

Summary
Astrocytes have emerged for playing important roles in brain tissue repair; however, the underlying mechanisms remain poorly understood. We show that acute injury and blood-brain barrier disruption trigger the formation of a prominent mitochondrial-enriched compartment in astrocytic endfeet, which enables vascular remodeling. Integrated imaging approaches revealed that this mitochondrial clustering is part of an adaptive response regulated by fusion dynamics. Astrocyte-specific conditional deletion of Mitofusin 2 (Mfn2) suppressed perivascular mitochondrial clustering and disrupted mitochondria-endoplasmic reticulum (ER) contact sites. Functionally, two-photon imaging experiments showed that these structural changes were mirrored by impaired mitochondrial Ca2+ uptake leading to abnormal cytosolic transients within endfeet in vivo. At the tissue level, a compromised vascular complexity in the lesioned area was restored by boosting mitochondrial-ER perivascular tethering in MFN2-deficient astrocytes. These data unmask a crucial role for mitochondrial dynamics in coordinating astrocytic local domains and have important implications for repairing the injured brain.
Keywords: mitochondrial dynamics, contact sites, brain repair, brain injury, calcium imaging, angiogenesis, proteomics, metabolism, Mitofusin 2, synthetic linker, perivascular endfeet
Graphical Abstract
Highlights
-
•
Reactive astrocytes remodel perivascular mitochondrial and ER networks in vivo
-
•
Mfn2 deletion disrupts astrocytic mitochondria-ER contact sites and calcium buffering
-
•
Mfn2 deletion in astrocytes prevents injury-induced vascular remodeling
-
•
Perivascular enrichment of mitochondria-ER contact sites restores vascular remodeling
Despite the importance of astrocytes in maintaining brain tissue homeostasis, it remains unclear how these cells may contribute to tissue remodeling after damage. Göbel et al. show that a significant enrichment of mitochondrial-ER contact sites in perivascular processes of reactive astrocytes is required to promote neovascularization in the injured area.
Context and Significance
Astrocytes are glial cells with central roles in maintaining brain energy metabolism by ensuring coupling between the vasculature and neurons. As demonstrated in recent years, in conditions of brain injury and disease, astrocytes acquire reactive cellular states, which underlie neurotoxic or protective roles. In this study, researchers of the University of Cologne and the Max Planck Institute for Biology of Aging have discovered that proper maintenance of functional contact sites between mitochondria and the endoplasmic reticulum in reactive astrocytes is required for neo-vascularization in the injured brain. These results demonstrate that astrocyte reactivity plays an active role in mediating vascular remodeling and lays the ground for future work aimed to understand the implications of astrocyte metabolic changes for brain repair.
Introduction
Astrocytes not only regulate essential aspects of brain energy metabolism (Bélanger et al., 2011) but also play important roles in the progression and resolution of numerous brain pathologies, including traumatic brain injury and stroke (Sofroniew, 2015). These types of injury often result in significant damage to the cerebrovasculature and are usually accompanied by blood-brain barrier breakdown, intracerebral hemorrhage, hypoxia, secondary inflammation, and neurodegeneration (Prakash and Carmichael, 2015, Salehi et al., 2017). Although a number of factors concerning the severity of the primary insult contribute to the extent of tissue damage and thus influence the subsequent attempt to repair, our understanding of the mechanisms underlying neovascularization in the injured area and which exact cellular components are recruited is still rudimentary.
Besides endothelial cells and pericytes, which constitute the actual blood-brain barrier, astrocytic endfeet functionally ensheathe most of the cerebrovascular network and serve as specialized dynamic exchange sites for ions, water, and energy substrates with brain parenchyma (Iadecola, 2017). Although maintenance of this tight coupling ensures the supply of metabolites across the gliovascular interface (Iadecola, 2017), the structural and functional changes experienced by astrocytic perivascular endfeet following injury are much less understood. In these settings, astrocytes acquire a reactive state that may underlie both beneficial and deleterious functions (Khakh and Sofroniew, 2015, Liddelow and Barres, 2017). Interestingly, some of these functions have been described for regulating angiogenesis via the secretion of trophic factors and molecules, which can ultimately lead to vascular remodeling (Salehi et al., 2017). Furthermore, a prominent physical association between astrocytes and vessels in the peri-lesioned area has been reported following acute injury, particularly at a time matching with the formation of new vessels (Horng et al., 2017, Villapol et al., 2014), thus suggesting that structural changes at the gliovascular interface may be critical in regulating vascular remodeling. Importantly, evidence suggests that alongside changes in morphology, reactive astrocytes also undergo significant metabolic adaptations (Chao et al., 2019, Polyzos et al., 2019). Supporting this notion, astrocytes can switch between oxidative phosphorylation (OXPHOS) and glycolysis for their energy metabolism (Ignatenko et al., 2018, Lovatt et al., 2007, Supplie et al., 2017), underscoring their capability to accommodate a significant metabolic rewiring depending on substrate availability and local energy needs. This important form of astrocytic plasticity is mirrored by reversible changes in mitochondrial network architecture following acute injury (Motori et al., 2013, Owens et al., 2015), suggesting a pivotal role for mitochondrial dynamics in regulating astrocyte reactivity.
In mammalian cells, the architecture of the mitochondrial network depends upon regulated fusion-fission events that are orchestrated by mitofusins (MFN1 and MFN2) (Chen et al., 2003) and optic atrophy-1 (OPA1) (Cipolat et al., 2004) for mitochondrial fusion, while dynamin-related protein-1 (DRP1) is the key player in outer mitochondrial fission (Ishihara et al., 2009). Together, the coordinated action of these molecules shapes mitochondrial morphology to match precise cellular metabolic needs (Gomes et al., 2011). Functionally, this mitochondrial remodeling is also mutually dependent upon a physical tethering with ER membranes (by forming so-called mitochondria-associated membranes, MAMs) where important metabolic functions take place (Scorrano et al., 2019), including lipid trafficking as well as Ca2+ and reactive oxygen species (ROS) signaling (Csordás et al., 2018). Intriguingly, evidence exists for complex mitochondrial and ER morphologies in astrocytes in situ, where these organelles have been found to reach fine perisynaptic and endfeet processes (Göbel et al., 2018, Jackson and Robinson, 2018, Lovatt et al., 2007, Mathiisen et al., 2010, Motori et al., 2013). This spatial distribution suggests the direct contribution of MAMs to specific astrocytic functions, yet whether a dynamic remodeling of these two organelles may effectively couple the acquisition of a reactive state with functional metabolic changes underlying tissue remodeling is unclear.
Results
Astrocyte Endfeet Are Naturally Enriched in Mitochondria-ER Contact Sites
In order to investigate how the architecture of mitochondrial and ER networks may match the morphological complexity of astrocytes, we utilized a virus-based strategy to specifically label these organelles in vivo (Figure 1A). Minimal amounts of either hGFAP promotor-driven adeno-associated viruses (AAVs) or modified EnvA-pseudotyped rabies viruses (RABVs) were stereotactically injected into the cortex of wild-type or hGFAP-TVA mice, respectively, to drive the expression of mitochondrial- or ER-targeted fluorophores (i.e., mitoRFP and ER-GFP). Both viral-based approaches have been previously shown to efficiently restrict the expression of transgenes to astrocytes in the adult brain (Motori et al., 2013, Shigetomi et al., 2013). Single-astrocyte analysis 1 week after virus delivery revealed a complex morphology of these organelles, which were found decorating the most peripheral astrocytic processes (Figure 1B). Interestingly, besides few primary branches originating from the soma, structures identified as perivascular endfeet (i.e., possessing a tube-like morphology and surrounding CD31+ vessels) were often enriched in ER and mitochondria (Figures 1C, S1A, and S1B). In these regions, the ER appeared to virtually delineate the shape of vessels, whereas mitochondria often formed a dense meshwork that was much similar to the one observed within primary branches rather than distally located fine perisynaptic processes (branchlets) (Figures 1C, S1A, and S1B). Experiments conducted by labeling the microvasculature via systemic dextran injection prior to sacrifice revealed that astrocytic ER and mitochondrial networks outlined the labeled vessels to the extent that often whole sections of the microvasculature appeared wrapped by a thin but discernible layer of astrocytic organelles (Figures 1D and 1E; Videos S1 and S2). In contrast, virus-mediated labeling of other organelles including peroxisomes and lysosomes yielded a much different distribution, being largely confined to the cell body and major branches (Figures S1C–S1F).
Figure 1.

Astrocytic Endfeet Are Enriched in Mitochondria-ER Contact Sites
(A) Experimental design.
(B) Example of an astrocyte co-transduced with ER-GFP and mitoRFP viruses. Yellow arrowheads: endfeet. Scale bar, 10 μm.
(C) Magnifications of the astrocyte shown in (B). Yellow arrowheads point to bundles of elongated mitochondria. Scale bar, 5 μm.
(D and E) Example of astrocytes transduced with ER-GFP or mitoYFP (in gray) wrapping around dextran-labeled vessels (in red). Insets show zooms of the perivascular endfoot. Side panels: 3D rendering of the same astrocytes. Scale bars, 10 and 25 μm.
(F) EM picture of a vessel cross-section showing the astrocytic endfoot (segmented black line) and its organelles (mitochondria, yellow; ER, red; contact sites, blue). Inset: mitochondria-ER contact sites lining the basal lamina. Scale bars, 2 and 1 μm.
(G) Perisynaptic astrocytic processes and their organelles. Scale bar, 2 μm.
(H) Quantification of mitochondrial parameters in branches (n = 21 cross-sections from 3 mice) and endfeet (n = 32 cross-sections from 3 mice; nonparametric Mann-Whitney t test). ∗∗∗p < 0.001. PC, pericyte; EC, endothelial cell; BL, basal lamina.
See also Figure S1.
Two-photon imaging scan through a portion of the intact cerebral cortex (vasculature labeled via dextran-red) following astrocyte labeling with ER-GFP
Two-photon imaging scan through a portion of the intact cerebral cortex (vasculature labeled via dextran-red) following astrocyte labeling with mitoYFP
At the ultrastructural level, astrocytic endfeet appeared enriched with ER membranes surrounding not only the basal lamina but also most of mitochondria located within the perivascular process (Figures 1F, S1G, and S1H). In particular, substantial portions of the mitochondrial perimeter were bordered by ER membranes and, at these contact sites, the two organelles maintained an average reciprocal distance of 18.9 ± 5.0 nm (Figure 1F). By comparison, both the size of mitochondria and the extent of ER membranes were smaller in perisynaptic astrocytic processes, resulting in visibly fewer contact sites, despite a similar mitochondria-ER average distance of 20.4 ±7.0 nm (Figure 1G). Accordingly, morphological quantification revealed a net enrichment in mitochondrial area and mitochondria-ER tethering domains within the endfeet (Figure 1H), suggestive of key metabolic functions being regulated by these two organelles at perivascular sites.
Marked Remodeling of Astrocyte Mitochondrial Networks following Cortical Injury
Astrocyte reactivity states are characterized by prominent changes in energy metabolism and mitochondrial network morphology (Castejón, 2015, Hamby et al., 2012, Motori et al., 2013, Zamanian et al., 2012), raising the question of whether perivascular organelle distribution may also become affected. To answer this question, we utilized a genetic approach to conditionally express mitoYFP in astrocytes and investigated in detail mitochondrial morphology after cortical stab-wound (SW) injury in vivo (Figure 2A). Human-GFAP-CreER mice (Chow et al., 2008) were crossed with mitoYFP floxed-stop mice (Sterky et al., 2011), and the resulting line was induced with tamoxifen at the age of 6–8 weeks. About 88% of cortical astrocytes (S100β+) underwent recombination (Figure S2A), allowing for a systematic analysis of the mitochondrial network in cells proximal to the lesion track (i.e., the area mostly enriched in extravasating pro-inflammatory CD45+ leukocytes) (Figures 2B and S2B). In particular, by 1 week following SW, astrocytes reacted by widespread mitochondrial fragmentation (Figures 2C and S2B) despite no major changes in the overall expression levels of mitochondrial fission-fusion proteins (Figures S2F and S2G), suggesting the occurrence of post-translational modifications of the existing fission-fusion machinery (Anton et al., 2013, Motori et al., 2013). Yet morphometric analysis of reconstructed mitoYFP+ astrocytes revealed that, irrespective of their “metabolic” state, the mitochondrial network was usually composed of highly heterogeneous morphologies, with both tubular and very long (up 8–10 μm) as well as much shorter organelles (less than 0.5 μm) (Figure 2C). This morphological diversity became apparent when plotting the length versus sphericity of the whole mitochondrial population of reconstructed astrocytes selected for their close proximity to the lesion track (Figure 2D). By 7 days post-SW, the mitochondrial network displayed a significant shift toward fragmentation with more than 60% of the whole mitochondrial population being <1 μm in length, in contrast to a 43.5% in control astrocytes (Figure 2D). Whole-cell, time course analysis during a period ranging from 3 days to 2 months after SW injury revealed that while the fraction of fragmented mitochondria sharply increased during the first week, the network was restored to levels comparable to control astrocytes by the third week (Figure 2E). This trend was mirrored by opposite changes in the proportion of tubular mitochondria, confirming that reactive astrocytes proximal to the lesion undergo a time-dependent remodeling of their mitochondrial network over the course of several weeks after injury (Motori et al., 2013).
Figure 2.

Dynamic Remodeling of Astrocyte Mitochondrial and ER Networks following Injury
(A) Experimental design.
(B) Example of an hGFAP::CreER x R26LSL-mitoYFP mouse at 7 days after cortical SW injury. Inset: extravasating CD45+ leukocytes in the lesion core. Scale bar, 150 μm.
(C) Surface rendering of mitoYFP+ control (uninjured animals) or reactive astrocytes proximal to the lesion track. Arrowheads point to the soma. Zooms depict the network morphology in branches. Scale bar, 15 μm.
(D) Density plots depicting the mitochondrial heterogeneity in resting (Ctrl, uninjured animals) or reactive astrocytes (SW 7 days). Threshold values for mitochondrial sphericity (0.8) and length (1 μm) are shown.
(E) Time course of mitochondrial fragmentation quantified as in (D) (n ≥ 3 mice/time point, 8–15 astrocytes/mouse; one-way ANOVA followed by Dunnett’s post hoc test).
(F) Volume reconstruction of mitoYFP+ reactive astrocytes (arrowheads) surrounding dextran-labeled vessels. Scale bar, 25 μm.
(G) Vessel cross-sections showing astrocytic mitoYFP in control (uninjured animals) and injured conditions. Scale bar, 10 μm.
(H) Quantification of perivascular mitoYFP (n ≥ 30 vessels/time point; nonparametric Kruskal-Wallis test).
(I) Experimental design for analyzing the astrocytic ER.
(J) 3D example of an astrocyte expressing ER-GFP (signal density shown in pseudocolors). Scale bars, 10 and 5 μm.
(K) Quantification of the ER-GFP perivascular g-ratio at the indicated time points (n ≥ 35 vessels/time point; nonparametric Kruskal-Wallis test). ∗∗p < 0.01, ∗∗∗p < 0.001.
See also Figure S2.
Interestingly, inspection of microvessels proximal to the lesion (labeled via either dextran injection or CD31 immunostaining) revealed a conspicuous accumulation of astrocytic mitochondria in perivascular endfeet (Figures 2F and S2C; Video S3). In particular, analysis of vessel cross-sections disclosed that the extent of perivascular mitochondria markedly increased by 7 and 28 days after SW (Figures 2G and 2H), the latter being a time when mitochondrial morphology had already normalized back to control levels (Figure 2E). In contrast, mitochondrial density in peripheral branches and total mitochondrial mass in astrocytes (examined via mitoYFP quantification and by label-free proteomic analysis of markers associated with mitochondrial biogenesis/mass in sorted astrocytes) appeared only mildly affected (Figures S2D, S2E, and S2H). We next assessed whether the ER may also undergo a similar extent of remodeling in response to injury. Reactive astrocytes expressing ER-GFP retained a significant amount of ER in perivascular endfeet surrounding CD31+ vessels (Figure S2C). Three-dimensional reconstruction of individual ER-GFP-expressing astrocytes in conjunction with dextran labeling revealed the whole distribution of the ER network across distinct astrocytic territories in uninjured hemispheres (Figures 2J and S2I). To investigate changes in perivascular ER dynamics and normalize these to putative variations in microvessel diameter, we calculated a perivascular ER-GFP “g-ratio” (Figures 2K and S2J). We found a time-dependent increase in the thickness of perivascular ER-GFP signal, which peaked by 7 days post-SW but reverted to near-basal conditions by 28 days (Figure 2K). These results corroborated volume distribution analysis of the ER-GFP signal (i.e., signal density) across astrocytic compartments (Figure S2I). In control astrocytes, perivascular endfeet accounted for 19.2% of all ER-GFP signal (Figure S2L). In contrast, in injury-induced reactive astrocytes, accumulation of ER-GFP signal was observed in the endfeet (35.9%) at the expense of main branches (where the relative ER-GFP proportion decreased from 39.1% in controls to 25.2% in injured samples) (Figures S2K–S2M). Interestingly, by 28 days after SW, the relative distribution of ER-GFP signal mostly normalized (Figures S2K–S2M), suggesting that in contrast to the enduring response of the mitochondrial network in perivascular endfeet (Figure 2H), remodeling of the ER compartment may only be temporary. Together, these data reveal that mitochondrial and ER networks undergo a regionalized morphological rearrangement in perivascular endfeet of astrocytes reacting to acute injury.
Two-photon imaging scan through a reconstructed cortical volume containing mitoYFP-expressing astrocytes and dextran-labeled vessels, showing the extent of perivascular mitochondrial clustering at 7 days post-SW
Conditional Deletion of Mfn2 Disrupts Perivascular Mitochondria-ER Contact Sites in Astrocytes
The reversible transition in mitochondrial network morphology (Figure 2E), together with the peculiar remodeling of mitochondria within the endfeet of reactive astrocytes (Figures 2G and 2H), suggests that mitochondrial dynamics may play an important role in astrocytes reacting to injury. We reasoned that preventing mitochondrial re-tubulation may provide a valid approach to dissect the specific role of this remodeling for astrocyte physiology (Figure 3A). We opted for the conditional deletion of MFN2, which is a key effector of mitochondrial fusion dynamics but also plays a role in maintaining mitochondria-ER tethering domains (de Brito and Scorrano, 2008). We generated astrocyte-specific Mfn2cKO mice by crossing Mfn2 floxed mice (Lee et al., 2012) with the inducible hGFAP-CreER × mitoYFP floxed-stop line (Figures S3A and S3B) and validated protein depletion via mass spectrometry analysis of astrocytes sorted from brain cortices following tamoxifen treatment (Figure 3B). In contrast to classic astrocytic markers (i.e., GLAST, GLT-1, ALDH1L1, and AQP4) or other reference mitochondrial proteins (OPA1 and TOMM40), MFN2 was specifically and markedly downregulated (more than 9-fold; Figure 3B). Transmission electron microscopy (TEM) revealed fewer and circular mitochondria of significant size within the endfeet of Mfn2cKO astrocytes, in net contrast to Mfn2WT samples, in which elongated and branched morphologies were observed lining the basal lamina of the microvasculature (average vessel diameter of 3.5 ± 0.6 μm in Mfn2WT and 3.6 ± 0.6 μm in Mfn2cKO) (Figures 3C and 3E). However, close inspection of perivascular endfeet revealed that the overall distribution of the ER was not overtly affected in Mfn2cKO astrocytes, with long stretches of ER tubule surrounding the basal lamina as in Mfn2WT astrocytes (Figure 3D). Interestingly, Mfn2cKO mitochondria were less enriched in ER contact sites despite the nearby presence of abundant ER membranes (Figures 3C–3E). Notably, deletion of Mfn2 did not visibly affect mitochondrial cristae morphology within the examined time frame (4 weeks post-tamoxifen treatment) (Figure 3C). Together, these results indicate that conditional deletion of Mfn2 in adult astrocytes leads to mitochondrial morphological changes and a concomitant reduction in mitochondria-ER contact sites within the endfeet.
Figure 3.

Mfn2 Deletion Affects Astrocytic Mitochondria-ER Tethering Domains
(A) Proposed model.
(B) Validation of Mfn2 knockout in astrocytes by MACS enrichment and proteomic analysis (n = 4 Mfn2cKO mice and 3 Mfn2WT mice).
(C) EM pictures of astrocytic endfeet at 4 weeks post-tamoxifen treatment. Organelles are highlighted in different colors. Right panels: zooms of mitochondrial cristae. EC, endothelial cell; BL, basal lamina. Scale bars, 1 μm and 200 nm.
(D) Perivascular distribution of ER tubules and their contact sites with mitochondria. Scale bars, 250 nm.
(E) Quantification of the indicated parameters in Mfn2WT (n = 85 cross-sections from 4 mice) and Mfn2cKO perivascular endfeet (n = 145 cross-sections from 3 mice; nonparametric Mann-Whitney t test). ∗∗∗p < 0.001.
See also Figure S3.
Astrocyte-Specific Mfn2 Deletion Abrogates Perivascular Remodeling of Still Functional Mitochondria
We next asked the question whether Mfn2 deletion would be sufficient to prevent astrocyte mitochondrial remodeling in response to acute injury. Histological and protein examination (i.e., via label-free proteomic analysis) of astrocytes derived from lesioned Mfn2cKO animals revealed no overt abnormalities in the extent of GFAP or Vimentin expression (i.e., classic markers of reactivity) as well as other markers of astrocytic reactivity (Liddelow et al., 2017) at 7 days post-SW (Figures 4A, S3C, and S3D). At the single-cell level, however, mitochondrial morphology appeared significantly fragmented even in uninjured conditions, confirming loss of MFN2 and the consequent lack of mitochondrial fusion, starting as soon as 1 week after tamoxifen-induced recombination (Figure S4A). In contrast, the ER network retained an overall intact morphology in the absence of MFN2 (Figure S4B). Interestingly, conditional deletion of Mfn1 resulted in somewhat heterogeneous and less pronounced morphological changes (Figure S4A), suggesting either differences in the relative expression levels of the two mitofusins or potential compensatory effects in the expression levels of MFN2 following Mfn1 deletion (Figure S3E) (Kulkarni et al., 2016). Single-cell, time course analysis of mitochondrial morphology revealed that both Mfn2cKO and Mfn1cKO astrocytes retained the capability to undergo further fragmentation following SW (Figure 4B). In particular, by 7 days post-SW, the overall proportion of fragmented versus tubular mitochondria was almost indistinguishable between examined groups (Figure 4B). However, while by 28 days post-SW wild-type and Mfn1cKO astrocytes efficiently reformed a tubular network, Mfn2cKO astrocytes lacked this ability and were left with visibly fragmented mitochondria (Figures 4B and 4C). Importantly, perivascular mitochondrial clustering was significantly impaired in Mfn2cKO astrocytes proximal to the lesion site, in contrast to wild-type and Mfn1cKO astrocytes, in which the extent of mitoYFP signal essentially doubled (Figures 4D, 4E, and S4C; Videos S4, S5, and S6). Conspicuously, TEM analysis of Mfn2cKO astrocytes confirmed a marked reduction in mitochondrial density and mitochondria-ER contact sites in perivascular endfeet despite intact mitochondrial cristae and abundant ER tubules (Figures S4D–S4F).
Figure 4.

Mfn2 Deletion Prevents Astrocytic Perivascular Clustering of Still Functional Mitochondria
(A) Example of cortical SW injury in Mfn2cKO mice at 7 days. Inset: CD45+ leukocytes in the lesion core. Scale bar, 100 μm.
(B) Time-course analysis of mitochondrial fragmentation in Mfn2cKO and Mfn1cKO astrocytes (n ≥ 3 mice/time point, 8–15 astrocytes/mouse; two-way ANOVA followed by Tukey’s post hoc test).
(C) Mitochondrial morphologies in astrocytes (arrowheads point to soma) proximal to the lesion site at 28 days post-SW. Zooms depict peripheral branches. Scale bar, 20 μm.
(D) Top view projections (100 μm deep) of whole-mount injured cortices (7 days) following tissue clearing. The injury site is indicated by a dashed yellow line. Middle panels: zooms of astrocytes proximal to the lesion track. Right panels: vessel cross-sections. Scale bars, 50, 10, and 10 μm.
(E) Quantification of mitoYFP perivascular density (n = 3 mice/condition, at least 80 vessel sections quantified; the contralateral sides were utilized as controls; one-way ANOVA followed by Holm-Sidak’s post hoc test).
(F) Experimental approach.
(G) Heatmaps of TCA cycle and associated enzymes in reactive Mfn2cKO and Mfn1cKO astrocytes. Asterisks indicate significant changes (−log10 of the p ≥ 1.3) (n = 4 Mfn2cKO mice, 4 Mfn1cKO mice and 3 Ctrl mice).
(H) Atom transition model of 13C labeling after 13C6-glucose supplementation.
(I and J) Graphs depicting the mass isotopomer enrichment analysis for the indicated metabolites (n = 5 Mfn2cKO mice, 6 Mfn1cKO mice, and 5 Ctrl mice; two-way ANOVA followed by Dunnett’s test).
(K) Heatmaps of proteins regulating Ca2+ transport. Asterisks indicate significant changes (−log10 of the p ≥ 1.3) (n = 4 Mfn2cKO mice, 4 Mfn1cKO mice, and 3 Ctrl mice). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
See also Figures S3 and S4.
Two-photon imaging scan through a portion of the injured cerebral cortex (vasculature labeled via dextran-red) containing mitoYFP-expressing control astrocytes at 7 days post-SW. Occasional blood cells are visible in the lumen as a result of auto-fluorescence overlapping with the GFP spectrum
Two-photon imaging scan through a portion of the injured cerebral cortex (vasculature labeled via dextran-red) containing mitoYFP-expressing Mfn2cKO astrocytes at 7 days post-SW. Occasional blood cells are visible in the lumen as a result of auto-fluorescence overlapping with the GFP spectrum
Two-photon imaging scan through a portion of the injured cerebral cortex (vasculature labeled via dextran-red) containing mitoYFP-expressing Mfn1cKO astrocytes at 7 days post-SW. Occasional blood cells are visible in the lumen as a result of auto-fluorescence overlapping with the GFP spectrum
In vivo 2-photon imaging of a representative GCaMP3-expressing astrocyte via cranial window implantation and following vasculature labeling. The recording (total duration 3 min, 1.16 frames per s) has been accelerated to fit into 20 s
The preserved cristae structure in reactive Mfn2cKO astrocytes raised the question of whether these mitochondria were still metabolically competent. Ingenuity pathway analysis (IPA) of our proteomic dataset (Figure 4F) disclosed the OXPHOS pathway among the Mfn2cKO-specific, downregulated hits in our samples (Figure S3F), yet detailed inspection of mitochondrial respiratory chain complexes indicated that only a few of the detected subunits in complexes I, III, IV, and V were significantly downregulated (Figure S3G). Likewise, proteins associated with mitochondrial stress responses revealed that only few of them were significantly upregulated in Mfn2cKO astrocytes (Figure S3H), suggesting that absence of MFN2 brings about only a modest mitochondrial dysfunction on top of potential changes induced by injury itself. Interestingly, we observed a general upregulation in the protein expression levels of enzymes associated to the tricarboxylic acid (TCA) cycle (Figure 4G) and the catabolism of amino acids and their derivatives (Figure S3F), which have emerged as hallmarks of mitochondrial metabolic rewiring in multiple cell types (Chen et al., 2018). Of note, a similar upregulation was found in Mfn1cKO astrocytes (Figures 4G and S3F). However, targeted metabolomics of sorted astrocytes following systemic infusion of 13C6-glucose (Figure 4F) revealed no changes in the incorporation of glucose-derived carbon into TCA cycle intermediates or amino acids between control, Mfn2cKO, and Mfn1cKO astrocytes (Figures 4H–4J), indicating that mitochondrial bioenergetics are not overtly compromised in reactive Mfn2cKO astrocytes up to 4 weeks post-SW.
Altogether, these results indicate that while conditional Mfn2 deletion in reactive astrocytes prevents perivascular enrichment of mitochondria and mitochondria-ER contact sites, mitochondrial cristae structure and function remain to a large degree unaffected after injury.
Lack of MFN2 Dampens Astrocytic Mitochondrial Ca2+ Uptake and Leads to Abnormal Perivascular Ca2+ Transients after SW Injury In Vivo
The lack of perivascular mitochondrial clustering and the reduction in mitochondria-ER contact sites in reactive Mfn2cKO astrocytes provides an opportunity for investigating potential functional consequences confined to the endfoot. Interestingly, a calcium transport pathway in our IPA analysis was selectively upregulated in Mfn2cKO astrocytes (Figure S3F). In particular, protein levels of Ca2+ channels and transporters in mitochondrial as well as plasma membranes were specifically increased in Mfn2cKO astrocytes (Figure 4K). We thus focused our analysis on local astrocytic Ca2+ dynamics (Volterra et al., 2014), as mitochondria-ER tethering domains play a major role in mitochondrial Ca2+ uptake and thus in shaping cytosolic Ca2+ transients (Csordás et al., 2018, Rizzuto et al., 2012).
We first delivered an astrocyte-specific AAV expressing the calcium indicator GCaMP6f targeted to the mitochondrial matrix (mitoGCaMP6) into the cortex of Mfn2cKO or control littermates and concurrently inflicted a SW lesion in the injected area (Figure 5A). We then conducted two-photon laser scanning microscopy (2PLSM) at 7 or 28 days after SW in freshly prepared brain slices. Imaging was carried out in sessions of 3 min each, during which mitochondrial dynamics—as examined via photoactivatable mito-GFP experiments—were negligible (Figures S5A–S5C). We also developed a dedicated algorithm (which we termed AstroSparks; see STAR Methods) permitting a semi-automated identification and quantification of spontaneous mitochondrial Ca2+ transients, including their activity, frequency, amplitude, and duration (Figure 5B). This allowed us to reveal that, in resting astrocytes, perivascular mitochondria are intrinsically more active but display a lower amplitude than mitochondria localized in branches (Figures 5C and 5D). Analysis of Mfn2cKO astrocytes (Figure 5E) disclosed an intrinsically lower mitochondrial Ca2+ activity within their endfeet (46.6% active regions of interest [ROIs] of all ROIs per cell) as compared with that in Mfn2WT astrocytes (68.2% active ROIs of all ROIs per cell) (Figure 5F). Interestingly, following SW injury, Mfn2WT astrocytes displayed a peculiar pattern of mitochondrial Ca2+ uptake that mirrored the morphological changes in mitochondrial network architecture described in Figure 2E: by 7 days (i.e., the peak of fragmentation), the extent of active mitochondria was visibly reduced (55.3% of all ROIs per cell), whereas by 28 days (when tubular morphology had been re-established), this percentage had reverted to levels comparable with uninjured conditions (62.8%) (Figure 5F). Likewise, most of the other parameters pertaining to Ca2+ events, particularly their frequency and duration, also followed a reversible pattern in Mfn2WT astrocytes (Figure 5G). In contrast, mitochondria in Mfn2cKO astrocytes were virtually unresponsive to injury (Figures 5F and 5G). In particular, the values of frequency, amplitude, and duration of Ca2+ transients were already diminished in absence of any injury and compared well with the 7-day time point of the Mfn2WT group (Figure 5G), suggesting that alterations in mitochondrial morphology (i.e., fragmentation) and mitochondria-ER tethering per se are, at least in part, responsible for the changes in mitochondrial Ca2+ uptake observed here.
Figure 5.

Compromised Mitochondrial Ca2+ Uptake and Abnormal Cytosolic Activity in Mfn2cKO Astrocytic Endfeet
(A) Experimental design.
(B) MitoGCaMP6-expressing astrocyte following AstroSparks processing and ROI detection. Inset displays cytosolic mCherry. Scale bar, 10 μm. Right panels: individual ROI traces and corresponding raster plot.
(C and D) Quantification of mitochondrial Ca2+ transient parameters, including active fraction, frequency, duration, and amplitude, in Mfn2WT astrocytes (n = 41–53 cells from 3 mice).
(E) Example of an Mfn2cKO astrocyte. Scale bar, 10 μm.
(F) Quantification of active mitochondria in Mfn2WT (n = 40–56 cells, 3 mice/condition) and Mfn2cKO (n = 36–73 cells, 2–3 mice/condition) astrocytic endfeet.
(G) Ca2+ transient parameters of the astrocytes in (F).
(H) Mitochondrial and cytosolic Ca2+ traces.
(I) Experimental in vivo setting.
(J) Example of GCaMP3-expressing astrocyte following AstroSparks processing. Scale bar, 20 μm.
(K) ROI traces and corresponding raster plots of Mfn2WT and Mfn2cKO astrocytes.
(L and M) Average frequency (endfeet) (L) and area of Ca2+ transients (M) in Mfn2WT (n = 35–111 cells, 2–3 mice/condition) and Mfn2cKO astrocytes (n = 51–73 cells, 2–3 mice/condition). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (nonparametric Mann-Whitney t test).
See also Figure S5.
Analysis of slices containing cytoGCaMP6-expressing astrocytes revealed plain differences with regard to cytosolic Ca2+ transients as compared with mitochondria (Figures 5H, S5D, and S5E). In particular, cytosolic transients in uninjured Mfn2WT astrocytes were markedly shorter in duration and, on average, higher in frequency than mitochondrial ones (Figures 5H, S5E, and S5F), consistent with a buffering role played by mitochondria (Rizzuto et al., 2012). SW injury in Mfn2WT astrocytes significantly modified perivascular cytosolic transients at 7 days (Figure S5F), yet these changes were not fully reversed by 28 days post-SW, suggesting the emergence of long-lasting alterations that may persist up to 1 month. Notably, the frequency of transients was significantly altered in resting astrocytes upon conditional deletion of Mfn2, but not Mfn1 (Figures S5E and S5F), and culminated in an exaggerated Ca2+ activity (i.e., frequency and amplitude of events) by 28 days post-SW (Figure S5F), thus validating our Mfn2cKO proteomic dataset (Figure 4C). Interestingly, similar changes in Ca2+ activity were also observed in astrocyte branches (Figure S5G), suggesting that lack of MFN2 affected mitochondrial and cytosolic Ca2+ frequency dynamics to a comparable extent in all astrocytic territories.
To examine in detail Ca2+ dynamics within an intact neurovascular unit, we next imaged astrocytes in anesthetized animals in vivo following cranial window implantation (Figure 5I). For these experiments, we utilized the inducible cytosolic reporter line GCaMP3 floxed-stop (Zariwala et al., 2012) and concurrently labeled the vasculature via systemic dextran injection to unambiguously identify perivascular endfeet in vivo (Figure 5J). Analysis of Ca2+ frequency confirmed that Mfn2WT astrocytes undergo substantial alterations in response to SW injury peaking at 7 days and persisting up to 28 days (Figures 5L and S5G). Importantly, by this time, Mfn2 deletion led to an abnormal frequency of Ca2+ events, which resulted in significantly higher rates of perivascular transients (0.64 ± 0.03 events/min/domain in Mfn2cKO astrocytes versus 0.46 ± 0.01 events/min/domain in controls) (Figures 5K and 5L). Although this phenotype was present both in endfeet and branches (Figure S5G), analysis of the spatial spreading of Ca2+ transients within astrocytic territories revealed that prominent and enduring changes (i.e., broader transients) up to 28 days post-SW were a unique feature of perivascular compartments in astrocytes lacking MFN2 (average transient size of 4.98 ± 0.67 μm2 in Mfn2cKO versus 3.25 ± 0.33 μm2 in Mfn2WT) (Figure 5M). This hallmark was masked at 7 days post-SW, when control astrocytes also showed broader transients presumably due to their conspicuous mitochondrial fragmentation and reduced mitochondrial Ca2+ uptake (Figure 5G), yet this specificity for the endfeet indicates that enrichment of mitochondria-ER contact sites at this location helps to demarcate a region of distinctive Ca2+ signaling and, possibly, metabolic supply.
Astrocyte Mitochondrial Fusion Dynamics Are Required for Vascular Remodeling following Injury
To understand if the observed structural and functional changes in perivascular endfeet of Mfn2cKO astrocytes may have direct consequences for vascular remodeling, we analyzed the vascular plexus following SW injury. Injured mice were intravenously infused with dextran-red shortly before sacrifice and their cortices processed for clearing and 2PLSM to obtain a complete overview of the vascular network architecture (Figure 6A). Top views of the first 600 μm deep into the cortex revealed that uninjured hemispheres were virtually undistinguishable among Mfn2cKO and Mfn2WT mice, showing comparable density and integrity of the labeled vasculature (Figure 6B). By 7 and 28 days post-SW, however, we found a prominent rarefication of the vasculature within the lesion core of Mfn2cKO mice as compared with Mfn2WT mice, suggesting an impairment in vascular remodeling (Figure 6B). To quantify these changes, we optimized a filament tracing analysis utilizing dextran labeling as a mask signal for our volumetric reconstructions (Figures 6C and S6A; STAR Methods) and performed a time-course analysis in injured Mfn2WT and Mfn2cKO mice. At the earliest analyzed time (3 days), we identified a similar reduction in the density of branch points, total length, and fractional vascular volume of the network immediately surrounding the injury track as compared with uninjured conditions (Figure 6D). Yet while by 7 days the Mfn2WT group showed partial recovery of these parameters (in particular branch points), which became conspicuous by 28 days, Mfn2cKO cortices failed in undergoing significant improvement (Figure 6D). Interestingly, injured Mfn1cKO mice displayed a recovery rate similar to wild-type mice (Figures S6B and S6C), in line with the fact that disruption of MFN1 expression did not prevent perivascular clustering of mitochondria (Figures 4K and 4L).
Figure 6.

Impaired Angiogenesis and Vascular Remodeling in Mfn2cKO Mice
(A) Experimental approach.
(B) Top views of reconstructed vascular networks. Arrowheads point to the lesion track. Insets: zooms of the lesioned core region (circled in white). Scale bar, 200 μm.
(C) Pipeline used for vasculature quantification. Scale bar, 30 μm.
(D) Vascular network analysis (n = 3 mice/condition; two-way ANOVA followed by Tukey’s post hoc test).
(E) Experimental protocol for EdU labeling. Lower pictures: views of injured cortices. Scale bar, 200 μm.
(F) Magnification showing the presence of proliferating endothelial cells (CD31+/ERG+).
(G) Quantification of proliferating ERG+ cells at 7 days post-SW (n = 4–5 mice/condition; nonparametric Mann-Whitney t test). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
See also Figures S6 and S7.
To gain insights into the phenotype of Mfn2cKO mice, we examined the angiogenic response to brain injury at 7 days post-SW. Mice were treated with EdU during the last 3 days before sacrifice (Figure 6E) and labeled cells examined for their positivity to the ETS-transcription factor ERG, an endothelial marker known to promote angiogenesis (Birdsey et al., 2008). In Mfn2WT mice, we found a number of EdU+/ERG+ cells along CD31+ vessels, indicative of neoformed vessels (Figure 6F). Importantly, in Mfn2cKO mice, both the overall extent of CD31+ vessels and the density of EdU+/ERG+ cells were markedly reduced (Figures 6E and 6G), despite unaffected numbers of total proliferating EdU+ cells as well as SOX2+ astrocytes (Figures S6D–S6F). Use of a Cre-dependent cytosolic tdTomato reporter further corroborated lack of changes in the density of astrocytes between injured Mfn2cKO and Mfn2WT mice (Figures S7A and S7C). Importantly, inspection of perivascular endfeet revealed no overt alterations as compared with that in Mfn2WT mice (Figure S7B), thus ruling out possible degenerative processes.
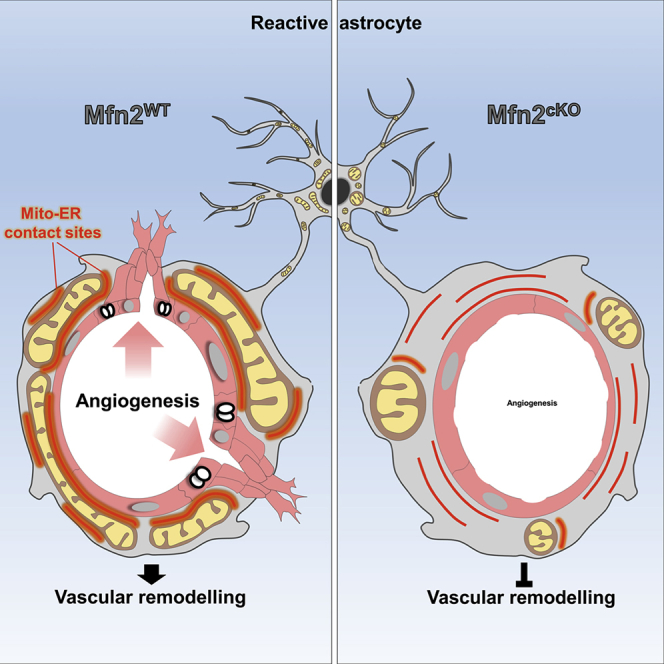
Together, these results indicate that lack of MFN2 in reactive astrocytes compromises injury-induced vascular remodeling by limiting angiogenesis.
Forced Enrichment of Mitochondria-ER Contact Sites in Perivascular Endfeet Rescues Vasculature Remodeling in the Absence of Mitochondrial Fusion
Although lack of MFN2 in reactive astrocytes is sufficient to impair the formation of new vessels, it remains unclear whether this effect is mediated by defective mitochondrial fusion rather than disrupted mitochondria-ER tethering. To address this question, we took advantage of a previously validated strategy to forcefully expand mitochondria-ER contact sites using a genetically encoded synthetic linker (Csordás et al., 2006) that we expressed in Mfn2cKO mice via an astrocyte-specific AAV (pAAV-hGfaABC1D-OMM-mRFP-ER) (Figure 7A). We reasoned that expression of this irreversible linker, which results in mRFP labeling of the outer mitochondrial membrane (OMM), may enhance the extent of contact sites irrespective of subsequent changes in morphology and fission-fusion dynamics. The construct encoding for this linker contains a monomeric RFP fused on one side to the OMM-targeting sequence of mAKAP1 and on the other to the ER membrane-targeting sequence of yUBC6 (Figure 7A) (Csordás et al., 2006). Few weeks after intracortical delivery of this AAV-linker (or its AAV control lacking the ER targeting sequence), mice were treated with tamoxifen to induce Mfn2 deletion followed by SW injury and mitochondrial network analysis (Figure 7A). At the single-astrocyte level, the amount of mRFP+ mitochondria decorating vessel cross-sections was visibly increased in astrocytes transduced with the AAV-linker as compared with that of controls in both resting and injured conditions (Figures 7B–7D). Importantly, this effect was independent of mitochondrial morphological changes within the endfeet, as AAV-linker expression was not able to restore tubular mitochondria (Figure S7D). To understand if this manipulation also functionally modified the perivascular microenvironment, we introduced a cassette encoding for mitoGCamp6f in the AAV-linker construct and performed Ca2+ imaging in brain slices following AAV cortical delivery in vivo (Figures S7E and S7F). Analysis of resting Mfn2cKO astrocytes revealed that AAV-linker transduction modified the extent of mitochondrial Ca2+ uptake by increasing both the percentage of active mitochondria and their frequency dynamics (Figures 7E, S7G, and S7H) to levels almost comparable to Mfn2WT astrocytes (Figures 5F and 5G), indicating that this forced tethering was sufficient to enhance mitochondrial Ca2+ uptake in absence of MFN2.
Figure 7.

Forced Astrocytic Mitochondria-ER Tethering Rescues Vascular Remodeling in Injured Mfn2cKO Mice
(A) Experimental approach.
(B) Example of an AAV-linker-expressing Mfn2cKO astrocyte (arrowhead points to the soma). Zooms depict the vessel cross-section. Scale bar, 10 μm.
(C) Examples of vessel cross-sections following AAV expression in Mfn2cKO astrocytes. Scale bar, 5 μm.
(D) Quantification of perivascular mitochondrial density (n ≥ 21 vessel sections; one-way ANOVA followed by Tukey’s post hoc test).
(E) Quantification of mitochondrial Ca2+ uptake in Mfn2cKO astrocytes (n ≥ 30 cells; nonparametric Mann-Whitney t test).
(F) Vasculature density in Mfn2WT and Mfn2cKO sections (dashed line: lesion track). Scale bar, 80 μm.
(G) Vasculature density in AAV-transduced Mfn2cKO cortices. Scale bar, 80 μm.
(H) Quantification of CD31 area fraction (n ≥ 3 mice/condition; one-way ANOVA followed by Tukey’s post hoc test). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
See also Figure S7.
We next analyzed the extent of vasculature remodeling induced by SW injury in the area subjected to AAV transduction. Visual inspection of CD31 immunoreactivity confirmed that non-transduced Mfn2cKO cortices were characterized by a less elaborated vascular network in the injured area as compared with Mfn2WT cortices (Figures 7F and 7H). Importantly, while injection of the AAV-ctrl did not overtly change the extent of CD31+ vessels by 7 days post-SW in Mfn2cKO mice, AAV-linker expression significantly enhanced vascular complexity to levels almost indistinguishable from those of Mfn2WT mice (Figures 7G and 7H). Accordingly, AAV-linker expression increased the number of branches and total vascular length in Mfn2cKO cortices as compared with AAV-ctrl expression (Figure S7I). Together, these results indicate that, within the examined time frames, forced enrichment of perivascular mitochondrial-ER tethering in Mfn2cKO astrocytic endfeet is sufficient to restore vascular remodeling following injury.
Discussion
We have shown that a profound reorganization of the mitochondrial network in astrocytes responding to acute injury underlies their ability to create a spatially defined mitochondrial-enriched domain in perivascular endfeet. Astroglial endfeet are naturally enriched in elaborated mitochondrial morphologies and bundles of ER tubules, which is in line with recent observations (Mathiisen et al., 2010, Moss et al., 2016), yet following injury, these cellular sites experience a further accumulation of mitochondria as a result of coordinated fusion-fission dynamics. Although mitochondrial biogenesis or trafficking are also likely to contribute to this process, mitochondrial fusion in particular was required for this localized clustering as deletion of Mfn2 not only prevented this response but also significantly altered the extent of contact sites with the ER, thus affecting local Ca2+ dynamics. Interestingly, we observed distinct temporal changes in the remodeling of mitochondrial and ER networks within perivascular processes of reactive astrocytes, with mitochondrial clustering persisting up to 4 weeks after injury, when the amount of ER membranes had already normalized (Figures 2H and 2K). Yet, the extent of mitochondria-ER contact sites remained conspicuous even at this late time point (Figure S4F), suggesting that remodeling of the ER might have functions exceeding the stabilization of contact sites with mitochondria (Joshi et al., 2017). Importantly, the extent of mitochondria-ER tethering in astrocytic endfeet had direct consequences for microvasculature remodeling. While depletion of contact sites impaired angiogenesis and vascular complexity in lesioned cortices, artificial tethering had opposite results and rescued vascular density even in absence of mitochondrial fusion. This finding is reminiscent of an equally enhanced accumulation of mitochondria to new axonal sprouts following axotomy experiments, a process which has implications for axon regeneration (Han et al., 2016, Mar et al., 2014, Misgeld et al., 2007). Along this line, our data support the notion that enrichment of mitochondria and mitochondria-ER contact sites in astrocytic endfeet does not simply identify a general trait of cellular reactivity but rather a mechanism that ensures the formation of an active metabolic compartment with direct implications for vascular remodeling.
Our experiments performed on astrocyte-specific Mfn2cKO mice were designed to reveal the acute effects resulting from the lack of mitochondrial fusion during injury. While this may explain the seemingly intact cristae morphology of Mfn2cKO astrocytes, in contrast to developmental knockout studies (Chen et al., 2007, Lee et al., 2012), it is of note that mitochondrial morphological changes were accompanied by a clear reduction in the extent of MAM domains in astrocytic endfeet. In cell lines, MFN2 regulates the extent of mitochondria-ER tethering, with a pro-tethering (de Brito and Scorrano, 2008, Naon et al., 2016) rather than an anti-tethering activity (Filadi et al., 2015) being validated also in vivo (Luchsinger et al., 2016, Schneeberger et al., 2013). In astrocytes, a reduction in MAMs and an increased mitochondria-ER distance supports a similar pro-tethering role of MFN2. However, we cannot entirely exclude that these may partly develop as secondary effects due to morphological changes of the mitochondrial network in perivascular endfeet.
One key finding of our study is that mitochondria-ER contact sites regulate the local environment surrounding microvessels in vivo and ex vivo, as revealed by Ca2+ imaging experiments. Astrocytes possess a remarkably complex Ca2+ activity, which underlies changes in synaptic transmission and vascular tone (Bindocci et al., 2017, Shigetomi et al., 2016). Besides the ER, mitochondria are also well known for being integral components of Ca2+ signaling given their significant buffering capacity via the mitochondrial calcium uniporter (MCU) complex (Baughman et al., 2011, De Stefani et al., 2011). Calcium uptake can potentially modify mitochondrial bioenergetics (Giorgi et al., 2018), but also the magnitude and spread of cytosolic Ca2+ transients and thus have important effects on key signaling events in cells, including astrocytes in vitro and in vivo (Agarwal et al., 2017, Göbel et al., 2018, Jackson and Robinson, 2018). As MCU exhibits low Ca2+ affinity, mitochondrial Ca2+ influx predominantly occurs at sites of elevated Ca2+ concentrations, i.e., mitochondria-plasma membrane and ER tethering domains (Hayashi et al., 2009, Rizzuto et al., 2012). Intriguingly, manipulation of MFN2 expression alters Ca2+ buffering capacity in cells as a consequence of its regulatory role on MAM domains (de Brito and Scorrano, 2008, Filadi et al., 2015, Luchsinger et al., 2016, Naon et al., 2016). Consistent with these earlier reports, here we find that Mfn2cKO astrocytes are virtually insensitive to the mitochondrial Ca2+ uptake dynamics induced by injury, but that Ca2+ uptake can be restored by forced expression of a synthetic linker even in the absence of mitochondrial fusion. Importantly, impaired mitochondrial uptake leads to long-term alterations in cytosolic Ca2+ activity, which, at the level of the endfeet, manifest as Ca2+ transients wider and more frequent than those observed in controls. It is thus tempting to speculate that these abnormal transients may affect astrocytic perivascular function, yet the exact consequences of this aberrant Ca2+ activity for vascular remodeling remain to be clarified. In future studies, it will be interesting to assess if astrocyte-specific manipulation of mitochondrial Ca2+ uptake, for instance via MCU deletion, may similarly affect vascular remodeling in injury settings.
Unexpectedly, we found that abrogation of astrocyte MFN2 and the ensuing disruption of mitochondria-ER contact sites were sufficient to impair angiogenesis after injury. While we suggest this effect to be mediated by a faulty metabolic domain at the gliovascular interface, at this stage, we can only argue what the exact signaling might be that facilitates a vascular response in physiological conditions. Interestingly, MFN2-mediated signaling has been implicated in regulating cell-autonomous proliferation in vascular smooth muscle cells (Chen et al., 2004); however, we did not find overt changes in astrocyte proliferation or survival in our system. In addition, the fact that astrocytes can efficiently sustain a glycolytic metabolism (Supplie et al., 2017) argues against a primary role of OXPHOS in this regard. In line with this notion, we were unable to reveal major changes in TCA cycle metabolism following 13C6-glucose administration in Mfn2cKO astrocytes. Although these data do not rule out potential local metabolic deficits restricted to the perivascular endfeet, mitochondrial cristae ultrastructure appeared intact in Mfn2cKO reactive astrocytes, suggesting that mitochondrial metabolism per se may not be strongly affected in our model. Moreover, AAV-linker expression alone was sufficient to restore vascular remodeling, providing additional evidence for the presence of still functional mitochondria. Thus, one intriguing possibility is that this dense supply of mitochondria-ER contact sites at the vascular interface may contribute to generate locally a metabolic signaling environment (Al-Mehdi et al., 2012, Booth et al., 2016, Lopez-Fabuel et al., 2016), which may act non-cell-autonomously in assisting the angiogenic response during the days that follow the initial insult (Wong et al., 2017). Alternatively, a local supply of key astrocytic biosynthetic intermediates, as those generated by the TCA cycle, or ATP itself may contribute to keep fueling the remodeling of the gliovascular interface (Boulay et al., 2017) as well as restore perivascular barrier (Voskuhl et al., 2009) or clearance functions, in particular of toxic metabolic by-products (Iliff et al., 2012). Ultimately, a combination of multiple factors, possibly converging onto the localized release of pro-angiogenic signaling molecules, are likely to participate in regulating astrocyte-mediated vascular remodeling following injury.
In conclusion, our study provides insights into the changes in mitochondrial structure and function experienced by astrocytes during their response to cerebrovascular damage, but it also identifies an important mechanism through which these cells directly contribute to vascular remodeling in the injured brain. Successful molecular dissection of the precise metabolic pathways playing a role in this process may therefore hold promise for therapeutic interventions to ameliorate tissue repair.
Limitations of Study
In this study, we were unable to detect significant changes in the metabolic profile of Mfn2cKO astrocytes, suggesting that in absence of Mfn2, astrocytic mitochondria retain a certain degree of metabolic competence. An obvious limitation of this study, however, is that we cannot draw conclusions on the precise metabolic state of mitochondria in perivascular processes, as these were underrepresented with our cell sorting strategy. Moreover, by performing metabolomics on isolated cells, we were prevented from interrogating the mitochondrial-specific metabolome, for which recently developed mitochondrial immunocapture approaches may be more appropriate (Bayraktar et al., 2019, Fecher et al., 2019). By taking advantage of further refined sorting procedures and imaging of innovative metabolic sensors, future studies will likely improve our understanding of the regionalized changes in astrocytes and brain tissue metabolism following injury.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| chicken polyclonal anti-GFP | Aves Labs | Cat# GFP-1020; RRID: AB_10000240 |
| rabbit polyclonal anti-RFP | Rockland | #600401379; RRID: AB_2209751 |
| mouse monoclonal anti-GFAP, clone GA5 | Millipore | MAB360; RRID: AB_11212597 |
| rabbit polyclonal anti-GFAP | Millipore | Cat# AB5804; RRID: AB_2109645 |
| rat monoclonal anti-CD45 | BD | #550539; RRID: AB_2174426 |
| rat monoclonal anti-CD31 | BD | #550274; RRID: AB_393571 |
| rabbit monoclonal anti-ERG | Abcam | Cat# ab92513; RRID: AB_2630401 |
| rabbit monoclonal anti-S100ß | Abcam | Cat# ab52642; RRID: AB_882426 |
| rabbit polyclonal anti-SOX2 | Abcam | Cat# ab97959; RRID: AB_2341193 |
| Donkey anti-chicken IgY (H+L), Alexa Fluor 488 | Jackson ImmunoResearch | Cat# 703-545-155; RRID: AB_2340375 |
| Goat anti-Rat IgG (H+L), Alexa Fluor 546 | Thermo Fisher Scientific | Cat# A-11081; RRID: AB_2534125 |
| Donkey anti-Rabbit IgG (H+L), Alexa Fluor 546 | Thermo Fisher Scientific | Cat# A10040; RRID: AB_2534016 |
| Donkey anti-Rat IgG (H+L), Alexa Fluor 647 | Jackson ImmunoResearch Labs | Cat# 712-605-150; RRID: AB_2340693 |
| Donkey anti-Mouse (H+L), Alexa Fluor 647 | Jackson ImmunoResearch Labs | Cat# 715-605-150; RRID: AB_2340862 |
| 4',6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI) | Thermo Fisher Scientific | Cat# D1306; RRID: AB_2629482 |
| CMC-Protein-A-gold | Cell Microscopy Core (Utrecht) | N/A |
| Click-iT EdU Imaging Kit | Thermo Fisher Scientific | Cat# C10338 |
| Bacterial and Virus Strains | ||
| RABV SAD ΔG ER-GFP (EnvA) | This paper | N/A |
| RABV SAD ΔG mito tag-RFP (EnvA) | This paper | N/A |
| RABV SAD ΔG mito-GFP (EnvA) | Motori et al., 2013 | N/A |
| RABV SAD ΔG mito-PA-GFP (EnvA) | This paper | N/A |
| AAV5-GFAP(0.7)-mCherry-peroxisome-2A-eBFP2-WPRE | This paper | N/A |
| AAV5-GFAP(0.7)-emerald-lamp1-2A-mCherry-WPRE | This paper | N/A |
| AAV5-GFAP(0.7)-mito-GCamp6f-P2A-mCherry-WPRE | This paper | N/A |
| AAV5-gfaABC1D-cyto-GCamp6f | Haustein et al., 2014 | N/A |
| AAV5-GFAP(0.7)-AKAP1-mRFP-2A-mitoGCamp6f-WPRE | This paper | N/A |
| AAV5-GFAP(0.7)-AKAP1-mRFP-ER-2A-mitoGCamp6f-WPRE | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tamoxifen | Sigma-Aldrich | Cat# T5648, CAS: 10540-29-1 |
| Corn-oil | Sigma-Aldrich | Cat# C8267, CAS: 8001-30-7 |
| Dextran, Texas Red, 70.000MW | Thermo Fisher Scientific | Cat# D1864 |
| Paraformaldehyde | Sigma-Aldrich | Cat# P6148, CAS: 30525-89-4 |
| Glutaraldehyde 25% | Merck | Cat# 354400, CAS: 113-30-8 |
| Deposited Data | ||
| Mass spectrometry dataset | Mendeley Data | https://doi.org/10.17632/ps8677yrjm.1 |
| Experimental Models: Organisms/Strains | ||
| Mouse: hGFAP-TVA | Holland and Varmus, 1998 | N/A |
| Mouse: Mfn1loxP | Lee et al., 2012 | N/A |
| Mouse: Mfn2loxP | Lee et al., 2012 | N/A |
| Mouse: Yme1lloxP | Anand et al., 2014 | N/A |
| Mouse: hGFAP-CreER | Chow et al., 2008 | N/A |
| Mouse: Glast-CreERT2 | Mori et al., 2006 | N/A |
| Mouse: Gt(ROSA26)SorStop-mito-YFP | Sterky et al., 2011 | N/A |
| Mouse: Gt(ROSA26)SorStop-GCaMP3 | Zariwala et al., 2012 | N/A |
| Mouse: Gt(ROSA26)SorStop-tdTomato | Madisen et al., 2010 | N/A |
| Recombinant DNA | ||
| Plasmid: mito-PAGFP | Addgene | #23348 |
| Plasmid: ER-GFP | Snapp et al., 2006 | N/A |
| Plasmid: mCherry-Peroxisomes-2 | Addgene | #54520 |
| Plasmid: Emerald-Lysosomes-20 | Addgene | #56476 |
| Plasmid: mito-GCamp6f-2A-mCherry | This paper | N/A |
| Plasmid: gfaABC1D-cyto-GCaMP6f | Addgene | #52925 |
| Plasmid: AKAP1-mRFP | Csordas et al., 2006 | N/A |
| Plasmid: AKAP1-mRFP-ER(ubc) | Csordás et al., 2006 | N/A |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/; RRID: SCR_003070 |
| Fiji | Max-Planck-Gesellschaft | http://fiji.sc; RRID: SCR_002285 |
| TurboReg | http://bigwww.epfl.ch/thevenaz/turboreg/ | RRID: SCR_014308 |
| moco plugin | https://github.com/NTCColumbia/moco | N/A |
| FindFoci | http://www.sussex.ac.uk/gdsc/intranet/microscopy/UserSupport/AnalysisProtocol/imagej/findfoci | N/A |
| FACS Aria Fusion cell sorter | BD Biosciences | N/A |
| FACS Diva software | BD Biosciences, version 8.0.1 | N/A |
| Igenuity Pathway Analysis (IPA) | Qiagen | http://www.ingenuity.com/products/pathways_analysis.html; RRID: SCR_008653 |
| GraphPad Prism 7.0 | GraphPad Software | https://www.graphpad.com/scientific-software/prism; RRID: SCR_002798 |
| Origin 2018 | Origin Lab | RRID: SCR_014212 |
| Huygens Professional | Scientific Volume Imaging | RRID: SCR_014237 |
| Adobe Illustrator CS6 | Adobe | RRID: SCR_014198 |
| Photoshop CC 2017 | Adobe | https://www.adobe.com/ |
| Imaris | Bitplane | https://imaris.oxinst.com/; RRID: SCR_007370 |
| Igor Pro v7.0.4.1 | WaveMetrics | http://www.wavemetrics.com/products/igorpro/igorpro.htm; RRID: SCR_000325 |
| AstroSparks | This Paper | N/A |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Matteo Bergami (matteo.bergami@uk-koeln.de). All unique/stable reagents generated in this study are available from the Lead Contact without restrictions. There are restrictions to the availability of mice due to MTA.
Experimental Model and Subject Details
Six to 8-week old C57BL/6 and transgenic mice of mixed genders were used for stereotactic injections, SW-injury, tamoxifen treatments, slice and in vivo imaging. Mice were housed in groups of up to 5 animals per cage supplied with standard pellet food and water ad libitum with a 12 h light/dark cycle, while temperature was controlled to 21-22°C. Mice carrying the loxP-flanked genes Mfn1fl/fl (Lee et al., 2012) and Mfn2fl/fl (Lee et al., 2012) were crossed with the inducible hGFAP-CreERTM (Chow et al., 2008) line and subsequently to the Cre-dependent mitochondrial-targeted mitoYFP (Sterky et al., 2011) or GCamp3 reporter (Zariwala et al., 2012). For validation experiments, Mfn2fl/fl mice were crossed with the astrocyte-specific Glast-CreERT2 line (Mori et al., 2006) in combination with the inducible tdTomato reporter (Madisen et al., 2010). For experiments involving the use of an EnvA-modified Rabies virus to express fluorescent indicators specifically in astrocytes, hGFAP-TVA mice (Holland and Varmus, 1998) expressing the avian membrane-bound TVA receptor under the control of human GFAP promoter were used. All experimental procedures were performed in agreement with the European Union and German guidelines and were approved by the State Government of North Rhine Westphalia.
Method Details
Tamoxifen Treatments
Mice were intraperitoneally injected with tamoxifen (40 mg/ml dissolved in 90% corn oil and 10% ethanol) once a day for a maximum of 5 consecutive days. All subsequent experiments were performed at least one week after the last tamoxifen injection. The exact time frames are indicated in the text for individual experiments.
Stereotactic Procedures and Viral Injections
Mice were anesthetized by intraperitoneal injection of a ketamine/xylazine mixture (130 mg/kg body weight ketamine, 10 mg/kg body weight xylazine), treated subcutaneously with Carprofen (5 mg/kg) and fixed in a stereotactic frame provided with a heating pad. A portion of the skull covering the somatosensory cortex (from Bregma: caudal: -2.0; lateral: 1.6) was thinned with a dental drill avoiding to disturb the underlying vasculature. For unilateral SW-injury, a stainless steel lancet was slowly inserted into the cortex to a depth of 0.8 mm, moved 1 mm caudally and then slowly removed. For virus injection, a finely pulled glass capillary was inserted through the dura (-0.6 to -0.3 from Bregma) and a total of 200-300 nl of virus were slowly infused via a manual syringe (Narishige) in multiple vertical steps spaced by 50-100 μm each during a time window of 10-20 minutes. After infusion, the capillary was left in place for few additional minutes to allow complete diffusion of the virus. After capillary removal, the scalp was sutured and mice were placed on a warm heating pad until full recovery. Physical conditions of the animals were monitored daily to improve their welfare before euthanize them. For cranial window implantation, anesthetized mice received a preemptive subcutaneous injection with Carprofen (5 mg/kg) and dexamethasone (0.25 mg/kg). The scalp was removed and the underlying connective tissue was cleared from the skull. A circular craniotomy (3 mm in diameter) was performed over the posterior parietal cortex using a dental drill and avoiding to disturb the underlying vasculature. During the whole procedure, a saline solution was flushed onto the area exposed with the craniotomy. A sterile 3 mm circular glass coverslip (#1 thickness, Warner Instruments) was gently implanted into the craniotomy site and sealed in place with a thin layer of Sylgard (Sigma) before applying dental cement (Dentalon plus, Heraeus Kulzer GmbH) to fix the coverslip and cover the surrounding exposed skull. An aluminium chamber plate (CP-1, Narishige) was fixed with cement on top of the cover glass to facilitate mouse head immobilization at the 2-photon microscope via a head holder (MAG-2, Narishige). A single tail vein injection of 50 μl Dextran Texas Red (70 kDa, Thermo Fisher, D1864) in saline was used to label the brain vasculature in anesthetized animals. The depth of anesthesia was assessed throughout the surgery and recording time (usually 1-2 hours) and eventually mice received one or more additional boluses of anesthetic each corresponding to one third of the initial dose.
Viral Production
Construction of the glycoprotein (G protein) gene-deleted RABV (SADΔG-mCherry) and virus rescue from pHH-SADΔG-mCherry SC has been described before (Ghanem et al., 2012). cDNAs encoding organelle-targeted fluorescent protein genes were used replace the mCherry ORF of using unique NheI/NotI restriction sites. RABVΔG-mito-tagRFP and RABVdG-mito-PA GFP contains the pre-peptide of human ornithine carbamoyltransferase fused to the N terminus of tagRFP or PA-GFP. ER-targeted GFP contains an N-terminal ER retention sequence (KDEL-GFP, kindly provided by E. Snapp). Viruses pseudotyped with the homologous SAD G glycoprotein were amplified in BSR MG-on cells complementing the G deficiency of the virus upon induction of G expression by doxycycline (Finke et al., 2003) and viruses pseudotyped with the EnvA protein in BHK-EnvARGCD cells expressing an ASLV-A envelope protein comprising the RABV G cytoplasmic tail (Wickersham et al., 2007). The G- or EnvA-coated virus was concentrated by ultracentrifugation and used for in vivo injection. Plaque-forming unit (pfu) number titration was performed by infecting BHK-wt cells and HEK293T-TVA cells with G-coated virus and EnvA-coated virus, respectively. Helper-free AAV vectors were either obtained from Vector Biolabs as custom projects or produced according to standard manufacturer’s instructions (Cell Biolabs). Briefly, 293AAV cells were transiently transfected with a transfer plasmid carrying the desired transgenes along with a packing plasmid encoding the AAV1 capsid proteins and a helper plasmid, using the calcium phosphate method. Crude viral supernatants were obtained via lysing cells in PBS by freeze-thaw cycles in a dry ice/ethanol bath. The AAV vectors were purified by discontinuous iodixanol gradient ultracentrifugation (24h at 32,000 rpm and 4°C) and concentrated using Amicon ultra-15 centrifugal filter unites. Genomic titres were determined by real-time qPCR.
In Vivo and Ex Vivo Imaging
Isolated brains were placed in ice-cold, carbogen-saturated (5% CO2, 95% O2, pH 7.4) artificial cerebrospinal fluid (ACSF) containing (in mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 Glucose, 0.5 CaCl2 and 3.5 MgCl2 (osmolarity of 310-330). 270-300 μm thick coronal slices were obtained using a vibratome (Micron, HM 650V) and transferred into a pre-incubation chamber maintained at room temperature and containing ACSF supplemented with 1 mM CaCl2 and 2 mM MgCl2. During imaging, slices were moved in a dedicated imaging chamber and experiments were conducted under continuous ACSF perfusion at a constant temperature of 32-33°C. Imaging in slices and in vivo was performed using a multiphoton laser-scanning microscope (TCS SP8 MP-OPO, Leica Microsystems) equipped with a Leica 25x objective (NA 0.95, water) and a Ti:Sapphire laser (Chameleon Vision II, Coherent). For Calcium imaging, detection of fluorescence changes of the GCaMP6f sensor in single astrocytes was achieved by tuning the laser to 920 nm. This wavelength also allowed simultaneous recording of Dextran Red signal in experiments in vivo. Two internal HyD detectors (FITC: 500-550 nm, TRITC: 565-605 nm) were utilized to monitor GCaMP6 and Dextran Red signals. Typical recording sessions consisted in 3-5 min of continuous imaging (resolution of 1024x1024 pixels and zoom of 1 or 5) with a frame rate of 1.16 frames /s (0.86 s/frame). Analysis of Ca2+ transients acquired with higher frame rates (up to 10 Hz) yielded comparable results in terms of frequency, amplitude and duration of events, but worsened the overall image quality. For mito-PA-GFP experiments, photoactivation of selected ROIs of individual astrocytes was carried out by tuning the 2-photon laser to 840 nm (10% of laser power for 10-20 seconds), while time-lapse imaging was performed utilizing GFP excitation (920nm) and an internal HyD detector (FITC: 500-550 nm). Usually 2-3 ROIs of identical size per astrocytes were selected in the endfeet and branches and, after photoactivation, the whole astrocyte volume (inter-stack interval of 1 μm) was imaged over the course of at least 1 h every 3 min. Only astrocytes located at least 20-30 μm below the slice surface, with a general healthy appearance throughout the recording time (i.e., absence of visibly fragmented mitochondria) and whose acquisitions displayed only no or a minor spatial drift in xyz during the whole imaging session were included in subsequent analysis. Acquired time points were then merged in a 4-D hyperstack in ImageJ and the resulting 3-D volumes registered utilizing the “Correct 3D drift” plugin in ImageJ. Quantification of fusion events was performed manually by inspecting the volumes including and surrounding the photoactivated ROIs. Fusion events were identified by the abrupt decrease in GFP intensity in directly photoactivated mitochondria due to GFP dilution into the newly appearing (fusing) mitochondria that were not initially photoactivated. In rare cases, mitochondria that simply moved away or through the photoactivated ROIs and did not satisfy these fusion parameters were not considered in our quantification.
Calcium Imaging Analysis
Time-lapse image sequences were drift-corrected by utilizing the “fast & rigid body” options of the TurboReg plugin (http://bigwww.epfl.ch/thevenaz/turboreg/) in ImageJ, aligning each frame to a median projection of eleven frames centered on the middle of the time series. In case of non-satisfactory results, the moco plugin (https://github.com/NTCColumbia/moco) was used alternatively. The image sequence was then cropped to exclude border regions that were not acquired throughout the whole recording period. The noise was reduced with an isotropic (σ = 2 px, xyt) Gaussian Blur filter. Next, only pixels with a median intensity or a peak intensity in a median filtered (radius: 5 px) and background corrected (Subtract Background plugin, options: "rolling=500 sliding disable") image exceeding the threshold of 5 (a.u.) were considered for further analysis. Based on a standard deviation (SD) projection, the FindFoci plugin identified regions of interest (ROIs). The threshold was set to the mean + 3x SD intensity of pixels identified as background by the “IsoData” auto-threshold. ROIs included all neighboring pixels with an intensity higher than a per ROI threshold of: (maximum intensity – background) x 0.4 + background, to compensate for the spreading of bright signals. ROIs smaller than 0.3 μm2 were excluded (in case of the mitoGCaMP6 script, the plugin was used on a median projection to include high, yet stable signals, meaning all mitochondrial ROIs). Next the ΔF/F was calculated based on a median projection reference. ROIs with a high ΔF/F were additionally identified by the FindFoci plugin on a mean filter (radius: 5 px) smoothed maximum projection. Finally, all ROIs were projected onto each other and overlapping ROIs were combined. Once ROIs were identified, the area and average intensity per ROI and time point were handed to IgorPro (v7.0.4.1, WaveMetrics, Lake Oswego, Oregon 97035, USA). Custom written routines identified the duration, amplitude, and frequency of events deviating from baseline. In order to correct for bleaching, all traces were averaged and fitted with an exponential decay function. Based on this reference all traces were corrected. The baseline was identified as follows: the average intensity per ROI was smoothed with a mean-sliding box algorithm (width: 3 time frames). The obtained values were sorted in ascending order and for each rank the standard deviation including all lower ranking values was calculated. To define the threshold at which the SD suddenly increases, i.e. when values start to deviate from baseline and thus increase the SD, the difference in SD (smoothed with a mean-sliding box algorithm (width: 3 frames)) between subsequent ranks was calculated. The rank, at which half maximal difference was reached for the first time, marks the threshold. If the threshold contained less than 15% of all values, the whole trace was defined as baseline. Its mean was used to calculate ΔF/F. In order to identify events, the 20% quantile (of 7 sliding frames) needed to exceed the 80% quantile (of 11 sliding frames) with a time lag of 2.58 s (3 frames) by 1.5 x the SD of the low-cut frequency-filtered (0.2 Hz) ΔF/F trace. For all those events, the end was defined as the earlier time point at which the ΔF/F trace crossed zero or crossed the ΔF/F level just prior to the start of the event.
Tissue Clearing
To assess the structure of the organelles and vasculature in intact cortices, the tissue was cleared using the short ScaleS protocol described previously (Hama et al., 2011). Following brain isolation and overnight post-fixation in 4% PFA at 4°C, the ventral portion of the brain was removed and the remaining dorsal part (including the somatosensory cortex) was placed in ScaleSQ(5) solution for 1 d at 37°C followed by incubation in ScaleS4(0) for another day at 37°C. ScaleSQ(5) was composed of 22.5% D-(-)-sorbitol (w/v), 9.1 M Urea, 5% Triton X-100 (w/v), pH 8.2 and ScaleS4(0) of 40% D-(-)-sorbitol (w/v), 10% Glycerol (w/v), 4 M Urea, 15-25% DMSO (v/v), pH 8.1. The next day, cleared cortices were placed in an imaging chamber filled with ScaleS4(0).
3D Reconstructions and Analysis
For analysis of ER-GFP-expressing astrocytes and vascular networks in 3D, 2PLSM (TCS SP8 MP-OPO, Leica Microsystems, 25x water immersion Objective, resolution of 1024x1024 pixels, zoom factor of 2, frame average of 2 and 1 μm inter-stack interval) was utilized to acquire the desired volumes in cleared cortices and the resulting z-stacks were imported into the Imaris software (version 8.3.1, Bitplane) to obtain a rendered 3-D volume utilizing the acquisition parameters. A surface mask was generated from the resulting volumes of the desired fluorescent signal. To analyse the fractional ER-GFP signal density, the created mask was then utilized to carefully segment the different cellular compartments, including the soma, endfeet (corresponding to the portion of ER-GFP signal surrounding dextran-labelled vessels) and by exclusion the branches. ER-GFP integrated signal density was then calculated for each of these sub-volumes per cell and condition. For analysis of vasculature density in cleared control and injured cortices as shown in Figure 6, z-stacks were acquired at a resolution of 1024x1024 pixels (whole fields of view, 590 x 590 μm scanning from surface of the cortex to the beginning of white matter) with a zoom factor 0.75 and 2 μm inter-stack interval. Magnifications were acquired with a zoom factor of 2, a frame average of 2 and a 1 μm inter-stack interval. Large volume acquisitions imported into Imaris were first cropped in xy on both sides of the lesion track in order to obtain a narrower area of 590 x 350 μm (175 μm on each side of the lesion along the whole track utilizing the dextran signal as reference), and then cropped in z to 600 μm to obtain a final cortical block of 590 x 350 x 600 μm. A surface mask was then generated from the dextran fluorescent signal and utilized to trace the vascular network via a filament tracing algorithm embedded in Imaris. Following filament tracing, volumes were thoroughly inspected for potential artifacts and eventually corrected (Figure S6A) before extracting vascular fractional volume, total length and branching points. For quantifications of vascular network complexity following expression of AAVs as shown in in Figures 7H and S7I, the AngioTool ImageJ plugin (Zudaire et al., 2011) was used.
Immunostainings
Following overnight post-fixation of isolated brains with PFA 4% in PBS, coronal brain sections (40 to 70 μm thick) were prepared using a vibratome (Leica, VT1000 S) and permeabilized in 1% Triton X-100 in PBS for 10 min at RT, followed by brief incubation in 5% BSA and 0.3% Triton X-100 in PBS before overnight immunodetection with primary antibodies diluted in blocking buffer at 4°C on an orbital shaker. The next day, sections were rinsed in PBS 3x 10 min and incubated for 2h at RT with the respective fluorophore-conjugated secondary antibodies diluted in 3% BSA. After washing and nuclear counterstaining with 4’,6-diamidino-2-phenylindole (DAPI, ThermoFisher, 3 μM), sections were mounted on microscopic slides using Aqua Poly/Mount (Polysciences). The following primary antibodies were used: chicken anti-GFP (1:500, Aves Labs, GFP-1020), rabbit anti-RFP (1:500, Rockland, #600401379), rabbit anti-GFAP (1:500, Millipore, ab5804), mouse anti-GFAP (1:500, Millipore, MAB360), rat anti-CD45 (1:500, BD, #550539), rat anti-CD31 (1:50, BD, #550274), rabbit anti-S100β (1:500, Millipore, ab52642). The following secondary antibodies were used (raised in donkey): Alexa Fluor 488-, Alexa Fluor 546-, Alexa Fluor 647- conjugated secondary antibodies to rabbit, mouse, chicken and rat (1:1000, Jackson ImmunoResearch). Images were acquired utilizing a SP8 Confocal microscope (Leica) equipped with a 20x (NA 0.75), 40x (NA 1.3), 63x (NA 1.4) or 100x (NA 1.3) oil immersion objective and further processed with Fiji.
Astrocyte Enrichment via MACS
For astrocyte enrichment the kit “Isolation and cultivation of astrocytes from adult mouse brain” (Miltenyi Biotec) was used according to the manual instructions. In brief, cortical brain tissue was extracted and dissociated enzymatically as well as mechanically. Myelin and cell debris were eliminated and in a subsequent step erythrocytes were removed. Using the anti-ACSA-2 microbeads with the autoMACS Pro Separator, astrocytes were magnetically separated from the suspension. Enriched astrocytes were further processed by mass spectrometry.
Astrocyte Enrichment via FACS
The cortical region exposed to SW-injury (typically 1 mm wide, 2 mm long and spanning the cortex depth but excluding the white matter) was microdissected and dissociated using the “Adult Brain Dissociation” kit from Miltenyi Biotec following the manufacturer’s instructions. Following astrocyte staining with ACSA-2-APC (Clone IH3-18A3, 1:200, Miltenyi Biotec), Hoechst to discriminate between cells and debris (10 μg/ml, Cell Signaling) and 7AAD for viability (2.5 μg, Affymetrix), 20.000 events of control samples were recorded to set appropriate gates and compensations. Cell sorting was performed with a BD FACSAria Fusion equipped with a 100 μm nozzle (20 psi) and five lasers (UV 355 nm, violet 405 nm, blue 488 nm, yellow 561 nm and red 640 nm). Sorted cells were collected in PBS and processed for mass spectrometry analysis.
Transmission Electron Microscopy
Anesthetized mice were transcardially perfused with a fixative solution containing 4% formaldehyde and 2.5% glutaraldehyde in 0.1 M cacodylate buffer. The brain was isolated, cut in 1 mm thick sagittal sections and small portions of upper layers of the cortex were dissected for further processing (for injured cortices, the examined area was dissected according to the location of the lesion track). For EPON embedding, the fixed tissue was washed with 0.1 M sodium cacodylate buffer, incubated with 2% OsO4 in 0.1 M cacodylate buffer (Osmium, Science Services; Caco Applichem) for 2 h at 4°C and washed again three times with 0.1 M cacodylate buffer. Subsequently, tissue was dehydrated using an ascending ethanol series with 15 min incubation at 4°C in each EtOH solution. Tissues were transferred to propylene oxide and incubated in EPON (Sigma-Aldrich) overnight at 4°C. Tissues were placed in fresh EPON at RT for 2 h, followed by embedding for 72 h at 62°C. Ultrathin sections of 70 nm were cut using an ultramicrotome (Leica Microsystems, UC6) with a diamond knife (Diatome, Biel, Switzerland) and stained with 1.5% uranyl acetate at 37°C for 15 min and lead citrate solution for 4 min. For immunogold staining (Tokuyasu technique), fixed tissue (4% PFA and 0.2% GA) was infiltrated with 2.3 M sucrose in 0.1 M phosphate buffer overnight at 4°C, mounted on aluminium pins for cryo-ultramicrotomy and snap-frozen in liquid nitrogen. Ultrathin cryo-sections of 70 nm were cut with a diamond knife (Diatome, Biel, Switzerland) using a Leica UC6 with FC7 at -90°C. Sections were picked up in a 1:1 mixture of 2% methylcellulose (Sigma-Aldrich) and 2.3 M sucrose. After rinsing 3x in PBS and incubation in 0.05 M glycine (Sigma-Aldrich), sections were blocked (2x3 min) with 1% BSA in PBS. For immuno-labelling, sections were incubated with antiserum specific for RFP (Rockland) in blocking buffer, followed by rinsing of 6x in PBS and 90 min incubation with protein A-gold (12 nm, CMC-Utrecht) in blocking buffer. After fixation with 2% glutaraldehyde (3 min), sections were washed in PBS and H2O and contrasted (5 min) with uranyl acetate (0.4% in 2% methylcellulose) on ice. Sections were picked up with a wire loop. Excess fluid was drained from the loop by gentle tapping to Whatman filter paper, and sections were embedded in the remaining thin film by air-drying. Electron micrographs were taken with a JEM-2100 Plus Transmission Electron Microscope (JEOL), equipped with Camera OneView 4 K 16 bit (Gatan) and software DigitalMicrograph (Gatan). For analysis, electron micrographs were acquired with a digital zoom of 5000x or 6000x. The area, perimeter and circularity of each mitochondrion was determined in ImageJ following manual drawing of single organelles. Mitochondrial density was assessed by quantifying the absolute number of mitochondria per measured astrocytic area or length of basal lamina. A similar approach was utilized to quantify the extent of mitochondria-ER contact sites (defined as sites of contact within a reciprocal distance of 50 nm) and minimal mitochondria-ER proximity. All parameters obtained from one field of view (usually containing several mitochondria and multiple contact sites) were averaged together.
Mass Spectrometry (MS) and Data Analysis
For proteomic analysis, MACS-enriched or FACS isolated astrocytes were lysed in SP3 lysis buffer (4% SDS in PBS) and chromatin was degraded using a Bioruptor (10 min, cycle 30/30 s). Samples were reduced with 5 mM Dithiothreitol (DTT) at 55°C for 30 min, alkylated with 40 mM Chloroacetamide (CAA) at RT for 30 min and protein amount was quantified using the Direct Detect Spectrometer from Merck. Protein digestion was performed using the Single-Pot Solid-Phase-enhanced Sample Preparation approach SP3. In brief, 2 μL of a 10 mg/mL mixture of hydrophilic and hydrophobic carboxylate coated paramagnetic beads (SeraMag Speed Beads, #44152105050250 and #24152105050250, GE Healthcare) were added to each sample. Acetonitrile was added to a final concentration of 50%. Bound proteins were washed with 70% ethanol and 100% acetonitrile. Beads were re-suspended in 5 μL 50 mM Triethylammoniumbicarbonate buffer containing 0.1 μg Trypsin (Sigma) and 0.1 μg LysC (Wako). Digestion was carried out at 37°C for 16 h in a PCR cycler. Recovered peptides were re-suspended in 1% formic acid / 5% DMSO and stored at -20°C prior MS analysis. All samples were analyzed on a Q-Exactive Plus (Thermo Scientific) mass spectrometer that was coupled to an EASY nLC 1000 UPLC (Thermo Scientific). Peptides were loaded with solvent A (0.1% formic acid in water) onto an in-house packed analytical column (50 cm × 75 μm I.D., filled with 2.7 μm Poroshell EC120 C18, Agilent). Peptides were chromatographically separated at a constant flow rate of 250 nL/min using the following gradient: 5-30% solvent B (0.1% formic acid in 80% acetonitrile) within 65 min, 30-50% solvent B within 13 min, followed by washing and column equilibration. The mass spectrometer was operated in data-dependent acquisition mode. The MS1 survey scan was acquired from 300-1750 m/z at a resolution of 70,000. The top 10 most abundant peptides were isolated within a 2 Da window and subjected to HCD fragmentation at a normalized collision energy of 27%. The AGC target was set to 5e5 charges, allowing a maximum injection time of 110 ms. Product ions were detected in the Orbitrap at a resolution of 17,500. Precursors were dynamically excluded for 20 s. All mass spectrometric raw data were processed with Maxquant (version 1.5.3.8) using default parameters. Briefly, MS2 spectra were searched against the Uniprot MOUSE.fasta database, including a list of common contaminants. False discovery rates on protein and PSM level were estimated by the target-decoy approach to 0.01% (Protein FDR) and 0.01% (PSM FDR), respectively. The minimal peptide length was set to 7 amino acids and carbamidomethyolation at cysteine residues was considered as a fixed modification. Oxidation (M) and Acetyl (Protein N-term) were included as variable modifications. The match-between runs option was enabled. LFQ quantification was enabled using default settings. The Maxquant output was processed as follows: Protein groups flagged as “reverse,” “potential contaminant,” or “only identified by site” were removed from the proteinGroups.txt. LFQ values were log2 transformed. Proteins with less than 2 valid values were removed. Missing values were replaced by imputation from a normal distribution (width 0.3, down shift 1.8). A two sample t test was used to determine significantly changing protein levels (S0 = 0.1), and a permutation-based FDR was calculated to correct for multiple testing. The obtained data was uploaded into the Ingenuity Pathway Analysis (IPA) software (Qiagen) utilizing a Benjamini adjusted p value of 0.05 or lower to investigate canonical pathways that were significantly changed. Heat map visualization of relative protein abundance was obtained calculating a z-score of the LFQ values for each protein.
13C-glucose Feeding in Mice
Mice were fasted overnight before being anesthetized and receiving a single tail vein injection of 150 μmol of 13C6-glucose (in saline) over the course of 30 seconds. After 30 min, mice were quickly sacrificed and the peri-lesioned cortical area extracted in PBS for astrocyte enrichment via MACS as described above. The resulting astrocytic fraction was homogenized in acetonitrile:methanol:water (40:40:20) for metabolite extraction.
LC-MS Analysis of Amino Acids
For amino acid analysis the benzoylchlorid derivatization method was used. In brief: One of the two dried metabolite pellets of each sample was re-suspended in 20 μl of the LC-MS-grade waters (Milli-Q 7000 equipped with an LC-Pak and a Millipak filter, Millipore). The re-suspended sample was mixed with 10 μl of 100 mM sodium carbonate (Sigma) followed by the addition of 10 μl 2% benzoylchloride (Sigma) in acetonitrile (Optima-Grade, Fisher-Scientific). Samples were vortexed before centrifuging them for 10 min 21.300x g at 20°C. Clear supernatants were transferred to fresh auto sampler tubes with conical glass inserts (Chromatographie Zubehoer Trott) and analyzed using an Acquity iClass UPLC (Waters) connected to a Q-Exactive HF (Thermo). For the analysis, 2 μl of the derivatized sample were injected onto a 100 x 1.0 mm HSS T3 UPLC column (Waters). The flow rate was set to 100 μL/min using a buffer system consisted of buffer A (10 mM ammonium formate (Sigma), 0.15% formic acid (Sigma) in Milli-Q water (Millipore)) and buffer B (acetonitrile, Optima-grade, Fisher-Scientific). The LC gradient was: 0% B at 0 min; 0-15% B 0-0.1 min; 15-17% B 0.1-0.5 min; 17-55% B 0.5-14 min, 55-70% B 14-14.5 min; 70-100% B 14.5-18 min; 100% B 18-19 min; 100-0% B 19-19.1 min, 19.1-28 min 0% B. The mass spectrometer was operating in positive ionization mode monitoring the mass range m/z 50-750. The heated ESI source settings of the mass spectrometer were: Spray voltage 3.5kV, capillary temperature 250°C, sheath gas flow 60 AU and aux gas flow 20 AU at a temperature of 250°C. The S-lens was set to a value of 60 AU. Data analysis of isotope ratios was performed using the TraceFinder software (Version 4.2, Thermo Fisher Scientific). Identity of each compound was validated by authentic reference compounds, which were analysed independently. For the isotope enrichment analysis the area of the extracted ion chromatogram (XIC) of each isotope [M + H]+ were determined with a mass accuracy (<5 ppm) before calculating the proportions of each detected isotope towards the sum of all isotopes of the corresponding compound. These proportions are given as percent values for each isotope and results are indicated as Molar Percent Enrichment (M.P.E.) as previously described (Divakaruni et al., 2017).
GC-MS Analysis of TCA Cycle Metabolites
Similar to the analysis of the isotope enrichment analysis in the amino acids, isotope enrichment analysis in TCA cycle metabolites were determined using GC-MS (Q-Exactive GC-Orbitrap, Thermo Fisher Scientific). For this purpose metabolites were derivatized using a two-step procedure starting with an methoxyamination (methoxyamine hydrochlorid, Sigma) followed by a trimethyl-silylation using N-Methyl-N-trimethylsilyl-trifluoracetamid (MSTFA, Macherey-Nagel). Dried samples were re-suspended in 5 μL of a freshly prepared (20 mg/mL) solution of methoxyamine in pyridine (Sigma) to perform the methoxyamination. These samples were then incubated for 90 min at 40°C on an orbital shaker (VWR) at 1500 rpm. In the second step additional 45 μL of MSTFA were added and the samples were incubated for additional 30 min at 40°C and 1500 rpm. At the end of the derivatisation the samples were centrifuged for 10 min at 21100x g and 40 μL of the clear supernatant was transferred to fresh auto sampler vials with conical glass inserts (Chromatographie Zubehoer Trott). For the GC-MS analysis 1 μL of each sample was injected using a PAL autosamplee system (Thermo Fisher Scientifc) using a Split/Splitless (SSL) injector at 300°C in splitless mode. The carrier gas flow (helium) was set to 2 ml/min using a 30 m DB-35MS capillary column (0.250-mm diameter and 0.25 μm film thickness, Agilent). The GC temperature program was: 2 min at 85°C, followed by a 15°C per min ramp to 330°C. At the end of the gradient the temperature is held for additional 6 min at 330°C. The transfer line and source temperature are both set to 280°C. The filament, which was operating at 70 V, was switched on 2 min after the sample was injected. During the whole gradient period the MS was operated in full scan mode covering a m/z range between 70 and 800 with a scan speed of 20 Hertz. For data analysis peak areas of extracted ion chromatograms of each isotope of compound-specific fragments [M - e−]+ were determined using the TraceFinder software (Version 4.2, Thermo Fisher Scientific) with a mass accuracy (<5 ppm). Subsequently proportions of each detected isotope towards the sum of all isotopes of the corresponding compound-specific fragment were determined. These proportions are given as percent values for each isotope. Details on the compound-specific fragments of the analysed compounds: citric acid was analysed from a five carbon-containing fragment (C11H21O4Si2) and a m/z of 273.09729; succinic acid was analysed from a four carbon-containing fragment (C9H19O4Si2) and a m/z of 247.08164; fumaric acid was analysed from a four carbon-containing fragment (C9H17O4Si2) and a m/z of 247.08164. The retention time and therefore identity of each compound was validated by authentic reference compounds which were analysed independently.
Mitochondrial and ER Morphological Analyses
For analysis of mitochondrial morphology in mitoYFP+ samples, serial z-stacks (0.3 to 0.5-μm steps) of individual astrocytes within the slice were acquired with an SP8 laser scanning confocal system (Leica) utilizing a 100x objective (NA 1.3) and digital zoom of 1.5. Acquired z-stacks were subjected to deconvolution (Huygens Professional software; Scientific Volume Imaging) utilizing the acquisition parameters and the resulting surface rendering images were utilized to extract mitochondrial morphological parameters (length, voxel volume and sphericity) via the object analyser plugin (Huygens). The length and sphericity of all quantified mitochondria (typically in the range of several hundreds) per astrocyte were plotted via the OriginPro software (OriginLab) and the resulting diagrams utilized to quantify the percentage of fragmented vs tubular mitochondria per astrocyte, utilizing as cut-off values 1 μm for the length and 0.8 for sphericity (where 1 would represent a sphere). At least 4-5 astrocytes (selected for their proximity to the lesion track) per mouse were analysed and the percentage of all individual astrocytes from the same mouse were pooled together. To estimate the perivascular density of mitochondria in astrocytic endfeet, a circular ROI exceeding the dextran-red signal by 5 μm (for cleared tissue) or 2 μm (for brain sections stained with CD31) was drawn around the labelled vessels in each analysed image. The acquired channel containing the mitochondrial signal (mitoYFP or mRFP depending on the experimental setup) was first thresholded and the resulting image utilized to calculate the ROI area fraction covered by mitochondrial signal. To calculate the perivascular ER-GFP g-ratio, the thickest sheet of ER-GFP signal in the endfeet in direct contact with the dextran-labelled vessel was measured and normalized to the vessel radius itself. The formula (Rlumen/(Rlumen + RER-GFP)) was utilized to obtain the g-ratio values per astrocyte.
Analysis of CD31 Immunoreactivity
To analyse the vasculature in 2D in sections labelled for CD31, a region of about 600x×600 μm in xy was cropped in the acquired z-stacks, its brightness adjusted with the same parameters for all images, smoothed in 3-D (sigma =1) and signal noise removed via a despeckle filter. Following z-projection (standard deviation, STD) and creation of a binary mask, the CD31 area fraction was measured for each image.
Quantification and Statistical Analysis
Data are represented as means ± SD or SEM. Graphical illustrations and significance were obtained with GraphPad Prism 7 (GraphPad) or with OriginPro (OriginLab). The statistical test used in each quantification is reported in the Figure legend. The levels of significance were set as ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Data and Code Availability
The mass spectrometry dataset have been deposited through Mendeley Data (https://doi.org/10.17632/ps8677yrjm.1) with the following identifiers: FACS time course astrocytes_reactive, MACS Mfn2cKO astrocytes_resting, FACS Mfn2cKO_Mfn1cKO astrocytes_reactive, FACS Mfn1cKO astrocytes_reactive_Mfn2_levels_assessment.
Acknowledgments
We thank N.G. Larsson for providing mitoYFP and Mfn1 and Mfn2 floxed mice; M. Götz and F. Kirchhoff for Glast::CreERT2 mice; L. Uhrbom and E. Holland for hGFAP-TVA mice; T. Langer for insightful comments; E.L. Snapp for ER-targeted probes; G. Hajnoczky for synthetic linkers; N. Toni for advice on EM; I. Atanassov for support with IPA analysis; B. Fernando, T. Öztürk, and S. Perin for excellent technical assistance; and the CECAD in vivo, imaging, and proteomics core facilities for excellent assistance. This work was supported by the Deutsche Forschungsgemeinschaft (SFB1218 and CECAD EXC 2030 - 390661388), European Research Council (ERC-StG-2015, grant number 67844), and Köln Fortune and UoC Advanced Postdoc Grant to M.B.; the Deutsche Forschungsgemeinschaft (SFB1218) to A.S.; and the Deutsche Forschungsgemeinschaft (SFB870) to K.-K.C. E.M. is a recipient of an Advanced Postdoc Grant (Deutsche Forschungsgemeinschaft, SFB1218). C.K. acknowledges the ISAC SRL Emerging Leaders Program.
Author Contributions
Conceptualization, E.M. and M.B.; Investigation, J.G., E.E., E.M., H.M.J., C.K.F., P.G., K.F.-D., and M.B.; Resources, V.S., M.J., A.G., K.-K.C., C.K., and A.S.; Software, P.P.; Writing – Original Draft, E.M. and M.B.; Writing – Review & Editing, E.M. and M.B.; Visualization, J.G., E.M., and M.B.; Funding Acquisition, M.B.; Supervision, E.M. and M.B.
Declaration of Interests
The authors declare no competing financial interests.
Published: March 26, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cmet.2020.03.005.
Contributor Information
Elisa Motori, Email: elisa.motori@age.mpg.de.
Matteo Bergami, Email: matteo.bergami@uk-koeln.de.
Supplemental Information
References
- Agarwal A., Wu P.H., Hughes E.G., Fukaya M., Tischfield M.A., Langseth A.J., Wirtz D., Bergles D.E. Transient opening of the mitochondrial permeability transition pore induces microdomain calcium transients in astrocyte processes. Neuron. 2017;93:587–605.e7. doi: 10.1016/j.neuron.2016.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Mehdi A.B., Pastukh V.M., Swiger B.M., Reed D.J., Patel M.R., Bardwell G.C., Pastukh V.V., Alexeyev M.F., Gillespie M.N. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signal. 2012;5:ra47. doi: 10.1126/scisignal.2002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand R., Wai T., Baker M.J., Kladt N., Schauss A.C., Rugarli E., Langer T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014;204:919–929. doi: 10.1083/jcb.201308006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anton F., Dittmar G., Langer T., Escobar-Henriques M. Two deubiquitylases act on mitofusin and regulate mitochondrial fusion along independent pathways. Mol. Cell. 2013;49:487–498. doi: 10.1016/j.molcel.2012.12.003. [DOI] [PubMed] [Google Scholar]
- Baughman J.M., Perocchi F., Girgis H.S., Plovanich M., Belcher-Timme C.A., Sancak Y., Bao X.R., Strittmatter L., Goldberger O., Bogorad R.L. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayraktar E.C., Baudrier L., Özerdem C., Lewis C.A., Chan S.H., Kunchok T., Abu-Remaileh M., Cangelosi A.L., Sabatini D.M., Birsoy K., Chen W.W. MITO-Tag mice enable rapid isolation and multimodal profiling of mitochondria from specific cell types in vivo. Proc. Natl. Acad. Sci. USA. 2019;116:303–312. doi: 10.1073/pnas.1816656115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger M., Allaman I., Magistretti P.J. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Bindocci E., Savtchouk I., Liaudet N., Becker D., Carriero G., Volterra A. Three-dimensional Ca2+ imaging advances understanding of astrocyte biology. Science. 2017;356 doi: 10.1126/science.aai8185. [DOI] [PubMed] [Google Scholar]
- Birdsey G.M., Dryden N.H., Amsellem V., Gebhardt F., Sahnan K., Haskard D.O., Dejana E., Mason J.C., Randi A.M. Transcription factor Erg regulates angiogenesis and endothelial apoptosis through VE-cadherin. Blood. 2008;111:3498–3506. doi: 10.1182/blood-2007-08-105346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth D.M., Enyedi B., Geiszt M., Várnai P., Hajnóczky G. Redox nanodomains are induced by and control calcium signaling at the ER-mitochondrial interface. Mol. Cell. 2016;63:240–248. doi: 10.1016/j.molcel.2016.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulay A.C., Saubaméa B., Adam N., Chasseigneaux S., Mazaré N., Gilbert A., Bahin M., Bastianelli L., Blugeon C., Perrin S. Translation in astrocyte distal processes sets molecular heterogeneity at the gliovascular interface. Cell Discov. 2017;3:17005. doi: 10.1038/celldisc.2017.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castejón O.J. Biopathology of astrocytes in human traumatic and complicated brain injuries. Review and hypothesis. Folia neuropathol. 2015;53:173–192. doi: 10.5114/fn.2015.54419. [DOI] [PubMed] [Google Scholar]
- Chao C.C., Gutiérrez-Vázquez C., Rothhammer V., Mayo L., Wheeler M.A., Tjon E.C., Zandee S.E.J., Blain M., de Lima K.A., Takenaka M.C. Metabolic control of astrocyte pathogenic activity via cPLA2-MAVS. Cell. 2019;179:1483–1498.e22. doi: 10.1016/j.cell.2019.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Detmer S.A., Ewald A.J., Griffin E.E., Fraser S.E., Chan D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K.H., Guo X., Ma D., Guo Y., Li Q., Yang D., Li P., Qiu X., Wen S., Xiao R.P. Dysregulation of HSG triggers vascular proliferative disorders. Nat. Cell Biol. 2004;6:872–883. doi: 10.1038/ncb1161. [DOI] [PubMed] [Google Scholar]
- Chen H., McCaffery J.M., Chan D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Chen Q., Kirk K., Shurubor Y.I., Zhao D., Arreguin A.J., Shahi I., Valsecchi F., Primiano G., Calder E.L., Carelli V. Rewiring of glutamine metabolism is a bioenergetic adaptation of human cells with mitochondrial DNA mutations. Cell Metab. 2018;27:1007–1025.e5. doi: 10.1016/j.cmet.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow L.M., Zhang J., Baker S.J. Inducible Cre recombinase activity in mouse mature astrocytes and adult neural precursor cells. Transgenic Res. 2008;17:919–928. doi: 10.1007/s11248-008-9185-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolat S., Martins de Brito O., Dal Zilio B., Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA. 2004;101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordás G., Renken C., Várnai P., Walter L., Weaver D., Buttle K.F., Balla T., Mannella C.A., Hajnóczky G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006;174:915–921. doi: 10.1083/jcb.200604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordás G., Weaver D., Hajnóczky G. Endoplasmic reticulum-mitochondrial contactology: structure and signaling functions. Trends Cell Biol. 2018;28:523–540. doi: 10.1016/j.tcb.2018.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brito O.M., Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- De Stefani D., Raffaello A., Teardo E., Szabò I., Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divakaruni A.S., Wallace M., Buren C., Martyniuk K., Andreyev A.Y., Li E., Fields J.A., Cordes T., Reynolds I.J., Bloodgood B.L. Inhibition of the mitochondrial pyruvate carrier protects from excitotoxic neuronal death. J. Cell Biol. 2017;216:1091–1105. doi: 10.1083/jcb.201612067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fecher C., Trovò L., Müller S.A., Snaidero N., Wettmarshausen J., Heink S., Ortiz O., Wagner I., Kühn R., Hartmann J. Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat. Neurosci. 2019;22:1731–1742. doi: 10.1038/s41593-019-0479-z. [DOI] [PubMed] [Google Scholar]
- Filadi R., Greotti E., Turacchio G., Luini A., Pozzan T., Pizzo P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. USA. 2015;112:E2174–E2181. doi: 10.1073/pnas.1504880112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finke S., Mueller-Waldeck R., Conzelmann K.K. Rabies virus matrix protein regulates the balance of virus transcription and replication. J. Gen. Virol. 2003;84:1613–1621. doi: 10.1099/vir.0.19128-0. [DOI] [PubMed] [Google Scholar]
- Ghanem A., Kern A., Conzelmann K.K. Significantly improved rescue of rabies virus from cDNA plasmids. Eur. J. Cell Biol. 2012;91:10–16. doi: 10.1016/j.ejcb.2011.01.008. [DOI] [PubMed] [Google Scholar]
- Giorgi C., Marchi S., Pinton P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018;19:713–730. doi: 10.1038/s41580-018-0052-8. [DOI] [PubMed] [Google Scholar]
- Göbel J., Motori E., Bergami M. Spatiotemporal control of mitochondrial network dynamics in astroglial cells. Biochem. Biophys. Res. Commun. 2018;500:17–25. doi: 10.1016/j.bbrc.2017.06.191. [DOI] [PubMed] [Google Scholar]
- Gomes L.C., Di Benedetto G., Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hama H., Kurokawa H., Kawano H., Ando R., Shimogori T., Noda H., Fukami K., Sakaue-Sawano A., Miyawaki A. Scale: a chemical approach for fluorescence imaging and reconstruction of transparent mouse brain. Nat. Neurosci. 2011;14:1481–1488. doi: 10.1038/nn.2928. [DOI] [PubMed] [Google Scholar]
- Hamby M.E., Coppola G., Ao Y., Geschwind D.H., Khakh B.S., Sofroniew M.V. Inflammatory mediators alter the astrocyte transcriptome and calcium signaling elicited by multiple G-protein-coupled receptors. J. Neurosci. 2012;32:14489–14510. doi: 10.1523/JNEUROSCI.1256-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S.M., Baig H.S., Hammarlund M. Mitochondria localize to injured axons to support regeneration. Neuron. 2016;92:1308–1323. doi: 10.1016/j.neuron.2016.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haustein M.D., Kracun S., Lu X.H., Shih T., Jackson-Weaver O., Tong X., Xu J., Yang X.W., O’Dell T.J., Marvin J.S. Conditions and constraints for astrocyte calcium signaling in the hippocampal mossy fiber pathway. Neuron. 2014;82:413–429. doi: 10.1016/j.neuron.2014.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T., Rizzuto R., Hajnoczky G., Su T.P. MAM: more than just a housekeeper. Trends Cell Biol. 2009;19:81–88. doi: 10.1016/j.tcb.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland E.C., Varmus H.E. Basic fibroblast growth factor induces cell migration and proliferation after glia-specific gene transfer in mice. Proc. Natl. Acad. Sci. USA. 1998;95:1218–1223. doi: 10.1073/pnas.95.3.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horng S., Therattil A., Moyon S., Gordon A., Kim K., Argaw A.T., Hara Y., Mariani J.N., Sawai S., Flodby P. Astrocytic tight junctions control inflammatory CNS lesion pathogenesis. J. Clin. Invest. 2017;127:3136–3151. doi: 10.1172/JCI91301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron. 2017;96:17–42. doi: 10.1016/j.neuron.2017.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatenko O., Chilov D., Paetau I., de Miguel E., Jackson C.B., Capin G., Paetau A., Terzioglu M., Euro L., Suomalainen A. Loss of mtDNA activates astrocytes and leads to spongiotic encephalopathy. Nat. Commun. 2018;9:70. doi: 10.1038/s41467-017-01859-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff J.J., Wang M., Liao Y., Plogg B.A., Peng W., Gundersen G.A., Benveniste H., Vates G.E., Deane R., Goldman S.A. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 2012;4:147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N., Nomura M., Jofuku A., Kato H., Suzuki S.O., Masuda K., Otera H., Nakanishi Y., Nonaka I., Goto Y. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- Jackson J.G., Robinson M.B. Regulation of mitochondrial dynamics in astrocytes: mechanisms, consequences, and unknowns. Glia. 2018;66:1213–1234. doi: 10.1002/glia.23252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A.S., Zhang H., Prinz W.A. Organelle biogenesis in the endoplasmic reticulum. Nat. Cell Biol. 2017;19:876–882. doi: 10.1038/ncb3579. [DOI] [PubMed] [Google Scholar]
- Khakh B.S., Sofroniew M.V. Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 2015;18:942–952. doi: 10.1038/nn.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni S.S., Joffraud M., Boutant M., Ratajczak J., Gao A.W., Maclachlan C., Hernandez-Alvarez M.I., Raymond F., Metairon S., Descombes P. Mfn1 deficiency in the liver protects against diet-induced insulin resistance and enhances the hypoglycemic effect of metformin. Diabetes. 2016;65:3552–3560. doi: 10.2337/db15-1725. [DOI] [PubMed] [Google Scholar]
- Lee S., Sterky F.H., Mourier A., Terzioglu M., Cullheim S., Olson L., Larsson N.G. Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum. Mol. Genet. 2012;21:4827–4835. doi: 10.1093/hmg/dds352. [DOI] [PubMed] [Google Scholar]
- Liddelow S.A., Barres B.A. Reactive astrocytes: production, function, and therapeutic potential. Immunity. 2017;46:957–967. doi: 10.1016/j.immuni.2017.06.006. [DOI] [PubMed] [Google Scholar]
- Liddelow S.A., Guttenplan K.A., Clarke L.E., Bennett F.C., Bohlen C.J., Schirmer L., Bennett M.L., Münch A.E., Chung W.S., Peterson T.C. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Fabuel I., Le Douce J., Logan A., James A.M., Bonvento G., Murphy M.P., Almeida A., Bolaños J.P. Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc. Natl. Acad. Sci. USA. 2016;113:13063–13068. doi: 10.1073/pnas.1613701113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovatt D., Sonnewald U., Waagepetersen H.S., Schousboe A., He W., Lin J.H., Han X., Takano T., Wang S., Sim F.J. The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J. Neurosci. 2007;27:12255–12266. doi: 10.1523/JNEUROSCI.3404-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchsinger L.L., de Almeida M.J., Corrigan D.J., Mumau M., Snoeck H.W. Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature. 2016;529:528–531. doi: 10.1038/nature16500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L., Zwingman T.A., Sunkin S.M., Oh S.W., Zariwala H.A., Gu H., Ng L.L., Palmiter R.D., Hawrylycz M.J., Jones A.R. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mar F.M., Simões A.R., Leite S., Morgado M.M., Santos T.E., Rodrigo I.S., Teixeira C.A., Misgeld T., Sousa M.M. CNS axons globally increase axonal transport after peripheral conditioning. J. Neurosci. 2014;34:5965–5970. doi: 10.1523/JNEUROSCI.4680-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathiisen T.M., Lehre K.P., Danbolt N.C., Ottersen O.P. The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia. 2010;58:1094–1103. doi: 10.1002/glia.20990. [DOI] [PubMed] [Google Scholar]
- Misgeld T., Kerschensteiner M., Bareyre F.M., Burgess R.W., Lichtman J.W. Imaging axonal transport of mitochondria in vivo. Nat. Methods. 2007;4:559–561. doi: 10.1038/nmeth1055. [DOI] [PubMed] [Google Scholar]
- Mori T., Tanaka K., Buffo A., Wurst W., Kühn R., Götz M. Inducible gene deletion in astroglia and radial glia--a valuable tool for functional and lineage analysis. Glia. 2006;54:21–34. doi: 10.1002/glia.20350. [DOI] [PubMed] [Google Scholar]
- Moss J., Gebara E., Bushong E.A., Sánchez-Pascual I., O'Laoi R., El M'Ghari I., Kocher-Braissant J., Ellisman M.H., Toni N. Fine processes of nestin-GFP-positive radial glia-like stem cells in the adult dentate gyrus ensheathe local synapses and vasculature. Proc. Natl. Acad. Sci. USA. 2016;113:E2536–E2545. doi: 10.1073/pnas.1514652113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motori E., Puyal J., Toni N., Ghanem A., Angeloni C., Malaguti M., Cantelli-Forti G., Berninger B., Conzelmann K.K., Götz M. Inflammation-induced alteration of astrocyte mitochondrial dynamics requires autophagy for mitochondrial network maintenance. Cell Metab. 2013;18:844–859. doi: 10.1016/j.cmet.2013.11.005. [DOI] [PubMed] [Google Scholar]
- Naon D., Zaninello M., Giacomello M., Varanita T., Grespi F., Lakshminaranayan S., Serafini A., Semenzato M., Herkenne S., Hernández-Alvarez M.I. Critical reappraisal confirms that mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. USA. 2016;113:11249–11254. doi: 10.1073/pnas.1606786113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens K., Park J.H., Gourley S., Jones H., Kristian T. Mitochondrial dynamics: cell-type and hippocampal region specific changes following global cerebral ischemia. J. Bioenerg. Biomembr. 2015;47:13–31. doi: 10.1007/s10863-014-9575-7. [DOI] [PubMed] [Google Scholar]
- Polyzos A.A., Lee D.Y., Datta R., Hauser M., Budworth H., Holt A., Mihalik S., Goldschmidt P., Frankel K., Trego K. Metabolic reprogramming in astrocytes distinguishes region-specific neuronal susceptibility in Huntington mice. Cell Metab. 2019;29:1258–1273.e11. doi: 10.1016/j.cmet.2019.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash R., Carmichael S.T. Blood-brain barrier breakdown and neovascularization processes after stroke and traumatic brain injury. Curr. Opin. Neurol. 2015;28:556–564. doi: 10.1097/WCO.0000000000000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R., De Stefani D., Raffaello A., Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- Salehi A., Zhang J.H., Obenaus A. Response of the cerebral vasculature following traumatic brain injury. J. Cereb. Blood Flow Metab. 2017;37:2320–2339. doi: 10.1177/0271678X17701460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger M., Dietrich M.O., Sebastián D., Imbernón M., Castaño C., Garcia A., Esteban Y., Gonzalez-Franquesa A., Rodríguez I.C., Bortolozzi A. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell. 2013;155:172–187. doi: 10.1016/j.cell.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorrano L., De Matteis M.A., Emr S., Giordano F., Hajnóczky G., Kornmann B., Lackner L.L., Levine T.P., Pellegrini L., Reinisch K. Coming together to define membrane contact sites. Nat. Commun. 2019;10:1287. doi: 10.1038/s41467-019-09253-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E., Bushong E.A., Haustein M.D., Tong X., Jackson-Weaver O., Kracun S., Xu J., Sofroniew M.V., Ellisman M.H., Khakh B.S. Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno-associated viruses. J. Gen. Physiol. 2013;141:633–647. doi: 10.1085/jgp.201210949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E., Patel S., Khakh B.S. Probing the complexities of astrocyte calcium signaling. Trends Cell Biol. 2016;26:300–312. doi: 10.1016/j.tcb.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapp E.L., Sharma A., Lippincott-Schwartz J., Hegde R.S. Monitoring chaperone engagement of substrates in the endoplasmic reticulum of live cells. Proc. Natl. Acad. Sci. USA. 2006;103:6536–6541. doi: 10.1073/pnas.0510657103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew M.V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015;16:249–263. doi: 10.1038/nrn3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterky F.H., Lee S., Wibom R., Olson L., Larsson N.G. Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc. Natl. Acad. Sci. USA. 2011;108:12937–12942. doi: 10.1073/pnas.1103295108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supplie L.M., Düking T., Campbell G., Diaz F., Moraes C.T., Götz M., Hamprecht B., Boretius S., Mahad D., Nave K.A. Respiration-deficient astrocytes survive as glycolytic cells in vivo. J. Neurosci. 2017;37:4231–4242. doi: 10.1523/JNEUROSCI.0756-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villapol S., Byrnes K.R., Symes A.J. Temporal dynamics of cerebral blood flow, cortical damage, apoptosis, astrocyte-vasculature interaction and astrogliosis in the pericontusional region after traumatic brain injury. Front. Neurol. 2014;5:82. doi: 10.3389/fneur.2014.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volterra A., Liaudet N., Savtchouk I. Astrocyte Ca2⁺ signalling: an unexpected complexity. Nat. Rev. Neurosci. 2014;15:327–335. doi: 10.1038/nrn3725. [DOI] [PubMed] [Google Scholar]
- Voskuhl R.R., Peterson R.S., Song B., Ao Y., Morales L.B., Tiwari-Woodruff S., Sofroniew M.V. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J. Neurosci. 2009;29:11511–11522. doi: 10.1523/JNEUROSCI.1514-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickersham I.R., Lyon D.C., Barnard R.J., Mori T., Finke S., Conzelmann K.K., Young J.A., Callaway E.M. Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron. 2007;53:639–647. doi: 10.1016/j.neuron.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong B.W., Marsch E., Treps L., Baes M., Carmeliet P. Endothelial cell metabolism in health and disease: impact of hypoxia. EMBO J. 2017;36:2187–2203. doi: 10.15252/embj.201696150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian J.L., Xu L., Foo L.C., Nouri N., Zhou L., Giffard R.G., Barres B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012;32:6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zariwala H.A., Borghuis B.G., Hoogland T.M., Madisen L., Tian L., De Zeeuw C.I., Zeng H., Looger L.L., Svoboda K., Chen T.W. A Cre-dependent GCaMP3 reporter mouse for neuronal imaging in vivo. J. Neurosci. 2012;32:3131–3141. doi: 10.1523/JNEUROSCI.4469-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zudaire E., Gambardella L., Kurcz C., Vermeren S. A computational tool for quantitative analysis of vascular networks. PLoS One. 2011;6:e27385. doi: 10.1371/journal.pone.0027385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Two-photon imaging scan through a portion of the intact cerebral cortex (vasculature labeled via dextran-red) following astrocyte labeling with ER-GFP
Two-photon imaging scan through a portion of the intact cerebral cortex (vasculature labeled via dextran-red) following astrocyte labeling with mitoYFP
Two-photon imaging scan through a reconstructed cortical volume containing mitoYFP-expressing astrocytes and dextran-labeled vessels, showing the extent of perivascular mitochondrial clustering at 7 days post-SW
Two-photon imaging scan through a portion of the injured cerebral cortex (vasculature labeled via dextran-red) containing mitoYFP-expressing control astrocytes at 7 days post-SW. Occasional blood cells are visible in the lumen as a result of auto-fluorescence overlapping with the GFP spectrum
Two-photon imaging scan through a portion of the injured cerebral cortex (vasculature labeled via dextran-red) containing mitoYFP-expressing Mfn2cKO astrocytes at 7 days post-SW. Occasional blood cells are visible in the lumen as a result of auto-fluorescence overlapping with the GFP spectrum
Two-photon imaging scan through a portion of the injured cerebral cortex (vasculature labeled via dextran-red) containing mitoYFP-expressing Mfn1cKO astrocytes at 7 days post-SW. Occasional blood cells are visible in the lumen as a result of auto-fluorescence overlapping with the GFP spectrum
In vivo 2-photon imaging of a representative GCaMP3-expressing astrocyte via cranial window implantation and following vasculature labeling. The recording (total duration 3 min, 1.16 frames per s) has been accelerated to fit into 20 s
Data Availability Statement
The mass spectrometry dataset have been deposited through Mendeley Data (https://doi.org/10.17632/ps8677yrjm.1) with the following identifiers: FACS time course astrocytes_reactive, MACS Mfn2cKO astrocytes_resting, FACS Mfn2cKO_Mfn1cKO astrocytes_reactive, FACS Mfn1cKO astrocytes_reactive_Mfn2_levels_assessment.