroxide (H2O2), and a second enzyme, catalase (CAT), to convert hydrogen peroxide, a potentially toxic molecule, into water and oxygen (Fig. 1b). The net effect of extracellular LOX and CAT combined with endogenous LDH is thus to transfer electrons from NADH to O2 to produce water, a function that the mitochondrial electron transport chain performs in healthy cells.
Figure 1. Effects of exogenous lactate oxidase (LOX) and catalase (CAT), alone or in combination, on intracellular NADH/NAD+.

(a) A schematic showing how enzymatic manipulation of the extracellular [lactate]/[pyruvate] ratio can impact the cytoplasmic [NADH]/[NAD+] ratio. (b) The enzymatic reactions of lactate oxidase (LOX) and catalase (CAT). The effect of a combination of extracellular 25 mU LOX and 5000 mU CAT on (c) the media [lactate]/[pyruvate] ratio (n = 10 total wells in antimycin-only treatment and n= 6 total wells in the rest of the treatments from 3 biologically independent experiments;), and (d) the total cellular [NADH]/[NAD+] ratio in HeLa cells treated with 1 µM antimycin A at 24 h (n = 6 total wells from 3 biologically independent experiments). Data are mean ± S.D. One-way ANOVA followed by Tukey’s multiple comparisons test. Molar ratios of lactate and pyruvate, and NADH and NAD+ are shown. ns, P = 0.58; **P = 0.002; ***P = 0.0002; and ****P < 0.0001.
To explore this idea, we began by separately adding commercially available LOX and CAT proteins to the media containing cells with chemically-induced mitochondrial ETC dysfunction. Cells treated for 24h with 1 µM antimycin A, a mitochondrial complex III inhibitor, exhibited a 3.7-fold (P < 0.0001) increase in the media lactate/pyruvate ratio (Fig. 1c) and a 2.1-fold (P < 0.0001) increase in the intracellular NADH/NAD+ ratio (Fig. 1d). A combination of 25 mU LOX and 5000 mU CAT supplemented in the media containing antimycin A decreased the extracellular lactate/pyruvate ratio to an extent that was comparable to the addition of 1mM of exogenous pyruvate (Fig. 1c). CAT was supplemented in excess to ensure effective detoxification of H2O2 produced in the LOX reaction. LOX alone killed most cells, likely because it produced toxic H2O2 (Supplementary Fig. 1), and CAT alone had no effect on the media lactate/pyruvate ratio (Fig. 1c). The combination of LOX and CAT in media lowered the total cellular NADH/NAD+ ratio by 43% (P < 0.0001) when compared to antimycin A-treated control cells (Fig. 1d). These experiments demonstrate that an extracellular combination of LOX and CAT can decrease the total intracellular NADH/NAD+ ratio by lowering the extracellular lactate/pyruvate ratio.
To ensure that the H2O2 detoxification activity of CAT remained in close proximity to H2O2 producing LOX in vivo, we constructed a single polypeptide consisting of LOX and CAT. Based on the crystal structures15,16 and previously published biochemical data17,18, we engineered a fusion construct, named “LOXCAT,” that contained catalase from Escherichia coli (molecular weight 84 kDa) at the N-terminus and lactate oxidase from Aerococcus viridans (molecular weight 40 kDa) at the C-terminus. These two enzymes were tethered using a 20-amino acid linker (L20 linker19), and an N-terminal His6-tag was appended for purification purposes (Fig. 2a). We also designed an enzymatically dead version of LOXCAT (termed LOXCATmut) in which the catalytically important histidine 265 and arginine 268 residues in LOX were mutated to alanine residues, to use as a negative control (Fig. 2a and Supplementary Fig. 2). Purified LOXCAT, but not LOXCATmut, converted lactate into pyruvate in buffer with a Km of 573 ± 101 µM, a Vmax of 160 ± 18 µmol/min/mg, a kcat/Km of 0.6 × 106 (M−1S−1) (Fig. 2b), and consumed oxygen (Supplementary Fig. 3). Importantly, purified LOXCAT did not produce detectable amounts of H2O2 even in the presence of 10 mM lactate, which is much higher than the normal physiological lactate concentration of 1–2 mM (Fig. 2c). H2O2 generation was restored in the presence of 5 mM sodium azide, which inhibits the catalase domain of LOXCAT (Fig. 2c). Moreover, both LOXCAT and LOXCATmut could eliminate exogenously added H2O2, indicating that CAT in both the constructs is active (Fig. 2d). These data demonstrate that LOXCAT, but not LOXCATmut, could effectively convert lactate into pyruvate without toxic byproduct generation.
Figure 2. Design and characterization of LOXCAT, a water-forming lactate oxidase.
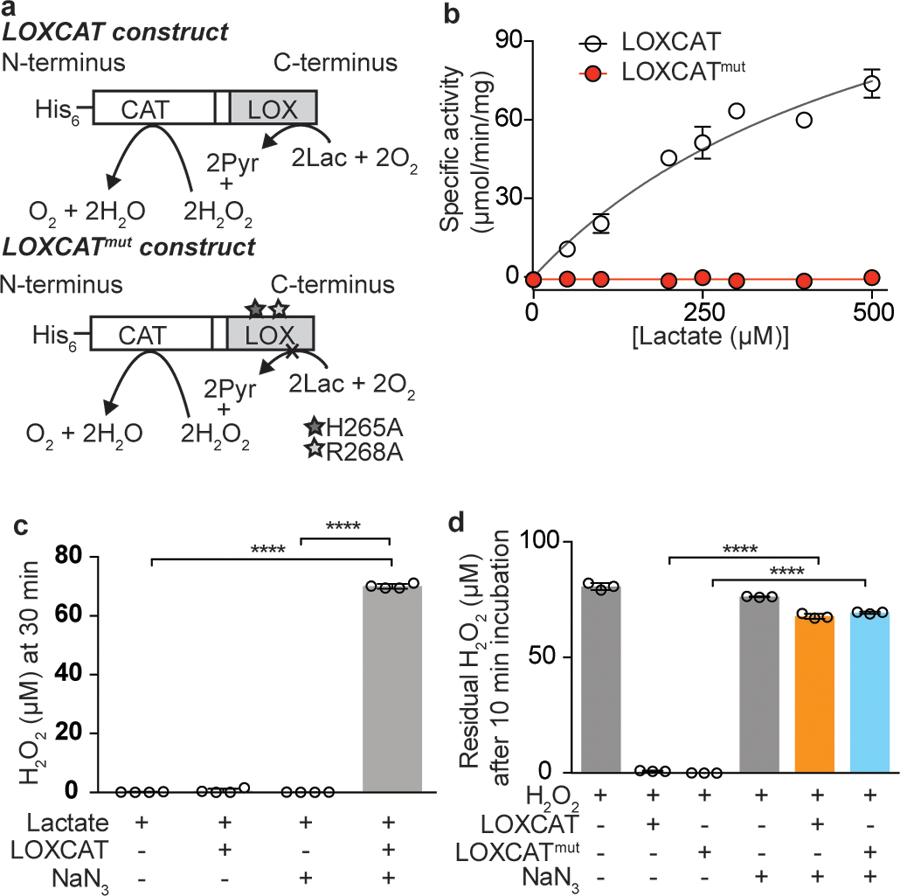
(a) Design of fusion constructs of catalytically active LOXCAT and catalytically dead LOXCATmut. (b) Michaelis-Menten kinetics for LOXCAT and LOXCATmut for the conversion of lactate to pyruvate (n = 3 replicates from 3 biologically independent experiments). (c) Measurement of H2O2 leakage from the reaction of 15 µg LOXCAT with 10 mM lactate at 30 min. 5 mM NaN3 was used to inhibit catalase enzyme activity in the control reaction (n= 4 biologically independent samples). (d) Capacity of 1µg LOXCAT and LOXCATmut to break down 100 µM of exogenous H2O2 at 10 min (n = 3 biologically independent samples). 5 mM NaN3 was used to inhibit catalase enzyme activity in the control reactions. Data are mean ± S.D. One-way ANOVA followed by Tukey’s multiple comparisons test.****P < 0.0001.
We next tested whether LOXCAT in media could decrease the NADH/NAD+ ratio in cells. Indeed, supplementation of the active fusion enzyme recapitulated the effects of separately-added LOX and CAT in lowering the extracellular lactate/pyruvate (Fig. 3a) and the total cellular NADH/NAD+ (Fig. 3b) ratios from antimycin A-treated HeLa cells. We verified the effect of LOXCAT on total cellular NADH/NAD+ ratios using two different analytical methods – an enzyme assay kit (Fig. 3b) and LCMS (Supplementary Fig. 4). Both methods showed comparable LOXCAT-mediated reductions in the total cellular NADH/NAD+ ratios. The inactive enzyme did not change the lactate/pyruvate or the NADH/NAD+ ratios (Fig. 3a–b). LOXCAT shifted the media lactate/pyruvate ratio by increasing media pyruvate concentrations compared to antimycin A-treated control cells (P < 0.0001) but did not affect the lactate concentrations (P > 0.99), whereas addition of 1 mM exogenous sodium pyruvate increased both media lactate (P = 0.0001) and pyruvate concentrations (P < 0.0001) (Supplementary Fig. 5). These data suggest that whereas exogenous sodium pyruvate therapy could further increase the lactate burden in mitochondrial dysfunction, LOXCAT may support extracellular pyruvate levels without a concomitant increase in lactate concentration.
Figure 3. Effect of adding LOXCAT to the media of cellular models of mitochondrial disease.

Effect of extracellular LOXCAT and LOXCATmut on (a) the media [lactate]/[pyruvate] ratio, (b) the intracellular [NADH]/[NAD+] ratio, (c) media glucose consumption, and (d) adenylate energy charge in HeLa cells treated with antimycin A for 24 h. (e) Growth curve for HeLa cells treated with antimycin A in the presence of LOXCAT, LOXCATmut or pyruvate. (f) Growth of NDUFA9 KO K562 cells at 72 h in the presence of LOXCAT, LOXCATmut or pyruvate. Effect of LOXCAT on (g) growth and (h) the media lactate/pyruvate ratio of mitochondrial complex I patient-derived fibroblasts. Data are mean ± S.D. Sample sizes: n = 16 total wells in antimycin-only treatment, n = 12 total wells in untreated and antimycin + pyruvate treatments, n = 10 total wells in antimycin + LOXCAT and antimycin+LOXCATmut treatments from 5 biologically independent experiments in panels (a) and (c); n= 8 total wells from 4 biologically independent experiments in panel (b); n = 4 total wells from 3 biologically independent experiments in panel (e); and n = 6 total wells from 3 biologically independent experiments in panels (d), (f), (g), and (h). One-way ANOVA followed by Tukey’s multiple comparisons test. Molar ratios of lactate and pyruvate, and NADH and NAD+ are shown. ns, P = 0.70, 0.40, and 0.84; *P = 0.013 and 0.019; **P = 0.001; ***P = 0.0004, 0.0001, and 0.0001; and ****P < 0.0001.
To determine whether LOXCAT-mediated normalization of cytoplasmic redox state could cause secondary changes in the intracellular bioenergetic state, we tested the effects of LOXCAT on media glucose consumption and the cellular adenylate charge, which is a measure of energetic state of the cell and is defined as [ATP+ 0.5ADP]/[ATP+ADP+AMP].20 Adenylate energy charge of a cell could theoretically vary from 0 (meaning fully discharged with 100% AMP) to 1 (meaning fully charged with 100% ATP). Most organisms maintain an adenylate energy charge between 0.7 to 0.95.21 LOXCAT increased media glucose consumption by 2-fold (P < 0.0001) and raised the adenylate energy charge from 0.89 to 0.93 (P < 0.0001) in antimycin A-treated HeLa cells at 24 h (Fig. 3c–d). LOXCATmut did not affect glycolytic ATP production in these experiments (Fig. 3c–d). In addition, we observed that LOXCAT could mitigate antimycin A-mediated accumulation of glycolytic intermediates upstream of GAPDH, most likely by providing NAD+ for the GAPDH reaction (Supplementary Fig. 6). Taken together, our experiments indicate that extracellular LOXCAT is capable of upregulating glycolysis in cells with ETC dysfunction by supporting the GAPDH reaction via restoration of the cytosolic NADH/NAD+ ratio.
It has been known for decades that cells with mitochondrial dysfunction exhibit proliferative defects, and that addition of pyruvate can alleviate these defects22. To test if LOXCAT rescues proliferative defects in cells with mitochondrial ETC dysfunction, we treated cells with antimycin A. As expected, these cells did not proliferate unless pyruvate was supplemented in the media (Fig. 3e). Adding LOXCAT to the media corrected proliferative defects in these antimycin A-treated HeLa cells for up to 4 days (Fig. 3e), whereas the control enzyme LOXCATmut failed to do so. Moreover, LOXCAT-rescued cells exhibited a similar morphology to that of the untreated wild-type HeLa cells (Supplementary Fig. 7). To test the generality of LOXCAT rescue, we treated cells with two other mitochondrial poisons: piericidin A, a complex I inhibitor, and oligomycin A, a complex V inhibitor. LOXCAT showed similar ability to rescue growth under these two conditions (Supplementary Fig. 8a). In addition, LOXCAT restored growth in cellular genetic models of disease-relevant complex I (NDUFA9)23, complex III (UQCRB)24 and complex V (ATP5A1)25 KO K562 cells (Fig. 3f and Supplementary Fig. 8b–c). Finally, we tested LOXCAT in fibroblasts derived from a patient with mutant NUBPL (c.311T>C) that leads to dysfunction in mitochondrial complex I.26 We observed that LOXCAT, but not LOXCATmut, improved growth in these fibroblasts (Fig. 3g). As shown in Fig. 3h NUBPL (c.311T>C) fibroblasts exhibited a 3.7-fold elevation (P < 0.0001) in the media lactate/pyruvate ratio compared to healthy control cells after 72 h incubation. LOXCAT reduced the media lactate/pyruvate ratio in NUBPL (c.311T>C) fibroblasts from an average of 94.1 to 0.5 (P < 0.0001) (Fig. 3h). Together, LOXCAT rescued proliferative defects in cells treated with three chemical models of mitochondrial dysfunction, three cell-based genetic models of mitochondrial disease, as well as in complex I dysfunction in patient-derived fibroblasts.
We next sought to determine if LOXCAT might be effective in vivo. Initial tail vein injections of LOXCAT into C57BL/6J mice showed that the blood lactate/pyruvate ratio was indeed lowered (by 48%, P = 0.001), however, the half-life of the enzyme was short (Supplementary Figs. 9, 10, and 11). To extend its in vivo half-life, we fused an albumin binding peptide (ABP) to the N-terminus of LOXCAT (Supplementary Fig. 10 and 11). This ABP has previously been utilized to extend half-life of therapeutic enzymes27,28. The resulting “ABP-LOXCAT” and LOXCAT showed comparable enzyme activities in quenching H2O2 (Fig. 2c–d and Supplementary Fig. 12) and abilities to rescue proliferative defects in cells with ETC dysfunction (Fig. 3e and Supplementary Fig. 13). ABP-LOXCAT lowered circulating lactate/pyruvate levels by 55% (P <0.0001) in 30 min in wild-type C57BL/6J mice (Fig. 4a) by decreasing blood lactate (Fig. 4b) and increasing blood pyruvate (Fig. 4c). We next tested ABP-LOXCAT in a model of drug-induced acute mitochondrial dysfunction using metformin, a mitochondrial complex I inhibitor29. Intraperitoneal (IP) injection of 300 mg/kg of metformin elevated blood lactate/pyruvate ratio by 1.5-fold (P = 0.0003) in 1 h (Fig. 4d). ABP-LOXCAT treatment lowered this ratio by 59% (P <0.0001), whereas ABP-LOXCATmut was ineffective (Fig. 4d). We found that our IP metformin dose raised the NADH/NAD+ ratios in the brain (2.2-fold, P <0.0001), heart (1.9-fold, P <0.0001), and kidney (1.8-fold, P = 0.015), but did not reach significance in liver despite trending up (1.3-fold, P = 0.16) (Fig 4e–f and Supplementary Fig. 14a–f). ABP-LOXCAT, operating in the circulation, effectively blunted this metformin-induced elevation in the heart and brain NADH/NAD+ ratios, but not in the kidney (Fig 4e–f and Supplementary Fig 14f). Furthermore, blood lactate/pyruvate ratios were positively correlated with both brain (r = 0.73, P < 0.0001) and heart (r = 0.6, P < 0.0001) NADH/NAD+ ratios (Fig. 4g–h). These results indicate that directly oxidizing elevated levels of lactate to pyruvate in the circulation is capable of normalizing tissue NADH/NAD+ redox balance in the brain and heart.
Figure 4. Impact of intravenous LOXCAT on blood lactate/pyruvate and tissue NADH/ NAD+ ratios.

Effect of ABP-LOXCAT and ABP-LOXCATmut on the circulating (a) [lactate]/[pyruvate] ratio, (b) lactate, and (c) pyruvate in wild-type mice at 30 min post injection (n = 7 mice). Effect of ABP-LOXCAT on the (d) blood [lactate]/[pyruvate] ratio, (e) brain [NADH]/[NAD+], and (f) heart [NADH]/ [NAD+] at 1 h in metformin-treated mice (n = 13 mice). Pearson correlation between (g) blood [lactate]/[pyruvate] and brain [NADH]/[NAD+] ratios, and (h) blood [lactate]/[pyruvate] and heart [NADH]/[NAD+] ratios in the metformin-treated mice at 1 h (two-tailed P values shown) (n = 13 mice). Data are mean ± S.D. One-way ANOVA followed by Tukey’s multiple comparisons test. Molar ratios of lactate and pyruvate, and NADH and NAD+ are shown. ns, P = 0.23, >0.99, 0.95, 0.89, and >0.99; *P = 0.026 and 0.027 ; **P = 0.001 and 0.003; ***P = 0.0003, 0.0006, 0.0007, and 0.0003; and ****P < 0.0001.
In summary, we have shown that direct manipulation of extracellular lactate and pyruvate levels by an engineered enzyme can alleviate intracellular redox imbalance in cells and tissues. Prior work by other groups has demonstrated that modulation of extracellular ratios of lactate/pyruvate, beta-hydroxybutyrate/acetoacetate, and sulfur containing metabolite pairs (cysteine/cystine and GSH/GSSG) by adding exogenous metabolites can impact intracellular redox homeostasis and physiology in cultured cells11,12,30,31. Clinical translation of this concept is challenging, as such an approach would require administration of stoichiometric amounts of the metabolite13. LOXCAT may overcome this limitation as it is catalytic. The blood lactate/pyruvate ratio has historically been used as a marker of the tissue redox status, but here we show that it can also be an effector, as directly manipulating blood lactate/pyruvate ratios restored NADH/NAD+ balance in several key organs. It is notable that there is ample precedent of injected bacterial enzymes eliciting therapeutic benefit: asparaginase isolated from Escherichia coli is used as an anticancer therapy and streptokinase isolated from Streptococci has historically been used to treat acute coronary syndromes32,33. Future variants of LOXCAT with optimized circulatory half-life and immunological properties may hold important therapeutic potential for the growing number of human diseases associated with systemic reductive stress.
ONLINE METHODS
Preparation of DNA constructs
The plasmids containing LOXCAT and LOXCATmut genes in vector pET30a were purchased from Genscript Corporation. Plasmids were sequenced independently for verification. Purified LOXCAT was also verified using mass spectrometric analysis.
Cell cultures and cell lines
All cultured cells were maintained at 37 °C in 5% CO2 and were tested for mycoplasma contamination once every three month using MycoAlert Mycoplasma Detection kit (Lonza, LT07–418) and all cell lines tested negative for mycoplasma contamination. Wild type HeLa and K562 cells originated from ATCC and were authenticated using DNA fingerprinting with small tandem repeat (STR) profiling using the facility available at Dana Farber Cancer Center, Boston. K562 knockout cells were generated in our lab and NDUFA9 KO was previously described34. For the other two cell lines, K562 cells transiently infected with CAS9 and sgRNA guides to UQCRB (GCGAGATGATACAATATACG), or ATP5A1 (AGTTTCTTCAAGATCAACAG) were single cell cloned. Complete biallelic loss of gene was confirmed with immunoblotting (Supplementary Figs 11 and 15). Patient (Catalog # GM24529) and control (Catalog # AG22586) fibroblasts were obtained from Coriell Institute.
Statistical Analysis
One way ANOVA followed by Tukey’s multiple comparisons test was used in GraphPad Prism 6 to evaluate whether a significant difference existed between three or more groups of samples. Data are presented as mean ± S.D.
Cell proliferation assays
50,000 HeLa or K562 cells/well were plated in a 6-well plate in 2ml of pyruvate-free DMEM media (5 mM glucose, no pyruvate, 200 µM uridine, penicillin/streptomycin, 10% dialyzed FBS). ETC inhibitor (1 µM) or pyruvate (1 mM) or LOXCAT/LOXCATmut/ABP-LOXCAT/ABP-LOXCATmut (20 µg) or 25 mU LOX from Aerococcus viridans (Sigma L9795) alone or in combination with 5000 mU catalase from bovine liver (Sigma SRE0041) were added to the media and cells were incubated at 37 °C in 5% CO2. At 72 h, cells were counted using a Beckman Coulter Z2 coulter counter. For the growth rate assay, cells were counted every day for 4 days.
Patient-derived (Catalog # GM24529) and control (Catalog # AG22586) fibroblasts were obtained from Coriell Institute and cultured in DMEM media (25 mM glucose, 1 mM pyruvate, 200 µM uridine, penicillin/streptomycin, 10% dialyzed FBS). For cell proliferation experiments, 10,000 cells/well were plated in a 24 well plate in 1ml of pyruvate-free DMEM media (5 mM glucose, no pyruvate, 200 µM uridine, penicillin/streptomycin, 10% dialyzed FBS). 15 µg of LOXCAT or LOXCATmut or 1 mM pyruvate were added as treatments. Fresh media and enzyme were replenished every 3 days and cells were counted on day 6, as described above.
NADH/NAD+ measurement in cell culture
Total cellular NADH/NAD+ were measured using NADH-glo assay (Promega, catalog number G9071). 200,000 HeLa cells/well were plated on a 6-well plate in 2ml of pyruvate-free DMEM media (5 mM glucose, no pyruvate, 200 µM uridine, penicillin/streptomycin, 10% dialyzed FBS). The next day, ETC inhibitor (1 µM) or pyruvate (1 mM) or 20 µg LOXCAT or 20 µg LOXCATmut, or 25 mU LOX from Aerococcus viridans (Sigma L9795) alone or in combination with 5000 mU catalase from bovine liver (Sigma SRE0041) were added to the media and cells were incubated for 24 h at 37 °C in 5% CO2. At 24 h cells were quickly washed with 1 ml of ice cold PBS and 500 µl of 1% DTAB (Sigma # D8638) solution was added to lyse cells. Manufacturer’s instructions were followed hereafter. Briefly, 50 µl of the lysate was then transferred to a PCR tube containing 25 µl 0.4 M HCl (for NAD+ measurement) or to an empty PCR tube (for NADH measurement). Both samples were heated at 60 °C for 15 min to deplete NAD+ or NADH. Samples were then left on the bench for 10 min to cool down to room temperature before adding neutralizing solutions (25 µl of 0.5 M Tris base to the NAD+ sample, and 50 µl of 1:1 = 0.4M HCl: 0.5 M Tris base to the NADH sample). Samples were then vortexed and 30 µl were added to 30 µl of NADH-glo detection reagent and incubated for 30 min at room temperature. Luminescence of samples were then measured on a Perkin Elmer Envision 2140 multiplate reader. Absolute concentrations of NADH and NAD+ were determined against standard curves.
Media lactate/pyruvate ratio and glucose measurements using LCMS
HeLa cells were grown as in the NADH/NAD+ ratio measurement assay. At 24 h post treatment 500 µl media was collected. Media was then immediately filtered through 10 kDa cutoff filter 500 µl centrifuge tubes (Amicon, Catalog number UFC501096) to remove any active enzyme. To 30 µl of this media, 160 µl of 100% acetonitrile and 10 µl of internal standards containing stable isotope labeled D3-lactate (1.5 mM), 13C6-glucose (1.5 mM) and 13C3-pyruvate (250 µM) were added and incubated on ice for 30 min. Samples were then centrifuged at 14800 rpm for 20 min and 150 µl of centrifuged sample was transferred to LCMS vials. For lactate and pyruvate measurements from the patient derived fibroblasts, 30 µl of media at 72 h (because media was exchanged every 72 h) was mixed with 160 µl of 100% acetonitrile and 10 µl of internal standards containing stable isotope labeled D3-lactate (1.5 mM), 13C6-glucose (1.5 mM) and 13C3-pyruvate (250 µM) and frozen immediately in liquid nitrogen and stored away at −80 °C. These samples were thawed on ice, incubated for another 30 min on ice, centrifuged at 14800 rpm for 20 min, and 150 µl supernatant was drawn for LCMS measurements. LCMS analysis was performed on a Dionex Ultimate 3000 UHPLC system coupled to Q Exactive Mass Spectrometer (ThermoFisher) using a XBridge BEH amide column (Waters, Part number 186006091). Absolute concentrations of lactate, pyruvate, and glucose were determined by comparing against standard curves after normalizing them to the corresponding internal standard.
Measurements of energy charge, NADH/NAD+ and glycolytic intermediates using LCMS
200,000 HeLa cells/well were plated on a 6-well plate in 2ml of pyruvate free DMEM media (25 mM glucose, no pyruvate, penicillin/streptomycin, 10% FBS). The next day, 1 µM antimycin A or 1 mM pyruvate or 40 µg of enzymes were added to the media (25mM glucose, no pyruvate, 200 µM uridine, penicillin/streptomycin, 10% dialyzed FBS) and cells were incubated for 24h at 37 °C in 5% CO2. At 24 h media was aspirated and 400 µl of lysis buffer (0.1 M Formic acid in 9:1 (75% acetonitrile: 25% methanol): water)) supplemented with 5 µl internal standard (5 µM 13C10 ATP, 2.5 µM 13C5 ADP and 0.5 µM 13C10,15N5 AMP) was added. After 2 minutes on ice, 35 µl of 15% NH4HCO3 was added and incubated for another 20 min on ice. Cells were scraped and lysate was transferred into a pre-chilled 1.5 ml centrifuge tube, vortexed briefly, and spun at 14000 rpm for 20 min at 4 °C. 200 µl of supernatant was then transferred into an LCMS vial and ATP, ADP, AMP, NADH, NAD+, and glycolytic intermediate levels were measured on a Dionex Ultimate 3000 UHPLC system coupled to Q Exactive Mass Spectrometer (ThermoFisher) using a ZIC-pHILIC column (EMD Millipore, Catalog number 150460). Absolute concentrations of AMP, ADP and ATP were measured by comparing against a standard curve.
Purification of LOXCAT and LOXCATmut in Escherichia coli
1 L culture of Escherichia coli BL21 (DE3) cells (Life Technologies # C6010–03) transformed with pET30a vector containing LOXCAT (LOX from Aerococcus viridans and catalase from Escherichia coli) or LOXCATmut genes were grown in 2.5 L flasks (IBI Scientific, Catalog number SS-8003) at 37 °C until O.D. (600 nm) of 0.4–0.6. The culture was then placed at 4 °C for 2 hours before moving it back to an 18 °C incubator for overnight induction with 400 µM 1-thio-β-d-galactoside (IPTG) (Sigma # I5502). Along with IPTG, cells were supplemented with 1mM 5-aminoluvilinic acid (Sigma # 08339) and 300 µM Ferrous Ammonium Sulfate (Sigma # 09719–250G) to assist heme biosynthesis. The next morning, cells were harvested and frozen away at −80 °C until purified. For purification, cells were resuspended in 40 ml lysis buffer (50 mM sodium phosphate (pH 7.0), 150 mM NaCl, 0.1 % Tween-20, 30 mM Imidazole, 2 mM DTT, 200 µM FMN, one Protease inhibitor complete EDTA-free (Roche # 11873580001), 2.5 µl benzonase and 1mg/ ml lysozyme) per liter of culture and lysed by ultra-sonication. After clarification of the lysate at 20,000 rpm for 30 min at 4 °C, the supernatant was bound to 5 ml Ni-sepharose beads/L of culture, washed with 10 column volume with washing buffer (50 mM sodium phosphate (pH 7.0), 100 mM NaCl, 0.1 % Tween-20, 30 mM Imidazole, 10 mM MgCl2, and 10 mM ATP) and eluted with elution buffer (50 mM sodium phosphate (pH 7.0), 150 mM NaCl, 300 mM Imidazole). Eluted fractions were then concentrated and passed through a HiTrapQFF ion exchange column (GE Healthcare # GE17–5156-01) with a NaCl gradient of 100 mM to 1 M. The pooled fractions from ion exchange were then passed through a Superdex 200 increase column (GE Healthcare) in buffer containing 50 mM sodium phosphate (pH 7.4) and 150 mM NaCl. Pure fractions from gel filtration were concentrated using 15 ml 100 kDa MWCO filter centrifuge tubes (Millipore # UFC910024), flash frozen in liquid nitrogen and stored at −80 °C. Ion-exchange and gel filtration were performed on an AKTA pure FPLC system (GE Healthcare).
Measurement of LOXCAT’s ability to quench H2O2
Production of H2O2 was monitored using the Amplex red assay. H2O2 reacts with amplex red in presence of horseradish peroxidase (HPR) to produce resorufin, a colored compound whose formation could be monitored by measuring fluorescence at 535 nm (ex)/587 nm (em). To measure catalase’s ability to quench H2O2 produced by the LOX domain in LOXCAT using lactate as a substrate, 10 mM lactate in 200 µl PBS was incubated with 15 µg LOXCAT or ABP-LOXCAT for 30 min at 37 °C. 5 mM sodium azide was added to the reaction mixture to inactivate catalase in the control reactions. At 30 min the reaction mixture was filtered through a 10 kDa cutoff filter centrifuge tubes (Amicon, Catalog number UFC501096) to remove any active enzyme. 50 µl of the filtrate was then transferred into a black 96 well plate and 50 µl of Amplex red reagent (1µl of 2.5 mg/ml Amplex red + 1 µl of 10 mU/µl HRP + 48 µl PBS) was added to it, incubated at room temperature for 10 min, and amplex red fluorescence was measured on a Perkin Elmer Envision 2140 multiplate reader. In another experiment, we tested if LOXCAT and LOXCATmut could break down exogenous H2O2. We incubated 100 µM of H2O2 with 1 µg LOXCAT, LOXCATmut, ABP-LOXCAT or ABP-LOXCATmut for 10 min at 37 °C and measured the remaining H2O2 using Amplex red reagent as described above. Absolute H2O2 concentrations were determined using a standard curve.
Measurement of LOXCAT’s ability to convert lactate to pyruvate
The ability of lactate oxidase in LOXCAT to convert lactate into pyruvate was assessed by incubating increasing concentrations of (0–500 µM) lactate with 8 µg of LOXCAT or LOXCATmut in 200 µl of buffer (50 mM potassium phosphate, pH 7.4 and 150 mM NaCl) for 30 min at 37 °C. At 30 min the reaction mixture was filtered through a 10 kDa cutoff filter centrifuge tubes (Amicon, Catalog number UFC501096) to remove any active enzyme. Concentrations of pyruvate produced in the reaction were then measured from the filtrate by using a pyruvate assay kit (Abcam ab65342) following the manufacturer’s instructions.
Measurement of LOXCAT’s ability to consume O2
Oxygen consumption by LOXCAT, LOXCATmut, and LOX were measured in a custom-built fluorimeter as described previously3. The setup includes a cuvette containing a RedEye oxygen patch (OceanOptics) and an optical probe that is connected to the fluorimeter. Oxygen consumption rate was measured by the rate of decrease in fluorescence signal in the RedEye patch. In our experiments, 0.5 ml of assay buffer (50 mM sodium phosphate pH 7.4 and 150 mM NaCl) was preincubated at 28 °C before adding 4 µg of LOXCAT or LOXCATmut or 25 mU LOX followed by 1 mM lactate. 5000 mU CAT was added to the LOX reaction to test if it increases oxygen concentrations by breaking down LOX-produced H2O2 into water and oxygen.
Measurement of the effect of LOXCAT and ABP-LOXCAT on blood lactate/ pyruvate ratios in wild-type mice
Animal studies followed protocols approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee. Wild-type C57BL/6J male mice 8–12 weeks of age were obtained from Jackson Laboratory and selected randomly for the studies. The studies were performed in a blinded fashion. Mice were anesthetized (8% sevoflurane with 100% oxygen for LOXCAT experiments, and 5% isoflurane with 100% oxygen for ABP-LOXCAT experiments) in a chamber. Animals were then transferred to mouse restrainer and maintained 3–4% sevoflurane with 100% oxygen (for LOXCAT experiments) or 2–2.5% isoflurane with 100% oxygen (for ABP-LOXCAT experiments) using a nose cone. 250 µl of vehicle (50 mM sodium phosphate (pH 7.4), and 150 mM NaCl), or 250 µl (41 mg/kg) of LOXCAT or 250 µl (150 mg/kg) of ABP-LOXCAT or 250 µl (41 mg/kg) of LOXCATmut or 250 µl (150 mg/kg) of ABP-LOXCATmut injected into the lateral tail vein. For the LOXCAT experiment animals were then kept under anesthesia (3–4% sevoflurane) for another 15 min. At 15 min blood was collected by cardiac puncture. For the ABP-LOXCAT experiments, animals were returned to the cage after injection and collected blood by cardiac puncture at 30 min under anesthesia (isoflurane). 30 µl of blood was immediately mixed with 160 µl of 100% acetonitrile and 10 µl of internal standards containing stable isotope labeled D3-lactate (1.5 mM) and 13C3-pyruvate (250 µM), frozen immediately in liquid nitrogen and stored away at −80 °C. For LCMS measurements, frozen samples were thawed on ice, vortexed to mix and incubated for another 30 min to precipitate out proteins. Samples were then centrifuged at 14000 rpm for 20 min at 4 °C. 150 µl of supernatant was then withdrawn for mass spectrometric analysis to determine blood lactate and pyruvate concentrations. Absolute concentrations of lactate and pyruvate were obtained by comparing them against standard curves and normalizing to the corresponding internal stanard.
Blood lactate/ pyruvate and tissue NADH/ NAD+ ratios in metformin-treated mice
Animal studies followed protocols approved by the MGH Institutional Animal Care and Use Committee. The studies were performed in a blinded fashion. Wild-type 8–12 week-old male C57BL/6J mice were anesthetized with 5% isoflurane in 100% oxygen in a chamber. Following induction, mice were transferred to a mouse restrainer (Kent Scientific) and anesthesia was maintained with 2–2.5% isoflurane in 100% oxygen using a nose cone. 150 mg/kg ABP-LOXCAT or ABP-LOXCATmut (in 150 µl) or 150 µl of vehicle (50 mM sodium phosphate (pH 7.4), and 150 mM NaCl) was injected into the lateral tail vein using a 30G needle (Becton Dickinson), polyethylene tubing 10 (Becton Dickinson), and a 0.3 ml insulin syringe (Becton Dickinson). Additional 30 μl of vehicle was injected to flash remaining fluid inside the catheter. After injection, the needle was removed and gentle compression was applied until bleeding had stopped and 300 μg/g in 6µl/g of metformin in PBS (Sigma # D150959) or 6 μl/g of PBS was injected intraperitoneally. Mice were returned to a cage and observed to make sure that bleeding did not resume. At 1 h, mice were anesthetized with 5% isoflurane in 100% oxygen. After induction, mice were transferred to an operating table, laid on their back, and anesthetized with 5% isoflurane in 100% oxygen using a nose corn. Lack of pedal withdrawal reflex was confirmed before skin incision. After the skin was soaked with 70% ethanol an incision was made from the belly to the neck. The abdominal cavity was opened and the diaphragm and ribs were cut to open the thoracic cavity. Blood was withdrawn from the right ventricle with a heparinized 1 ml syringe (Covidien) with a 22G needle (Becton Dickinson) and transferred to an EDTA tube (RAM Scientific). 30 μl of blood was mixed with 160 µl of 100% acetonitrile and 10 µl of internal standards containing stable isotope labeled D3-lactate (1.5 mM) and 13C3-pyruvate (250 µM) were immediately added. LCMS samples were snap frozen in liquid nitrogen and lactate/pyruvate ratios were measured as described above. Organs were harvested in the following order and snap-frozen in liquid nitrogen: right and left medial lobe of the liver, right and left kidneys, left and right ventricles of the heart, and right and left cerebral hemispheres. The right medial lobe of the liver, the right kidney, both ventricles of the heart, and the right cerebral hemisphere were used for NADH/NAD+ measurement. Tissue NADH/NAD+ ratios were measured within a week of harvesting, using Biovision NAD/NADH quantitation colorimetric kit (Catalog number K337) following manufacturer’s instruction. Absolute concentrations of NADH and NAD+ were determined using a standard curve and normalized against protein concentrations.
Detection of LOXCAT in mouse blood using western blot
LOXCAT, LOXCATmut, ABP-LOXCAT, and ABP-LOXCATmut were detected in whole blood by western blot by using antibodies against N-terminal S-tag that these protein sequences contain. 2 µl of 6X SDS loading dye was added to 5 µl of EDTA-treated whole blood. The sample was then diluted by adding 25 µl of 1X SDS loading dye, flash frozen and stored at −80 °C until use. Samples were boiled at 95 °C before loading onto a 4–20% SDS-PAGE gel and blotted against anti-S tag antibody (Abcam # 19324). Albumin was used as a loading control and blotted using mouse serum albumin antibody (Abcam # ab19195).
Supplementary Material
Acknowledgements.
We thank Olga Goldberger, Kimberli J Kamer, and Jason D Arroyo for technical assistance. This work was supported in part by grants from the Marriott Foundation and the National Institutes of Health (R35GM122455). A.P. was supported by the Helen Hay Whitney Postdoctoral Fellowship and the Tosteson & Fund for Medical Discovery award. O.S.S was supported by a F32 Fellowship from the NIGMS (1F32GM133047–01). V.K.M. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Competing Financial Interests. Massachusetts General Hospital has filed a patent on the technology described in this paper listing V.K.M., A.P. and X.R.B as inventors. O.S.S. is a paid consultant for Proteinaceous Inc. V.K.M. is a paid advisor to Janssen Pharmaceuticals and 5AM Ventures, and is a founder of Raze Therapeutics.
References
- 1.Vafai SB & Mootha VK. Mitochondrial disorders as windows into an ancient organelle. Nature 491, 374–383, doi: 10.1038/nature11707 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Thompson Legault J. et al. A Metabolic Signature of Mitochondrial Dysfunction Revealed through a Monogenic Form of Leigh Syndrome. Cell reports 13, 981–989, doi: 10.1016/j.celrep.2015.09.054 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Titov DV. et al. Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science 352, 231–235, doi: 10.1126/science.aad4017 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao W & Loscalzo J. Metabolic Responses to Reductive Stress. Antioxid Redox Signal, doi: 10.1089/ars.2019.7803 (2019). [DOI] [PMC free article] [PubMed]
- 5.Gores GJ. et al. Swelling, reductive stress, and cell death during chemical hypoxia in hepatocytes. Am J Physiol 257, C347–354, doi: 10.1152/ajpcell.1989.257.2.C347 (1989). [DOI] [PubMed] [Google Scholar]
- 6.Tilton WM, Seaman C, Carriero D & Piomelli S. Regulation of glycolysis in the erythrocyte: role of the lactate/pyruvate and NAD/NADH ratios. J Lab Clin Med 118, 146–152 (1991). [PubMed] [Google Scholar]
- 7.Williamson JR. et al. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes 42, 801–813 (1993). [DOI] [PubMed] [Google Scholar]
- 8.Williamson DH, Lund P & Krebs HA. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. The Biochemical journal 103, 514–527 (1967). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poole RC & Halestrap AP. Transport of lactate and other monocarboxylates across mammalian plasma membranes. The American journal of physiology 264, C761–782 (1993). [DOI] [PubMed] [Google Scholar]
- 10.Shaham O. et al. A plasma signature of human mitochondrial disease revealed through metabolic profiling of spent media from cultured muscle cells. Proceedings of the National Academy of Sciences of the United States of America 107, 1571–1575, doi: 10.1073/pnas.0906039107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bucher T. et al. State of oxidation-reduction and state of binding in the cytosolic NADH-system as disclosed by equilibration with extracellular lactate-pyruvate in hemoglobin-free perfused rat liver. Eur J Biochem 27, 301–317 (1972). [DOI] [PubMed] [Google Scholar]
- 12.Hung YP, Albeck JG, Tantama M & Yellen G. Imaging cytosolic NADH-NAD(+) redox state with a genetically encoded fluorescent biosensor. Cell metabolism 14, 545–554, doi: 10.1016/j.cmet.2011.08.012 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujii T. et al. Efficacy of pyruvate therapy in patients with mitochondrial disease: a semi-quantitative clinical evaluation study. Molecular genetics and metabolism 112, 133–138, doi: 10.1016/j.ymgme.2014.04.008 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Saito K. et al. Pyruvate therapy for mitochondrial DNA depletion syndrome. Biochimica et biophysica acta 1820, 632–636, doi: 10.1016/j.bbagen.2011.08.006 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Li SJ. et al. Crystallographic study on the interaction of L-lactate oxidase with pyruvate at 1.9 Angstrom resolution. Biochemical and biophysical research communications 358, 1002–1007, doi: 10.1016/j.bbrc.2007.05.021 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Melik-Adamyan W. et al. Substrate flow in catalases deduced from the crystal structures of active site variants of HPII from Escherichia coli. Proteins 44, 270–281 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Taurino IR, R.; Richterb M; Fairheadb M; Thöny-Meyerb L; De Michelia G; Carrara S. Comparative study of three lactate oxidases from Aerococcus viridans for biosensing applications. Electrochimica Acta 93, 72–79 (2013). [Google Scholar]
- 18.Andre C, Kim SW, Yu XH & Shanklin J. Fusing catalase to an alkane-producing enzyme maintains enzymatic activity by converting the inhibitory byproduct H2O2 to the cosubstrate O2. Proceedings of the National Academy of Sciences of the United States of America 110, 3191–3196, doi: 10.1073/pnas.1218769110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin A, Baker TA & Sauer RT. Rebuilt AAA + motors reveal operating principles for ATP-fuelled machines. Nature 437, 1115–1120, doi: 10.1038/nature04031 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Atkinson DE & Walton GM. Adenosine triphosphate conservation in metabolic regulation. Rat liver citrate cleavage enzyme. The Journal of biological chemistry 242, 3239–3241 (1967). [PubMed] [Google Scholar]
- 21.De la Fuente IM. et al. On the dynamics of the adenylate energy system: homeorhesis vs homeostasis. PloS one 9, e108676, doi: 10.1371/journal.pone.0108676 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.King MP & Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246, 500–503 (1989). [DOI] [PubMed] [Google Scholar]
- 23.Baertling F. et al. NDUFA9 point mutations cause a variable mitochondrial complex I assembly defect. Clin Genet 93, 111–118, doi: 10.1111/cge.13089 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Haut S. et al. A deletion in the human QP-C gene causes a complex III deficiency resulting in hypoglycaemia and lactic acidosis. Hum Genet 113, 118–122, doi: 10.1007/s00439-003-0946-0 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Jonckheere AI. et al. A complex V ATP5A1 defect causes fatal neonatal mitochondrial encephalopathy. Brain : a journal of neurology 136, 1544–1554, doi: 10.1093/brain/awt086 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Johnson SC. et al. mTOR inhibitors may benefit kidney transplant recipients with mitochondrial diseases. Kidney Int 95, 455–466, doi: 10.1016/j.kint.2018.08.038 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mukherjee J. et al. Prolonged prophylactic protection from botulism with a single adenovirus treatment promoting serum expression of a VHH-based antitoxin protein. PloS one 9, e106422, doi: 10.1371/journal.pone.0106422 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen A. et al. The pharmacokinetics of an albumin-binding Fab (AB.Fab) can be modulated as a function of affinity for albumin. Protein Eng Des Sel 19, 291–297, doi: 10.1093/protein/gzl011 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Gui DY. et al. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell metabolism 24, 716–727, doi: 10.1016/j.cmet.2016.09.006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nocito L. et al. The extracellular redox state modulates mitochondrial function, gluconeogenesis, and glycogen synthesis in murine hepatocytes. PloS one 10, e0122818, doi: 10.1371/journal.pone.0122818 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramirez A. et al. Extracellular cysteine/cystine redox potential controls lung fibroblast proliferation and matrix expression through upregulation of transforming growth factor-beta. Am J Physiol Lung Cell Mol Physiol 293, L972–981, doi: 10.1152/ajplung.00010.2007 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Muller HJ & Boos J. Use of L-asparaginase in childhood ALL. Crit Rev Oncol Hematol 28, 97–113 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Laffel GL & Braunwald E. Thrombolytic therapy. A new strategy for the treatment of acute myocardial infarction (2). The New England journal of medicine 311, 770–776, doi: 10.1056/NEJM198409203111205 (1984). [DOI] [PubMed] [Google Scholar]
- 34.Arroyo JD. et al. A Genome-wide CRISPR Death Screen Identifies Genes Essential for Oxidative Phosphorylation. Cell metabolism 24, 875–885, doi: 10.1016/j.cmet.2016.08.017 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.