

Abstract
Background
Tyrosine kinase inhibitors (TKIs), such as gefitinib, are widely used as standard treatments for non-small cell lung cancer (NSCLC) patients with epidermal growth factor receptor (EGFR) mutations. However, the subsequent inevitable drug resistance has become a major challenge in clinical treatment. The aim of this study was to investigate the role of tissue-type plasminogen activator (PLAT) in gefitinib resistance in NSCLC.
Methods
The function of PLAT was determined using gefitinib-resistant cells and a nude mouse model. The gene knockdown was achieved by Lentivirus based RNA silence technique. Expression of relevant genes and proteins, cell viability, proliferation, apoptosis, cell cycle, reactive oxygen species levels, mitochondrial membrane potential and differential gene expression was detected by RT-qPCR, western blot, cell counting kit-8 assay, EdU incorporation, flow cytometry, JC-1 dye assay and complementary DNA arrays. The effects of PLAT knockdown on tumorigenesis was analyzed in vivo.
Results
Gefitinib-resistant cells expressed higher levels of PLAT and that knockdown of PLAT in resistant cells restored gefitinib sensitivity. Tumor proliferation was limited in vivo following PLAT knockdown. Moreover, PLAT knockdown affected mitochondrial function, caused caspase activation and cell cycle arrest, and activated TNF-α signaling, leading to apoptosis of gefitinib-resistant PC9 cells.
Conclusions
Our results suggest that PLAT reduces apoptosis of NSCLC cells and knockdown of PLAT enhances anticancer effect of gefitinib by upregulating TNF-α signaling.
Keywords: Tissue-type plasminogen activator (PLAT), non-small cell lung cancer (NSCLC), gefitinib resistance, apoptosis
Introduction
Lung cancer is the leading cause of cancer incidence and mortality worldwide, with 1.8 million people diagnosed and 1.6 million deaths due to the disease annually (1). Non-small cell lung cancer (NSCLC) accounts for nearly 85% of all cases of lung cancer (2,3). Epidermal growth factor receptor (EGFR), an extensively studied protein that promotes tumor proliferation, angiogenesis, metastatic potential, and chemo-resistance and inhibits cell apoptosis, is highly expressed on the surface of NSCLC cells and commonly mutated (4,5). Nearly 90% of EFGR mutations in NSCLC patients are attributed to the deletion of exon 19 and the L858R substitution in exon 21 (6-8). Thus, tyrosine kinase inhibitors (TKIs) targeting EGFR, such as gefitinib and erlotinib, have become an efficient first-line anti-tumor approach for such patients. However, patients receiving TKI therapy inevitably develop resistance to these drugs after about 10–14 months. Mechanisms of resistance include additional EGFR mutations (T790M or Cys797Ser), c-MET amplification, human epidermal growth factor receptor 2 (HER2) amplification, epithelial-mesenchymal transition (EMT), small cell lung cancer transformation, or AXL activation (7-11). Meanwhile, other mechanisms of resistance remain to be discovered.
Tissue-type plasminogen activator (PLAT) encodes tissue-type plasminogen activator (t-PA), a serine protease that converts the proenzyme plasminogen to plasmin and is effective in treating thromboembolic diseases (12). Despite its function in fibrinolysis, many studies have revealed the role of t-PA in cancer progression and prognosis. Evidence has shown that breast cancer patients with low levels of t-PA tend to have poor prognosis, while in melanoma, neuroblastoma, acute myeloblastic leukemia, and pancreatic cancer, high levels of t-PA are linked to invasive or metastatic behavior (13,14). Moreover, t-PA promoted angiogenesis in a pancreatic cancer model (12). In another study, PLAT was commonly expressed on invasive hepatocellular carcinoma cells, and its upregulation promoted the proliferation, migration, and invasion of cancer cells (15). Furthermore, NSCLC patients with high expression of t-PA tend to have poor prognosis, and the activation of t-PA contributes to lung cancer metastasis (16). Oncology drug therapy is of great significance to cancer patients while drug resistance might be fatal to them. Since PLAT is associated with cancer progression and prognosis, we hypothesize that PLAT might also be involved in TKI resistance of NSCLC, which has not been studied before. In this study, we used gefitinib-resistant cells and a nude mouse tumor model to evaluate the effect of PLAT on gefitinib resistance in NSCLC.
Methods
Cell culture and lentiviral transfection
Human lung adenocarcinoma cell lines PC9 and HCC827 were obtained from ATCC (Manassas, VA, USA). The cells were grown in RPMI-1640 medium (Hyclone, UT, USA) with 10% fetal bovine serum (Gibco, Waltham, MA, USA) and 100 U/mL penicillin/streptomycin at 37 °C in a 5% CO2 culture chamber. Escalating doses of gefitinib (Selleckchem, TX, USA) ranging from 0.01 to 10.0 mM were added to the medium in order to induce drug resistance, and 1 µM gefitinib was added every 2 weeks to maintain resistance of gefitinib-resistant PC9 (PC9R) cells. Lentiviral particles were collected by transfecting 293T cells with plasmids (GeneChem, Shanghai, China) expressing negative control short hair RNA (shNEG) or short hair RNA against PLAT (shPLAT, RNAi Consortium). Lentivirus overexpressing human PLAT (PC9-PLAT, GenBank accession number NM_000930) was obtained from GeneCopoeia (Germantown, MD, USA). After cells reached 60% confluence in 6-well plates, they were infected with lentiviral particles at a multiplicity of infection of 10 and incubated with 4 µg/mL polybrene in medium.
Cell viability assay
A total of 100 µL medium containing about 1,500 cells were seeded each well in 96-well plates for 24 h. Then, 10 µL of cell counting kit-8 (CCK8) (Dojindo Laboratories, Kumamoto, Japan) was added to each well. The plates were protected from light and incubated for 1–2 h at 37 °C. The optical density (OD) at 450 nm was measured by a microplate reader (Thermo Fisher Scientific, MA, USA).
5-ethynyl-2’-deoxyuridine (EdU) staining
Cells transfected with shRNA were stained with EdU (Thermo Fisher Scientific, MA, USA) and Hoechst 33342 (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s protocols. Images were captured by a confocal laser scanning microscope (Nikon, Tokyo, Japan).
Apoptosis and cell cycle detection
Cell samples were detected by flow cytometer (BD Biosciences, USA). About 5×105 cells were plated into each well in 6-well plates. After exposure to 1 µM gefitinib for 24 h, cells were trypsinized, collected and washed with phosphate buffer saline (PBS) 3 times. For apoptotic analysis, cells were resuspended in 500 µL binding buffer before incubation at room temperature in the dark with 5 µL fluorescein isothiocyanate (FITC)-conjugated annexin V and 3 µL propidium iodide (Thermo Fisher Scientific, MA, USA) for 15 min. For cell cycle detection, collected cells were fixed at 4 °C for 24 h with 70% ethanol and were then stained with propidium iodide and protected from light for 30 min at room temperature.
Mitochondrial membrane potential analysis
Procedures were performed strictly under the manufacturer’s protocols. Mitochondrial membrane potential was measured using 5,5',6,6'-tetrachloro-1,1',3,3'-tetraethyl-benzimidazolyl carbocyanine iodide (JC-1) (Thermo Fisher Scientific, MA, USA), an indicator of mitochondrial potential, which shows mitochondrial depolarization through a decrease in the red/green fluorescence intensity ratio. Cell samples were assessed by flow cytometry.
Reactive oxygen species level measurement
To examine reactive oxygen species (ROS) levels, about 5×105 cells were plated into each well in 6-well plates. After exposure to 1 µM gefitinib for 24 h, cells were trypsinized and incubated at 37 °C with 5 µM CellROX Orange reagent (Thermo Fisher Scientific, MA, USA) for 30 min. Then cells samples were washed with PBS three times and assessed by flow cytometry.
Enzyme linked immunosorbent assay (ELISA)
The TNF-α ELISA kit (R&D Systems, MN, USA) was used to measure the level of TNF-α in the culture supernatant of cells according to the manufacturer’s instructions. The absorbance was detected by the microplate reader (Thermo Fisher Scientific, MA, USA)
Animal experiments
Balb/c male nude mice (4 to 6 weeks old) were obtained from the Shanghai Experimental Animal Center of Chinese Academy of Sciences and were housed under specific pathogen-free conditions. Mice were randomly divided into two groups and subcutaneously implanted with 1×107 PC9R-shNEG or PC9R-shPLAT-1 cells into the flank of each mouse. Tumor length (a) and width (b) were measured once per week, and tumor volume was calculated by the formula: Tumor volume = a×b2/2. Mice were sacrificed on the 35th day, and tumors were removed and weighed. The paraffin-embedded tumors were immunostained with Ki67 to stain proliferating cells, which were then quantified by ImageJ.
Western blotting analysis
Cells were lysed in radio immunoprecipitation assay (RIPA) buffer (Beyotime Biotechnology, China) to extract total protein, and the concentration was measured with a BCA Protein Assay Kit (Beyotime Biotechnology, China). Equivalent amounts of protein lysates were loaded into each well of polyacrylamide gels and then transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were blocked with blocking buffer at room temperature for 1 h and incubated with anti-PLAT (1:1,800, Novus Biologicals, CO, USA), anti-TNF-α (1:1,000, Abcam, MA, USA), anti-FADD (1:1,000, Cell Signaling Technology (CST), Danvers, USA), anti-cleaved caspase-8 (1:1,000, Asp391, CST), anti-caspase-8 (1:1,000, D35G2, CST), and anti-actin (1:2,000, CST) antibodies at 4 °C overnight. The membranes were incubated with corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature. The blots were visualized using electro-chemiluminescence (ECL) reagents (Thermo Fisher Scientific, MA, USA) and imaged using a Bio-Rad (Hercules, CA, USA) system.
Real-time quantitative PCR
TRIzol Reagent (Thermo Fisher Scientific, MA, USA) was used to lyse cells. RNA was reverse-transcribed into cDNA using the cDNA kit (TOYOBO, Japan) according to the manufacturer’s protocols. Then, 20 ng cDNA was used as template and amplified using the SYBR® Premix Ex Taq™ (TaKaRa Bio, Osaka, Japan). The cycling conditions were as follows: 40 cycles of 95 °C for 15 s, 60 °C for 15 s, and 72 °C for 45 s. β-actin was used as an internal control, and the relative quantification of each PCR product was calculated using the 2−ΔΔCq method (17). The sequences of the primes were as follows:
PLAT: Forward: 5'-GGT CTG GAG AAG TCT GTA GAG-3'; Reverse: 5'-CCT AGA CTG GAT TCG TGA CAA-3; TNF-α: Forward: 5'-CCC GAG TGA CAA GCC TGT AG-3'; Reverse: 5'-TGA GGT ACA GGC CCT CTG AT-3; β-actin: Forward: 5'-CTG GCA CCC AGC ACA ATG-3'; Reverse: 5'-CCG ATC CAC ACG GAG TAC TTG-3
cDNA array screening
RNeasy Plus Mini Kit (Qiagen, USA) was used to extract cell RNA. RNA was then reverse-transcribed into cDNA and was amplified using the OneArray Plus RNA Amplification Kit (Phalanx Biotech Group, Hsinchu, Taiwan). Then the amplification products were dyed with Cyanine 5 and hybridized to Human Whole Genome OneArray (Phalanx Biotech Group, Hsinchu, Taiwan). The signal was captured by a G2505C Agilent Microarray Scanner (Agilent Technologies, Santa Clara, CA, USA) and raw data extraction was generated by GenePix™ 4 (Molecular Devices, Sunnyvale, CA, USA). Data was processed with the error model of Rosetta Resolver System (Rosetta Bio software, Seattle, WA, USA). The log2(Ratio) were calculated by pair-wise combination and error weighted average. Significant differentially expressed genes (DEGs) were selected according to log2(Ratio) ≥1 and P value (differentially expressed) <0.05.
TCGA analysis
Lung adenocarcinoma and lung squamous-cell carcinoma datasets were downloaded from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov). A total of 1,014 tumor samples and 108 non-tumor samples were included in our study. The gene expression level was normalized as transcripts per million (TPM) before performing analysis.
Statistical analysis
Results are shown as the mean ± standard deviation. Comparisons of two groups were assessed by Student’s t-test, while comparisons among multiple groups were assessed by one-way analysis of variance with Bonferroni’s multiple comparisons test as a post hoc test. Results with P<0.05 were considered statistically significant. All tests were repeated in triplicate.
Results
PLAT is overexpressed in NSCLC
We first intended to observe the expression of PLAT in NSCLC patients. A total of 1014 lung tumor tissues and 108 non-tumor lung tissues from TCGA were included in our study. Results showed that PLAT was overexpressed in lung tumor tissues compared to non-tumor lung tissues (P=0.0006, Figure 1).
Figure 1.
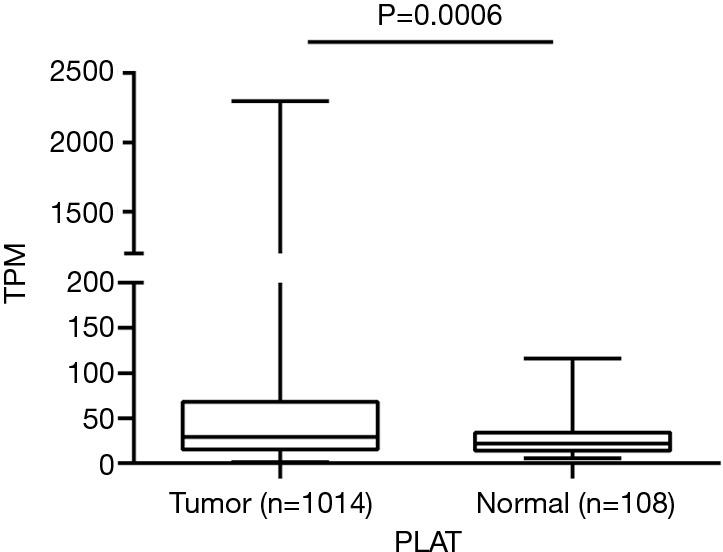
PLAT is overexpressed in NSCLC. The expression of PLAT in lung tumor tissues was higher than that in non-tumor lung tissues (P=0.0006). TPM represents transcripts per million. NSCLC, non-small cell lung cancer; TPM, transcripts per million.
PLAT is overexpressed in gefitinib-resistant cells
Human NSCLC were treated with gefitinib to screen for gefitinib-resistant cells. We use RT-qPCR and western blotting to compare the expression levels in PC9 and PC9R cells (Figure 2A,B) as well as in HCC827 and gefitinib-resistant HCC827 (HCC827R) cells (Figure 2C,D). Results showed that the expression of PLAT and t-PA in PC9R and HCC827R cells were higher than that in PC9 and HCC827 cells.
Figure 2.
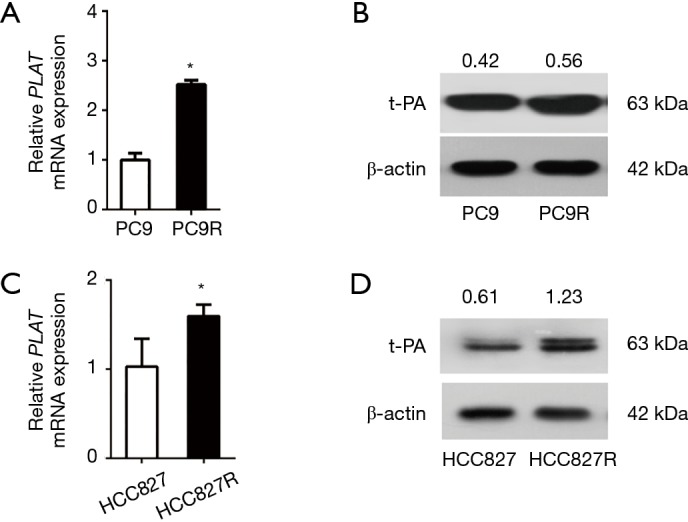
PLAT is overexpressed in gefitinib-resistant PC9R and HCC827R cells. (A,B) The expression of PLAT/t-PA in PC9 cells and gefitinib-resistant PC9R cells was examined by (A) RT-qPCR and (B) western blotting. (C,D) The expression of PLAT/t-PA in HCC827 cells and gefitinib-resistant HCC827R cells was examined by (C) RT-qPCR and (D) western blotting. Each data point represents the mean ± SD of three independent experiments. *, P<0.05 vs. control.
The occurrence of gefitinib resistance is mediated by PLAT
We further investigated the effect of PLAT on gefitinib resistance by silencing or overexpressing it in cells. PC9R cells were transfected with shPLAT-1 or shPLAT-2 to knock down PLAT expression, with PC9R cells transfected with shNEG used as a control. RT-qPCR and western blotting results verified that compared with levels in control cells, PLAT and t-PA was decreased in shPLAT-1 or shPLAT-2 cells (Figure 3A,B). The viability of the silenced cells was reduced under gefitinib treatment at concentrations ranging from 0.1 to 10 µM (Figure 3C). Moreover, after gefitinib treatment of 1 µM for 96 h, as observed under the microscope, numbers of shPLAT-1 and shPLAT-2 cells were significantly reduced in comparison with that of shNEG cells, and morphological changes were also apparent (Figure 3D).
Figure 3.

The occurrence of gefitinib resistance is mediated by PLAT. (A,B) After transfection with shNEG, shPLAT-1, or shPLAT-2, the expression of PLAT/t-PA in PC9R cells was examined by (A) RT-qPCR and (B) Western blotting; (C) viability of shNEG, shPLAT-1, and shPLAT-2 cells treated with different doses of gefitinib were examined by CCK8 assay; (D) cells were treated with 1 µM gefitinib for 96 h, and the morphology of PC9R cells expressing shNEG or shPLAT was observed under a microscope; (E,F) after overexpressing PLAT in PC9 cells, the mRNA and protein levels in control PC9-vector cells and PC9-PLAT cells were verified by (E) RT-qPCR and (F) western blotting; (G) viability of PC9-vector and PC9-PLAT cells treated with different doses of gefitinib were examined by CCK8 assay. Each data point represents the mean ± SD of three independent experiments. *, P<0.05 vs. control. NEG, negative control.
We also established PLAT-overexpressing PC9 cells (PC9-PLAT). As shown in Figure 3E and F, the mRNA and protein expression levels of PLAT and t-PA in PC9-PLAT cells were significantly higher than those in control cells (PC9-vector). In the presence of gefitinib, PC9-PLAT cells showed higher viability than PC9-vector cells (Figure 3G). Taken together, these results confirm that the silence of PLAT enhances gefitinib effects in vitro. PLAT may contribute to gefitinib resistance in NSCLC cells.
Silencing of PLAT leads to reduction in proliferation and induction in apoptosis and cell cycle arrest
EdU staining showed that the proliferation of silenced PC9R cells was clearly inhibited after gefitinib treatment relative to that in control cells (Figure 4A,B). Moreover, FITC-annexin V/propidium iodide staining and flow cytometric analysis demonstrated that the apoptosis of shPLAT-1 and shPLAT-2 cells was enhanced (Figure 5A,B). This was consistent with the observation of cell cycle arrest in silenced PC9R cells, as evidenced by the accumulation of cells in the G2/M phase and the reduction of cells in the G0/G1 and S phase (Figure 5C,D).
Figure 4.
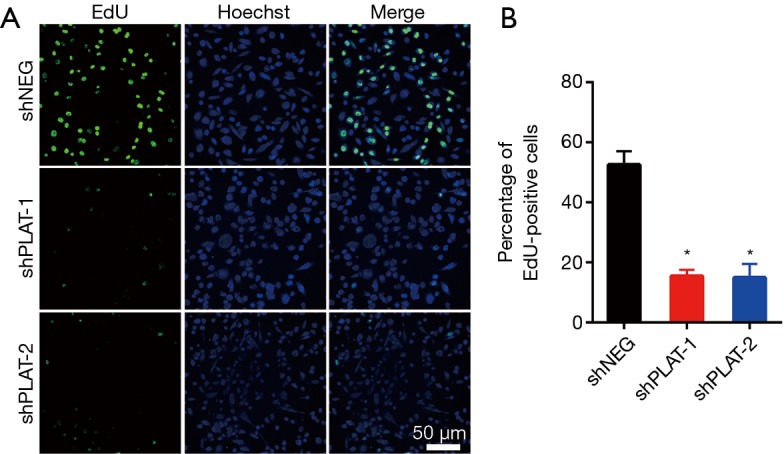
Silencing of PLAT leads to reduction in proliferation in vitro. (A) PC9R cells transfected with shNEG, shPLAT-1, or shPLAT-2 were treated with 1 µM gefitinib and stained with EdU and Hoechst 33342; (B) the percentage of EdU-positive cells was calculated. Each data point represents the mean ± SD of three independent experiments. *, P<0.05 vs. control.
Figure 5.

Silencing of PLAT leads to induction in apoptosis and cell cycle arrest. (A) PC9R cells transfected with shNEG, shPLAT-1, or shPLAT-2 were treated with 1 µM gefitinib and detected by flow cytometry; (B) the percentage of apoptotic cells was analyzed; (C) cell cycle phase was detected after treatment with 1 µM gefitinib by flow cytometry; (D) cell cycle distribution was quantified. Each data point represents the mean ± SD of three independent experiments. *, P<0.05 vs. control.
Silencing of PLAT leading to reduction in cell proliferation was also confirmed in vivo. The animal models were established successfully by injecting PC9R cells transfected with shPLAT-1 and shNEG into nude mice. We measured tumor volume once a week for 5 weeks and visible tumors were observed 2 weeks later. Tumors in mice injected with shPLAT-1 cells were smaller and weighed significantly less than those in mice injected with shNEG cells (Figure 6A,B). In addition, Ki67 staining showed that the ratio of proliferating cells in shPLAT-1 tumors was clearly lower than that in shNEG tumors (Figure 6C,D).
Figure 6.
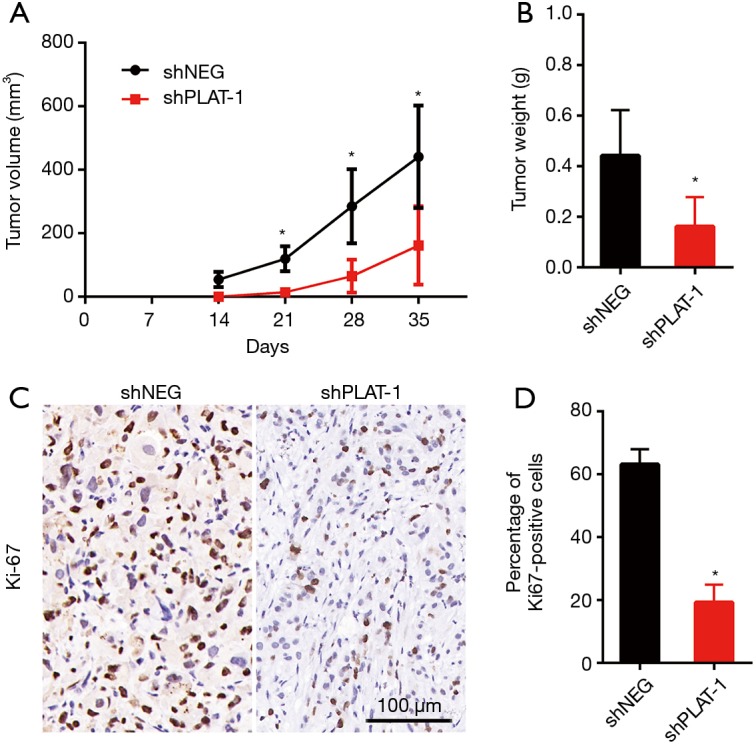
Silencing of PLAT leads to reduction in proliferation in vivo. PC9R cells transfected with shNEG or shPLAT-1 were injected into nude mice. (A) Tumor volume was measured every week; (B) tumors were weighed after sacrificing; (C,D) immunohistochemical staining of Ki67 was performed on tumor samples, and positive cells were quantified. n=6 in shNEG group and n=5 in shPLAT-1 group. *, P<0.05 vs. control.
Silencing of PLAT leads to damage of mitochondria and activation of caspase
JC-1 staining was used to examine the integrity of the mitochondria. We found that the JC-1 monomer ratio in PLAT-silenced cells was higher than that in control cells, indicating a high percentage of damaged mitochondria (Figure 7A,B). CellROX Orange staining showed that PLAT-silenced cells generated twofold more reactive oxygen species (ROS) than control cells (Figure 7C,D). Western blotting results showed that apoptotic proteins, cleaved caspase-3, and cleaved PARP were upregulated in shPLAT-1 and shPLAT-2 cells relative to levels in control cells (Figure 7E). These results indicate that silence of PLAT promotes the damage of mitochondria and the activation of caspase, which may results in the induction of apoptosis in PLAT-silenced cells.
Figure 7.

Silencing of PLAT leads to damage of mitochondria and activation of caspase. (A) JC-1 staining was performed after treatment with 1 µM gefitinib and was assessed by flow cytometry; (B) the JC-1 monomer ratio was calculated; (C,D) ROS generation after treatment with 1 µM gefitinib was measured by flow cytometry, and was quantified; (E) the protein levels of cleaved caspase-3 and cleaved PARP in cells transfected with shNEG, shPLAT-1, or shPLAT-2 were examined by western blotting. Each data point represents the mean ± SD of three independent experiments. *, P<0.05 vs. control.
Silencing of PLAT induces apoptosis of PC9R cells through TNF-α signaling
The mechanism by which PLAT knockdown induces apoptosis was further investigated. We used a whole human cDNA array to explore the differential expression of genes in shNEG and shPLAT-1 cells, and found that knockdown of PLAT upregulated the gene expression of TNF-α (Figure 8A). As TNF-α can induce a variety of cellular responses, including inflammatory cytokine production, cell death, and cell proliferation, we validated the expression of TNF-α in PC9R cells expressing shNEG, shPLAT-1, and shPLAT-2 using RT-qPCR, ELISA, and western blotting. The results of RT-qPCR showed that the mRNA levels of TNF-α in shPLAT-1 and shPLAT-2 cells were significantly higher than those in shNEG cells (Figure 8B). The secretory levels of TNF-α in the culture supernatants of shPLAT-1 and shPLAT-2 cells were also markedly higher than that in shNEG cells (Figure 8C). It is reported that upon binding to TNFR-1, TNF-α induces apoptosis via the recruitment of FADD and caspase-8 (18). Thus, we assessed levels of FADD, caspase-8, and cleaved caspase-8 in cells by western blotting. Compared to levels in shNEG cells, FADD, caspase-8, and cleaved caspase-8 were more highly expressed in shPLAT-1 and shPLAT-2 cells (Figure 8D). These results suggest that knockdown of PLAT induces apoptosis of PC9R cells via the TNF-α/FADD/caspase-8 pathway.
Figure 8.

Silence of PLAT induces apoptosis of PC9R cells through TNF-α signaling. (A) PC9R cells transfected with shNEG or shPLAT-1 were analyzed by whole human cDNA array; (B,C) the expression of TNF-α in shNEG, shPLAT-1, and shPLAT-2 cells incubated with gefitinib was measured by (B) RT-qPCR and (C) ELISA; (D) the protein levels of TNF-α, FADD, cleaved caspase-8, and caspase-8 were measured by western blotting. Each data point represents the mean ± SD of three independent experiments. *, P<0.05 vs. control.
Discussion
The current study investigated the role of PLAT in gefitinib resistance. We found that gefitinib-resistant PC9 and HCC827 cells expressed higher levels of PLAT. Overexpression of PLAT in PC9 cells enhances gefitinib resistance, while knockdown of PLAT in PC9R cells restored gefitinib sensitivity. In addition, tumor proliferation was limited in vivo after knockdown of PLAT. We further demonstrated that knockdown of PLAT affected mitochondrial function, caused caspase activation and cell cycle arrest, and activated TNF-α signaling. Therefore, our findings suggest that PLAT regulates proliferation and apoptosis and confers gefitinib resistance via TNF-α signaling.
Targeted therapy, aimed at targeting various molecularly defined cancer subtypes, represents a revolution in anti-cancer therapy, improves patient outcomes, and reduces side effects (19). The administration of gefitinib to NSCLC patients with EGFR inhibitor-sensitizing mutations not only reveals the potential of targeted therapy in lung cancer but also provides insight into the genetic complexity of this disease (19). Inevitably, resistance develops after the initial response to EGFR-TKIs. Mechanisms of resistance include the additional EGFR T790M mutation (50–60%), MET amplification (5%), HER2 amplification (8–13%), PIK3CA amplification (1–2%), KRAS mutations (6.7%), and small cell lung cancer transformation (6%) (20-22). Thus, unknown mechanisms account for at least 10–20% of acquired resistance. Here, we established gefitinib-resistant PC9 cells by exposing PC9 cells to increasing concentrations of gefitinib. It has been reported that the t-PA protein is upregulated in erlotinib-resistant PC9 cells based on mass spectrometry-based secretome analysis (23). Comparing gefitinib-resistant PC9 cells with gefitinib-sensitive PC9 cells, we found that the mRNA and protein levels of PLAT/t-PA in resistant cells were elevated. Overexpression of PLAT in gefitinib-sensitive PC9 cells improved cell viability when exposed to gefitinib, suggesting the important role of PLAT in resistance.
PLAT is necessary for the growth of pancreas tumor cell lines in vitro and in vivo, maintaining pancreas tumor cells in a state of proliferative competence (13). In our study, knockdown of PLAT in PC9R cells reduced proliferation in vitro. Our in vivo experiment also verified that the proliferation of PC9R-shPLAT cells was limited. Proliferation is tightly connected with the cell cycle, and tumorigenesis is closely linked with the dysregulation of the cell cycle (24,25). We observed the accumulation of PC9R-shPLAT cells in the G2/M phase and a reduction of cells in the G0/G1 and S phases. Moreover, knockdown of PLAT interfered with mitochondrial function in our study. Mitochondria play a crucial role in cellular apoptosis. The loss of mitochondrial membrane potential and excess generation of mitochondrial ROS, followed by the release of cytochrome C and the activation of caspase-3, signals the progression of apoptosis (26-28). After incubating PC9R-shPLAT cells with gefitinib, the mitochondrial membrane potential decreased, while ROS generation and caspase-3 and PARP expression increased. These results indicate that knockdown of PLAT inhibits cell proliferation and promotes cell apoptosis via mitochondrial damage and caspase activation.
We also found an elevated level of TNF-α in PC9R-shPLAT cells in our study. TNF-α is an important cytokine involved in inflammatory cytokine production, cell survival, proliferation, and apoptosis (29). TNF-α functions mainly via its receptor TNFR-1, which is a dual receptor in that it is involved in signaling both apoptosis as well as survival (18). When signaling survival, it forms complex I and activates NF-κB, which then drives the transcription of several cytokines and anti-apoptotic proteins to inhibit apoptosis. When signaling apoptosis, complex I recruits FADD and caspase-8 to form complex II and initiate a protease cascade that leads to apoptosis (18,30). A previous study reported that TNF-α induced signaling cross-talk between TNFR and EGFR, subsequently activating the Akt/NF-κB cascade and increasing the expression of anti-apoptotic proteins in gefitinib-sensitive PC9 cells but not in gefitinib-resistant PC9 cells (31). In our study, we found that levels of TNF-α, FADD, and cleaved caspase-8 were elevated in PC9R-shPLAT cells, indicating that knockdown of PLAT may lead to apoptosis in gefitinib-resistant cells via TNF-α signaling.
In summary, this represents the first report elucidating the role of PLAT in gefitinib resistance in NSCLC. Further investigation in the clinic is needed to determine the predictive value of PLAT for gefitinib resistance.
Acknowledgments
We thank Xiaoyan Chen for her kind help in bioinformatics analysis.
Funding: This work was supported by Natural Science Foundation of China (No. 81570028, 81490533, 81770039 and 81400018), National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program”, China (No. 2018ZX09201002-006), Shanghai Science and Technology Committee Grant (15DZ1941103), and Zhongshan Hospital Clinical Research Foundation (No. 2016ZSLC05).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All procedures performed in studies involving animals were in accordance with the ethical standards of the Institutional Animal Care and Use Committee at Zhongshan Hospital of Fudan University, Shanghai, China.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Hirsch FR, Scagliotti GV, Mulshine JL, et al. Lung cancer: current therapies and new targeted treatments. Lancet 2017;389:299-311. 10.1016/S0140-6736(16)30958-8 [DOI] [PubMed] [Google Scholar]
- 2.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394-424. 10.3322/caac.21492 [DOI] [PubMed] [Google Scholar]
- 3.Bui KT, Cooper WA, Kao S, et al. Targeted Molecular Treatments in Non-Small Cell Lung Cancer: A Clinical Guide for Oncologists. J Clin Med 2018;7:E192. 10.3390/jcm7080192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma W, Xu M, Liu Y, et al. Safety profile of combined therapy inhibiting EFGR and VEGF pathways in patients with advanced non-small-cell lung cancer: A meta-analysis of 15 phase II/III randomized trials. Int J Cancer 2015;137:409-19. 10.1002/ijc.29377 [DOI] [PubMed] [Google Scholar]
- 5.Wang S, Gao A, Liu J, et al. First-line therapy for advanced non-small cell lung cancer with activating EGFR mutation: is combined EGFR-TKIs and chemotherapy a better choice? Cancer Chemother Pharmacol 2018;81:443-53. 10.1007/s00280-017-3516-1 [DOI] [PubMed] [Google Scholar]
- 6.Murakami H, Nokihara H, Hayashi H, et al. Clinical activity of ASP8273 in Asian patients with non-small-cell lung cancer with EGFR activating and T790M mutations. Cancer Sci 2018;109:2852-62. 10.1111/cas.13724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loong HH, Kwan SS, Mok TS, et al. Therapeutic Strategies in EGFR Mutant Non-Small Cell Lung Cancer. Curr Treat Options Oncol 2018;19:58. 10.1007/s11864-018-0570-9 [DOI] [PubMed] [Google Scholar]
- 8.Tomasello C, Baldessari C, Napolitano M, et al. Resistance to EGFR inhibitors in non-small cell lung cancer: Clinical management and future perspectives. Crit Rev Oncol Hematol 2018;123:149-61. 10.1016/j.critrevonc.2018.01.013 [DOI] [PubMed] [Google Scholar]
- 9.Yoda S, Dagogo-Jack I, Hata AN. Targeting oncogenic drivers in lung cancer: Recent progress, current challenges and future opportunities. Pharmacol Ther 2019;193:20-30. 10.1016/j.pharmthera.2018.08.007 [DOI] [PubMed] [Google Scholar]
- 10.Lim SM, Syn NL, Cho BC, et al. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Treat Rev 2018;65:1-10. 10.1016/j.ctrv.2018.02.006 [DOI] [PubMed] [Google Scholar]
- 11.Ke EE, Wu YL. EGFR as a pharmacological target in EGFR-mutant non-small-cell lung cancer: Where do we stand now? Trends Pharmacol Sci 2016;37:887-903. 10.1016/j.tips.2016.09.003 [DOI] [PubMed] [Google Scholar]
- 12.Kruithof EKO, Dunoyer-Geindre S. Human tissue-type plasminogen activator. Thromb Haemost 2014;112:243-54. 10.1160/TH13-06-0517 [DOI] [PubMed] [Google Scholar]
- 13.Díaz VM, Planaguma J, Thomson TM, et al. Tissue plasminogen activator is required for the growth, invasion, and angiogenesis of pancreatic tumor cells. Gastroenterology 2002;122:806-19. 10.1053/gast.2002.31885 [DOI] [PubMed] [Google Scholar]
- 14.Corte MD, Vérez P, Rodríguez JC, et al. Tissue-type plasminogen activator (tPA) in breast cancer: relationship with clinicopathological parameters and prognostic significance. Breast Cancer Res Treat 2005;90:33-40. 10.1007/s10549-004-2624-x [DOI] [PubMed] [Google Scholar]
- 15.Lin ZY, Chuang WL. Genes responsible for the characteristics of primary cultured invasive phenotype hepatocellular carcinoma cells. Biomed Pharmacother 2012;66:454-8. 10.1016/j.biopha.2012.04.001 [DOI] [PubMed] [Google Scholar]
- 16.Lin CY, Chen HJ, Huang CC, et al. ADAM9 promotes lung cancer metastases to brain by a plasminogen activator-based pathway. Cancer Res 2014;74:5229-43. 10.1158/0008-5472.CAN-13-2995 [DOI] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCq Method. Methods 2001;25:402-08. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 18.Van Horssen R, Ten Hagen TL, Eggermont AM. TNF-alpha in cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist 2006;11:397-408. 10.1634/theoncologist.11-4-397 [DOI] [PubMed] [Google Scholar]
- 19.Mayekar MK, Bivona TG. Current Landscape of Targeted Therapy in Lung Cancer. Clin Pharmacol Ther 2017;102:757-64. 10.1002/cpt.810 [DOI] [PubMed] [Google Scholar]
- 20.Ernani V, Steuer CE, Jahanzeb M. The End of Nihilism: Systemic Therapy of Advanced Non-Small Cell Lung Cancer. Annu Rev Med 2017;68:153-68. 10.1146/annurev-med-042915-102442 [DOI] [PubMed] [Google Scholar]
- 21.Chiu CF, Chang YW, Kuo KT, et al. NF-κB-driven suppression of FOXO3a contributes to EGFR mutation-independent gefitinib resistance. Proc Natl Acad Sci U S A 2016;113:E2526-35. 10.1073/pnas.1522612113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res 2013;19:2240-47. 10.1158/1078-0432.CCR-12-2246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bosse K, Haneder S, Arlt C, et al. Mass spectrometry-based secretome analysis of non-small cell lung cancer cell lines. Proteomics 2016;16:2801-14. 10.1002/pmic.201600297 [DOI] [PubMed] [Google Scholar]
- 24.King KL, Cidlowski JA. Cell cycle regulation and apoptosis. Annu Rev Physiol 1998;60:601-17. 10.1146/annurev.physiol.60.1.601 [DOI] [PubMed] [Google Scholar]
- 25.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature 2001;411:342-8. 10.1038/35077213 [DOI] [PubMed] [Google Scholar]
- 26.Hsiao CJ, Hsiao G, Chen WL, et al. Cephalochromin induces G0/G1 cell cycle arrest and apoptosis in A549 human non-small-cell lung cancer cells by inflicting mitochondrial disruption. J Nat Prod 2014;77:758-65. 10.1021/np400517g [DOI] [PubMed] [Google Scholar]
- 27.Liu WB, Xie F, Sun HQ, et al. Anti-tumor effect of polysaccharide from Hirsutella sinensis on human non-small cell lung cancer and nude mice through intrinsic mitochondrial pathway. Int J Biol Macromol 2017;99:258-64. 10.1016/j.ijbiomac.2017.02.071 [DOI] [PubMed] [Google Scholar]
- 28.Gottlieb E, Armour SM, Harris MH, et al. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ 2003;10:709-17. 10.1038/sj.cdd.4401231 [DOI] [PubMed] [Google Scholar]
- 29.Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008;133:693-703. 10.1016/j.cell.2008.03.036 [DOI] [PubMed] [Google Scholar]
- 30.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003;114:181-90. 10.1016/S0092-8674(03)00521-X [DOI] [PubMed] [Google Scholar]
- 31.Ando K, Ohmori T, Inoue F, et al. Enhancement of sensitivity to tumor necrosis factor alpha in non-small cell lung cancer cells with acquired resistance to gefitinib. Clin Cancer Res 2005;11:8872-9. 10.1158/1078-0432.CCR-05-0811 [DOI] [PubMed] [Google Scholar]