


Abstract
Transcription start sites (TSS) in eukaryotes are characterized by a nucleosome-depleted region (NDR), which appears to be flanked upstream and downstream by strongly positioned nucleosomes incorporating the histone variant H2A.Z. H2A.Z associates with both active and repressed TSS and is important for priming genes for rapid transcriptional activation. However, the determinants of H2A.Z occupancy at specific nucleosomes and its relationship to transcription initiation remain unclear. To further elucidate the specificity of H2A.Z, we determined its genomic localization at single nucleosome resolution, as well as the localization of its chromatin remodelers Swr1 and Ino80. By analyzing H2A.Z occupancy in conjunction with RNA expression data that captures promoter-derived antisense initiation, we find that H2A.Z’s bimodal incorporation on either side of the NDR is not a general feature of TSS, but is specifically a marker for bidirectional transcription, such that the upstream flanking −1 H2A.Z-containing nucleosome is more appropriately considered as a +1 H2A.Z nucleosome for antisense transcription. The localization of H2A.Z almost exclusively at the +1 nucleosome suggests that a transcription-initiation dependent process could contribute to its specific incorporation.
INTRODUCTION
At the most basic level of chromosome structure, eukaryotic genomes are packaged into chromatin in the form of nucleosomal arrays. The positioning of nucleosomes along DNA depends on the intrinsic DNA sequence binding preferences of histones and the actions of ATP-dependent chromatin remodelers, which can incorporate, evict, or reposition histones and nucleosomes. The best-characterized nucleosome arrangement is one found at the transcription start site (TSS). Here, a wide nucleosome-depleted region (NDR) is bordered by two well-positioned nucleosomes, referred to as +1 for the nucleosome immediately downstream of the NDR in the direction of transcription, and −1 for the upstream nucleosome. These NDR-proximal nucleosomes set the pattern for a periodicity in nucleosome positioning extending upstream and downstream (1). The yeast TSS is typically found at about one helical turn into the +1 nucleosome (2).
Within individual nucleosomes, canonical histones may be replaced by histone variants. The H2A histone variant H2A.Z is highly conserved throughout eukaryotes, displaying sequence conservation of ∼70–90% (3). In contrast, the Saccharomyces cerevisiae protein sequences for H2A.Z and canonical H2A share 61% identity, with H2A.Z containing additional amino acids at both the N-terminal and C-terminal ends. H2A.Z-containing nucleosomes are found predominantly at the two nucleosomes bordering the NDR (4). H2A.Z has also been reported to be incorporated at the 3′ end of genes (5). Incorporation of H2A.Z is catalyzed by the ATP-dependent chromatin remodeler Swr1 (6). The exchange takes place by removal of an H2A/H2B dimer and replacement with an H2A.Z/H2B dimer. In the opposite direction the ATP-dependent chromatin remodeler Ino80 exchanges H2A.Z/H2B dimers for H2A/H2B dimers (7). H2A.Z can also likely be evicted by the progression of RNA polymerase, as depletion of components of the pre-initiation complex cause an increase in H2A.Z occupancy whereas depletion of Ino80 does not (8,9). How H2A.Z is targeted specifically to the TSS-proximal nucleosomes is unclear. Swr1 shows a preference for binding NDRs (10,11), and its H2A/H2A.Z exchange activity is additionally stimulated by H4 and H2A acetylation (12). However, as a small but significant number of genes do not incorporate H2A.Z, and as there is variation in the extent of incorporation, these factors fail to fully explain H2A.Z localization (2). In particular, the differences in H2A.Z incorporation at the two nucleosomes flanking the NDR, commonly regarded as the +1 and −1 nucleosomes (13–17), and its relationship to transcription, are not well understood.
S. cerevisiae tolerates loss of H2A.Z with few transcriptional changes and little fitness defect under optimal growth conditions (18). This result is puzzling given the important TSS-proximal positions H2A.Z occupies. However, localization to the TSS implies that H2A.Z has some relationship to transcription. Studies of H2A.Z’s effect on nucleosomes have provided conflicting evidence for both more and less stable structures (19) as well as no significant effect on stability (20). The region of H2A.Z that contacts the nucleosome core displays lower conservation, perhaps suggesting that H2A.Z’s effect on nucleosome stability is somewhat species specific (21). Incorporation of H2A.Z has the potential to decrease the barrier posed by the +1 nucleosome (22) and its presence is associated with high nucleosome turnover rates (23) and rapid transcriptional activation. H2A.Z contributes to the prompt induction of cell-cycle promoting genes (24) and is required for the transcription of some genes. At the GAL1-10 promoter it is required for recruitment of RNA Pol II and TBP (25), as well as Mediator, SAGA, and Swi/Snf (26), and cells lacking H2A.Z therefore display a Gal- phenotype. In contrast to yeast, for most metazoans H2A.Z is essential. In mammals, H2A.Z is also present at active enhancers that recruit RNA polymerase and produce enhancer RNAs (eRNAs) (27). Additionally, H2A.Z has been shown to be important for RNA Pol II pausing (28) and cell differentiation (29), and it may even be involved in memory formation (30–32).
In an attempt to resolve the relationship between H2A.Z occupancy, transcription initiation, and the occupancy of H2A.Z remodelers, we mapped the incorporation of H2A.Z at promoters at single-nucleosome resolution, along with the localization of its remodelers Swr1 and Ino80 and analyzed this data in conjunction with RNA-seq data. Our results show that H2A.Z incorporation flanking the promoter NDR is a marker for bidirectional transcription such that even the -1 nucleosome upstream of the NDR is a +1 H2A.Z-containing nucleosome for an upstream antisense non-coding transcript. Moreover, the binding of its remodelers Swr1 and Ino80 is also dependent on H2A.Z, suggesting an interplay between processes governing the normal localization of H2A.Z and its chaperones.
MATERIALS AND METHODS
Strains and growth conditions
The yeast strains used were all from the WT haploid BY4741 background (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) and the yeast haploid deletion strain collection (Open Biosystems/GE Dharmacon) (33). The gene deletions were confirmed via PCR and were also remade. The primary strain used for MNase ChIP-seq experiments contained a TAP-tagged copy of the histone protein H2A.Z from the TAP-tagged protein collection (34). SWR1- and INO80-Myc-tagged strains were created by transformation of yeast with 3Myc-His3MX6 cassettes amplified with primers targeted to the C-terminus coding region of SWR1 or INO80 from the pYM5-3Myc-His3MX6 plasmid (35). The strains were verified for the correct genomic replacement using PCR and for functional complementation by the epitope-tagged proteins by growth assays in selective conditions (Supplementary Figure S1). For molecular experiments, yeast cells were grown up in liquid culture in yeast extract peptone dextrose (YPD) at 30°C until the cells reached a concentration measured via A600 O.D. of ∼0.8 and harvested by centrifugation.
Mononucleosome isolation
We followed a previously described protocol to isolate nucleosomes (36). Samples were crosslinked with 1% formaldehyde for 30 min and then treated with 250 μg of zymolyase (MP Biomedicals Catalog # IC320921) to permeabilize the cell wall. The cells were then washed and resuspended in NP buffer (1 M Sorbitol, 50 mM NaCl, 10 mM Tris pH 7.4, 5 mM MgCl2, 0.075% NP-40, 1 mM 2-mercaptoethanol, 500 μM spermidine). The cells were then subjected to increasing concentrations of MNase (Worthington Biochemical Corp. Catalog # LS004797) at 25, 50, 75, and 100 U/ml for 10 min at 37°C. Reactions were stopped by addition of 10 mM EDTA and 1% SDS. Crosslinks were reversed by a 65°C overnight incubation with Proteinase K. RNA was then removed by RNase treatment. DNA was extracted by phenol-chloroform treatment followed by ethanol precipitation. The DNA was then run on an E-gel cassette (Invitrogen), and the fraction of DNA running at ∼150 bp was extracted.
Chromatin immunoprecipitation
Yeast cells were fixed by adding formaldehyde to cultures at a final concentration of 1% and incubating for 30 min at 30°C in a shaking incubator. Cells were then spun down, washed, and resuspended in chilled lysis buffer and subjected to bead beating at 4°C. Samples were then sonicated using a Branson Sonifier, spun down, and the supernatant was isolated. A portion of the supernatant was reserved for an input sample and the remainder was subjected to immunoprecipitation using either IgG Sepharose 6 Fast Flow beads (GE Healthcare Life Sciences) or anti-Myc conjugated agarose beads (Sigma Aldrich).
MNase chromatin immunoprecipitation
The MNase ChIP-seq protocol was largely adapted from a published protocol (37). Yeast cells were fixed by adding formaldehyde to cultures at a final concentration of 1% and incubating for 30 minutes at 30°C in a shaking incubator. Cells were then spun down, washed, and resuspended in chilled NP buffer and subjected to bead beating at 4°C. The cell lysate mixture was then collected and treated with MNase at increasing concentrations of 25, 50, 75 and 100 U/ml for 10 min at 37°C. The reaction was then terminated by addition of EDTA to a final concentration of 10 mM and incubating on ice for 10 min. The samples were then spun down at 4°C and the supernatants from the different MNase concentrations pooled and collected. A portion of the supernatant was reserved as an input sample, and the rest was used for chromatin immunoprecipitation. TAP-tagged proteins were pulled down by an overnight incubation at 4°C with IgG Sepharose 6 Fast Flow beads (GE Healthcare Life Sciences).
SMORE-seq
Simultaneous mapping of RNA ends (SMORE-seq) provides a method for determining the 5′ ends of transcripts (38). This technique allows for precise mapping of transcription start sites, and, can also allow the detection of short antisense RNAs that are difficult to detect by standard RNA-seq methods. In particular, this technique can be used to identify antisense transcripts arising from between tandemly arranged genes, which we term UAN-RNAs (for sites of (promoter) upstream antisense non-coding RNA transcription). SMORE-seq was performed as previously described (38). Briefly, RNA was incubated with tobacco acid pyrophosphatase (TAP, Epicentre) to remove 5′ caps. TAP was inactivated by heating at 65°C and RNA was purified. A control reaction omitting TAP was also carried out. Purified RNAs were then used for construction of Illumina RNA-seq libraries using the NEBNext Small RNA kit following manufacturer's instructions and as previously described (38). For our analysis, we combined data from our previously published study (GSE49026) with newly generated datasets.
Library preparation and sequencing
MNase and ChIP libraries were prepared using the NEBNext ChIP-Seq library preparation kit for Illumina sequencing (NEB Catalog # E6240L) with adapters from Bioo. RNA SMORE-seq libraries were prepared with the NEBNext Small RNA library preparation kit, also for Illumina sequencing (NEB Catalog # E7300S). The libraries were then sequenced either at the University of Texas at Austin Genome Sequencing and Analysis Facility (UT GSAF) or at the M.D. Anderson Next-Generation Sequencing Facility at Science Park.
Analysis of sequencing data
Sequencing reads were aligned against the SacCer3 reference genome (from the Saccharomyces Genome Database) using BWA (39). Nucleosome positions from MNase-seq data and ChIP-seq peaks from ChIP-seq data for Swr1 and Ino80 were analyzed as previously described (36,40,41). The ChIP-seq peaks and nucleosome positions (which we also refer to as ‘peaks’) and their associated scores were then used to create a per-gene score matrix file which consisted of a window of 10 bp bins centered either on the transcription start site (TSS), the transcription termination site (TTS) or other genomic loci of interest, containing nucleosome scores or ChIP-seq peak scores for each gene and bin. We called TSS and TTS positions using either SMORE-seq data, previously available annotation data (42), or a reasonable estimate based on average TSS and TTS positions. These files were then normalized by in-matrix normalization to allow comparison across files regardless of sequencing depth. This, in effect, is a normalization system centered around ORF regions, and bypasses having to take into account the large variability in nucleosome enrichment seen at repetitive regions. These normalized files were then used to create an average TSS nucleosome profile and nucleosome heatmaps based on the matrix, and visualized using Java TreeView. ChIP-seq data was corrected by subtracting out the signal from matched input samples. For MNase ChIP-seq, input samples correspond to the background mononucleosome profiles (Figure 1).
Figure 1.
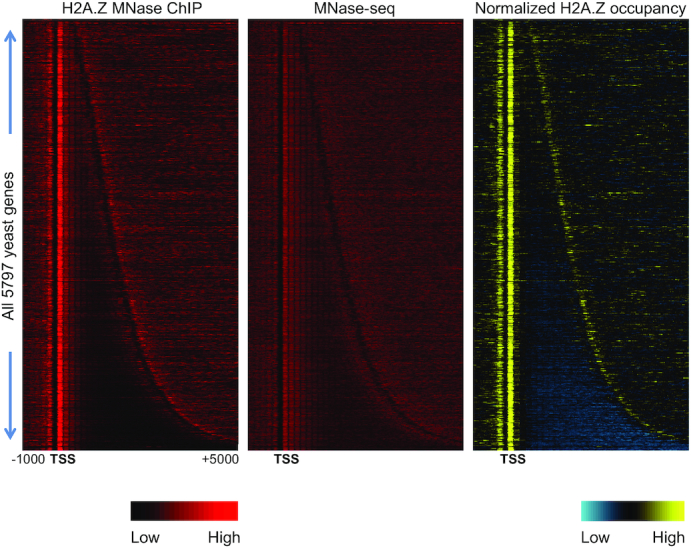
Genome wide H2A.Z incorporation. Left: Heatmap of H2A.Z occupancy as measured by MNase ChIP-seq without input correction. Middle: Background nucleosome profiles in the input using standard MNase-seq. Right: Input-corrected MNase ChIP-seq data, with yellow indicating higher H2A.Z nucleosomal occupancy relative to background nucleosomes and blue indicating depletion of H2A.Z-containing nucleosomes relative to background nucleosomes. Genes are arranged by increasing length, revealing a peak of H2A.Z at the 5′ end and a less pronounced peak at the 3′ end, corresponding to the 5′ end of the nearest downstream gene.
Gene lists used for sorting
Gene expression data was obtained from publicly available RNA sequencing data derived from WT cells (accession numbers: SRX046708 and SRX046709) (43). Reads mapping to annotated transcripts were counted and then normalized for gene length. This deeply sequenced data set was used to assign a gene expression level to each gene in the yeast genome. UAN-RNA counts were obtained from a combination of previously published SMORE-seq data from our lab (38) and newly generated SMORE-seq data. Reads mapping between −50 bp and −300 bp upstream of the TSS on the antisense strand for annotated coding genes were counted to give a measure of upstream antisense transcription. For tandemly arranged transcripts, these antisense reads are referred to as UAN-RNAs. Data from nascent elongating transcription and sequencing (NET-seq) analysis (44) (SRR2005997, SRR2005998) was used additionally for a similar analysis. TATA and TATA-less gene lists were obtained from a published study (45). A ribosomal protein coding gene list was obtained from The Ribosomal Protein Gene Database (46). Tandem and divergent gene orientations were determined computationally by annotating each TSS with whether the nearest upstream gene end was a transcription start or termination site. H2A.Z enrichment values for +1 and −1 nucleosomes were determined by taking the maximum signal across 150 bp within a specified range for each feature (−350 to −100 for the −1 nucleosome and −50 to +150 for the +1 nucleosome) (Supplementary Figure S2). RNA Pol II Ser-5 phosphorylation ChIP-seq data was obtained from a data set previously published by our lab (GSE51251) (47).
RESULTS
Swr1 localization to its targets is dependent on H2A.Z
In order to explore H2A.Z incorporation at nucleosomes and its relationship with Swr1 and Ino80 binding across the S. cerevisiae genome, we first performed micrococcal nuclease chromatin immunoprecipitation (MNase ChIP-seq) to pull down TAP-tagged H2A.Z. H2A.Z occupancy has been measured using ChIP-seq in earlier studies (9,11,48), but it was important to measure its occupancy along with RNA 5′ ends and Swr1 and Ino80 occupancy in parallel grown cultures under the same conditions. As a control, to normalize for background bulk nucleosomes, we generated input samples in parallel, which were also treated with MNase but not subjected to ChIP-seq. Here, we refer to the binding of H2A.Z relative to background bulk nucleosomes as H2A.Z enrichment, which is a measure of specific H2A.Z incorporation. As expected, H2A.Z is sharply localized to the +1 nucleosome, with somewhat lower occupancy at the −1 nucleosome flanking the NDR (Figure 1). We used the average of two consistent replicates for further analysis (Supplementary Figure S3). As the chromatin remodelers Swr1 and Ino80 govern the incorporation dynamics of H2A.Z, we also performed ChIP-seq for Swr1 and Ino80 in both WT and in htz1Δ backgrounds (HTZ1 is the gene encoding H2A.Z).
We found that Swr1 and Ino80 occupied promoters in the vicinity of the +1 nucleosome, though the signal was fairly low and consistent across most promoters, with only a handful of genes showing strong binding peaks. This pattern of occupancy of Swr1 and Ino80 is consistent with previous studies (11,49,50). Swr1′s strongest peak was localized at its own promoter (Figure 2A and Supplementary Figure S4). This result was also consistent with previously published Swr1 ChIP-exo data (11), and similar observations from microarray data have been made before (51). The strong binding of Swr1 to its own promoter prompted us to examine H2A.Z incorporation at this promoter as well. ChIP-seq data revealed a modest peak of H2A.Z at the promoter of RQC1, the gene upstream of SWR1, but rather weak incorporation directly at the Swr1 binding site in the SWR1 promoter (Figure 2A). Interestingly, deletion of HTZ1 dramatically reduced Swr1 binding at its own promoter (Figure 2A and Supplementary Figure S4). Thus, Swr1 recruitment to the NDR is itself strongly dependent on the presence of H2A.Z. The significance of the strong binding of Swr1 to its own promoter is not clear at present. Although suggestive of feedback regulation, the strong reduction in Swr1 binding to its promoter in htz1Δ did not lead to changes in SWR1 transcript levels (not shown) or protein (Supplementary Figure S5), indicating that a simple feedback loop is not operational. The promoter regions with the strongest Swr1 binding signal did not show high correlation with the promoter regions showing the strongest H2A.Z occupancy signals at the +1 nucleosome (Figure 2B and Supplementary Figure S6A–D). To verify if the low correlations were due to the quality of our ChIP-seq data for Swr1, we examined the correlation of signal strength at the promoter between H2A.Z occupancy and another recently published ChIP-exo dataset for Swr1 (50). The correlation was only marginally improved (Supplementary Figure S6E). As an alternative way to measure genome-wide correlations between ChIP-seq datasets, we calculated the rank correlation between bins along the length of the chromosome for the entire genome. This analysis revealed that our single replicate of Swr1 ChIP-seq was as well correlated with other Swr1 datasets as individual Swr1 replicates were with one another in recent published datasets (Supplementary Figure S6F).
Figure 2.
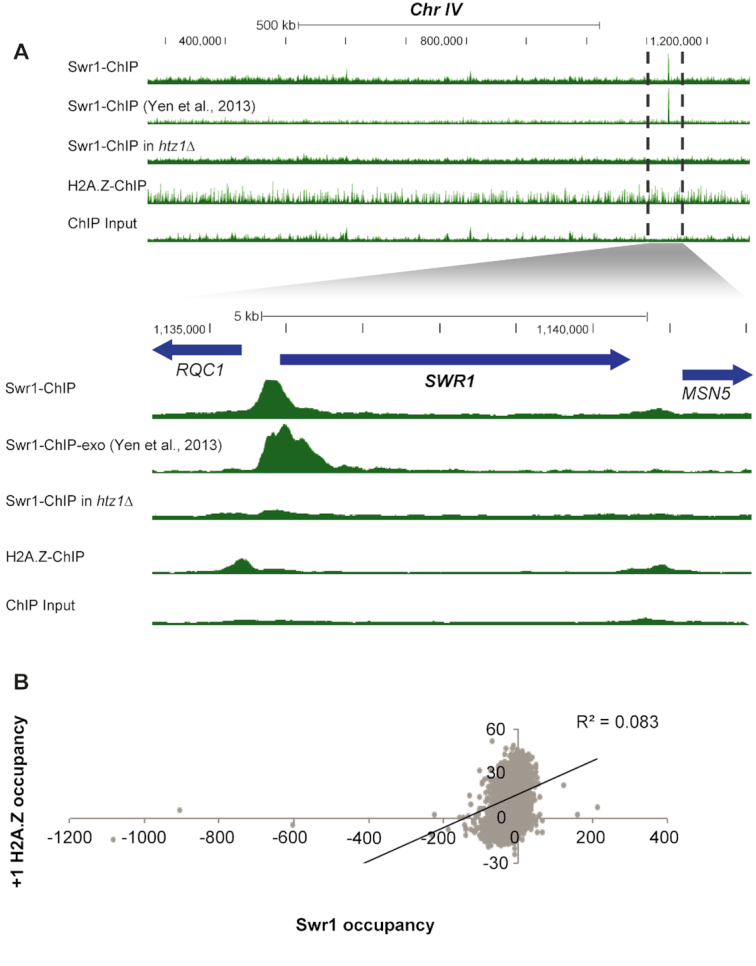
Relationship of H2A.Z and Swr1 binding. (A) Swr1 binding depends on H2A.Z. ChIP-seq data tracks visualized on the UCSC Genome Browser, showing ∼850 kb of Chr IV containing the most prominent peak of Swr1 binding at its own promoter. The set of tracks below show a close-up of SWR1 indicated by dotted lines. Tracks have been scaled to normalize for differences in sequencing read depth. Swr1 binding at this strong site is greatly reduced in the htz1Δ mutant. (B) Correlation between Swr1 ChIP-seq signal between −350 to +150 relative to the TSS and the H2A.Z signal at the +1 nucleosome. The correlation is low, at ∼0.08.
The dense gene packing within the S. cerevisiae genome means that neighboring genes can affect the nucleosome patterns observed at any particular gene. In order to distinguish nucleosome patterns at closely juxtaposed promoters and isolated promoters, we separated genes into divergent (head-to-head) and tandem (head-to-tail) categories based on the orientation of their transcripts and examined the relationship of Swr1 binding and H2A.Z incorporation. While tandem and divergent genes showed similar levels of Swr1 localization (Supplementary Figure S7A), their incorporation patterns for H2A.Z differed markedly. Tandem and divergent genes had equivalent incorporation of H2A.Z at the +1 nucleosome but divergent genes showed significantly more H2A.Z at the −1 nucleosome (Supplementary Figure S7B). Moreover, the −1 nucleosome at tandem genes was primarily a single peak and was not followed by a regularly spaced array of nucleosomes upstream. Thus, the differential incorporation of H2A.Z at the −1 nucleosome at tandem genes cannot be explained solely by the binding of Swr1, which binds similarly at tandem and divergent genes.
Given the strong dependence of the most prominent Swr1 binding peak on H2A.Z (Figure 2A), we analyzed the genome-wide binding pattern of Swr1 in the H2A.Z deletion compared to the WT strain, which has not been examined in previous studies. Promoters with the highest levels of Swr1 binding in the WT strain showed a strong reduction in htz1Δ, indicating again that Swr1 binding to its most prominent targets in the genome is strongly dependent on H2A.Z (Figure 3A). This reduction in Swr1 binding to its usual targets was not due to reduced expression of Swr1 in the htz1Δ background (Supplementary Figure S5). To quantify this effect and examine the fate of Swr1 in the htz1Δ background in more detail, we generated average binding profiles for the top 500 most strongly Swr1-bound targets, the bottom 500 least bound targets, as well as the middle set. These plots revealed that at a small proportion of target promoters (∼10%), Swr1 is strongly localized at the +1 nucleosome, while at the majority of genes, its binding levels do not exceed the background values seen in the input samples (Figure 3B). This strong binding of Swr1 to its targets was clearly lost in the absence of H2A.Z (Figure 3B, top right panel). When we ranked the Swr1 ChIP-seq data by the strength of its signal in the htz1Δ strain, it was apparent that Swr1 gets redistributed to a different set of promoters in htz1Δ, showing that ChIP-seq was able to detect Swr1 binding even in the htz1Δ strain (Figure 3B bottom right panel). Similar results were observed for the Ino80 remodeler, although its redistribution was more pronounced, because both the least and most strongly occupied promoters changed in htz1Δ in the case of Ino80 (Figure 3C and D). By contrast, sites with minimal Swr1 binding in the WT remained depleted in the htz1Δ strain (Figure 3A and B). Swr1 and Ino80 showed a modest correlation in their occupancy profiles (Supplementary Figure S8).
Figure 3.

Swr1 and Ino80 are redistributed in the absence of H2A.Z. (A) Heat map of Swr1 binding in WT and htz1Δ strains. ChIP-seq data was sorted by the signal in the WT strain between −200 and +200 bp surrounding the TSS, but the interval from −1000 to +1000 is displayed. (B) Average binding profiles of Swr1 across the TSS. Gene groups are determined as follows: Top: the top 500 genes by binding signal, Bottom: the bottom 500 genes by binding signal, Middle: the remaining 4797 annotated genes. The lighter shaded envelope around the average line is the 95% confidence interval. The top row of plots shows data grouped after sorting by the ChIP-seq signal in the WT strain. The bottom row plots contain data grouped after sorting by the ChIP-seq signal in the htz1Δ strain. (C) Same as A, but for Ino80 binding. (D) Same as B, but for Ino80 binding.
H2A.Z incorporation at the +1 nucleosome does not correlate with gene expression
Previous studies using low-resolution microarrays have indicated that H2A.Z might be inhibitory to transcriptional initiation, as its occupancy was shown to correlate negatively with transcription rate (51,52). However, when H2A.Z is acetylated it is associated with actively transcribed genes (53). There is also evidence that H2A.Z promotes transcriptional elongation. In its absence, nucleosome occupancy increases over the GAL10-VPS13 gene locus, and the elongation rate of RNA Pol II decreases by ∼24% (54).
We began by examining whether there was a correlation between the transcript levels of a gene and H2A.Z incorporation at its +1 nucleosome. Ranking transcripts based on their expression level did not show any overall correlation with H2A.Z incorporation (Figure 4 and Supplementary Figure S9A). Instead, we found that genes with very high expression or very low expression both showed low levels of H2A.Z incorporation, while the majority of genes displayed fairly uniform levels of H2A.Z over a broad range of expression/transcript levels (Figure 4). Because steady-state transcript levels reflect a combination of transcription rate and RNA turnover rates, and H2A.Z might be expected to correlate with only transcription rate, we also examined the relationship of H2A.Z occupancy with the occupancy of RNA Pol II Ser-5P, which is an indirect measure of transcription rate. In yeast RNA Pol II Ser-5P levels are fairly well correlated with RNA levels (41). However, similar to transcript levels, RNA Pol II Ser-5P occupancy also did not show any overall correlation with H2A.Z incorporation (Supplementary Figure S9B).
Figure 4.
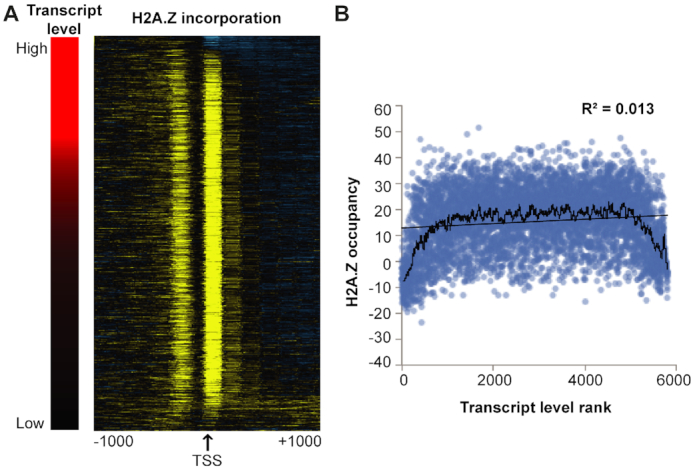
H2A.Z incorporation at the +1 nucleosome does not correlate with gene expression. (A) Heat map displaying H2A.Z incorporation across gene TSS regions (right) when sorted by transcript level as measured by read counts mapping to a transcript, normalized by transcript length (left). The H2A.Z occupancy data on the right is sorted according to the ranked transcript levels on the left. (B) H2A.Z occupancy at the +1 nucleosome plotted against ranked gene expression values. Ranking was used to accommodate outliers that would otherwise skew the plot. The black line represents a 50-gene moving average of H2A.Z occupancy levels.
H2A.Z incorporation corresponds to a +1 nucleosome in the direction of transcription, even at the ‘−1’ nucleosome flanking the NDR
A number of studies have described high levels of antisense transcription occurring upstream of gene promoters (55). As promoter antisense transcription might also contribute to the chromatin structure at the promoter, we investigated the relationship between H2A.Z incorporation and upstream antisense transcription. For this analysis we combined antisense transcription data previously generated in our lab using SMORE-seq (38) with newly performed SMORE-seq to obtain high quality antisense transcription measurements for each gene. The upstream antisense noncoding (UAN) RNAs produced at promoters are analogous to previously described bidirectional noncoding RNAs (or BNCs) (38). Here, we focused on upstream antisense transcription at tandemly arranged genes because such transcription is not confounded by the presence of full-length transcripts from divergent promoters (Supplementary Figure S10).
We separated genes by TSS orientation and ranked them by upstream antisense transcription levels within a window suitable for their detection (from −50 to −300 bp upstream of the TSS). At tandem genes, the H2A.Z-containing nucleosome upstream of the TSS can be unambiguously identified as one independent of the upstream gene promoter, and this nucleosome (at the −1 position) showed H2A.Z incorporation proportional to the UAN-RNA transcription level which was used to rank the genes (Figure 5A). Although divergent genes also appeared to show a similar trend, they displayed detectable amounts of H2A.Z incorporation in nucleosomal arrays extending further upstream from the NDR. Arranging these divergent genes by distance to the upstream TSS revealed that much of the diffuse ‘−1’ nucleosome signal was a result of upstream TSS that were farther away (Figure 5A). Thus, genes with the highest H2A.Z incorporation at the ‘−1’ nucleosome correspond to those where two divergent coding genes share an NDR, and presumably a bidirectional promoter. For the remaining tandem genes and divergent genes with well-separated promoters, it is the antisense transcription of UAN-RNAs that corresponds to the strong incorporation of H2A.Z signal at the −1 nucleosome, which is therefore more properly regarded as the +1 nucleosome for the UAN-RNA. We investigated this association at tandem genes by separating genes based on UAN-RNA levels and determined that there were significant differences in H2A.Z incorporation among the groups (Figure 5B and C). In general, the extent of H2A.Z incorporation into the −1 nucleosome was indicative of the transcription of promoter-associated UAN-RNAs. We obtained similar results when we used data from nascent elongating transcription and sequencing (NET-seq) analysis (44) in place of SMORE-seq (Supplementary Figure S11).
Figure 5.

H2A.Z incorporation at the −1 nucleosome increases with increasing antisense transcription. (A) H2A.Z occupancy data measured by MNase ChIP-seq is plotted for genes segregated based on the orientation of the upstream gene at the TSS. Tandem and divergent genes are first arranged by antisense transcription level in decreasing order (two left panels). The apparent correlation between antisense transcription and H2A.Z incorporation at the −1 nucleosome at divergent genes reflects differences in the intergenic distance between transcript TSS. At divergent genes, it is difficult to distinguish the nucleosome at the 5′ end of the upstream transcript from a nucleosome corresponding to the UAN-RNA transcript. To resolve these, in the second to last heatmap, divergent genes were sorted based on the distance to the upstream TSS. Finally, divergent genes with a TSS-to-TSS distance >350 bp were re-sorted based on the level of UAN-RNA transcription (right-most panel). (B and C) Tandem transcripts were grouped into quartiles according to their UAN-RNA transcription levels. (B) Average profiles are plotted for the quartiles. Overall, H2A.Z occupancy at the −1 nucleosome increases in concert with increasing UAN-RNA transcription level. (C) Boxplots of H2A.Z occupancy at the −1 nucleosome. Welch's t-tests and ANOVA were performed to compare the averages between the four groups, and all comparisons yielded P-values indicating that the differences were significant (P < 2.2 × 10−16).
The increased H2A.Z incorporation at divergent genes cannot be explained by increased Swr1 targeting to the NDR, given that levels of Swr1 binding were roughly equivalent between tandem and divergent genes (Supplementary Figure S7A). Divergent transcripts show some H2A.Z incorporation at the −2 and −3 nucleosomes, whereas tandem genes only display this incorporation at the −1 nucleosome (Supplementary Figure S12). This pattern is consistent with increased nucleosomal displacement occurring at divergent genes where upstream transcripts are coding and longer. By contrast, antisense upstream transcripts associated with tandem gene promoters are short, and transcription at these regions could, therefore, be less likely to displace H2A.Z in the direction of transcription.
H2A.Z has also previously been reported to be incorporated at the 3′ end of genes, where it has been reported to be associated with antisense transcripts originating from these 3′ ends (5). However, the dense packaging of the yeast genome makes it possible for UAN-RNA transcription emanating from a downstream promoter to be mistaken for a general class of 3′ originating transcripts. To establish the true nature of the 3′ H2A.Z signal, we separated genes based on their orientation at the transcription termination site (TTS) and arranged them by the distance to the next downstream TSS (Figure 6). This clearly showed that convergent genes (tail-to-tail orientation) lacked a 3′ H2A.Z signal altogether. In contrast, while tandem TTS genes showed an apparent 3′ H2A.Z enrichment, this signal was clearly aligned with the downstream TSS rather than remaining localized at the primary TTS, indicating that the apparent 3′ H2A.Z incorporation signal originates from the UAN-RNA transcript of the downstream promoter. These results are in agreement with previous microarray-based observations which lacked our resolution and genome wide scope (4).
Figure 6.
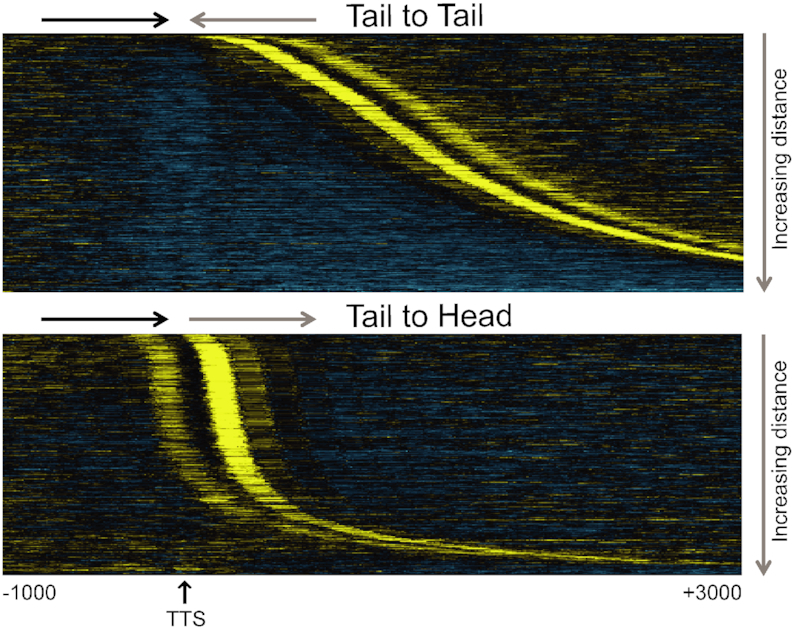
H2A.Z localization by gene orientation at the TTS. Heat map showing H2A.Z localization at Tail-to-Tail (convergent) and Tail-to-Head (divergent) TTS orientation genes. The annotated TTS of the left-hand transcript is indicated, and the plot is sorted by increasing distance to the nearest downstream TSS of the gene on the right. Distances shown are in bp.
H2A.Z incorporation at TATA box-containing promoters is disorganized
In general, genes with TATA box-containing promoters exhibit distinct transcriptional characteristics. On average, they produce higher transcript levels and lower levels of upstream antisense transcription (Supplementary Figure S13) (38). Genes with TATA-containing promoters also exhibit more dynamic expression levels and are enriched for genes up-regulated during environmental stress and depleted for housekeeping genes (56). Since H2A.Z marks sites of bidirectional transcription, we wanted to explore H2A.Z incorporation at TATA box-associated TSS. A previous study using microarray hybridization and an 80-gene sliding window analysis showed that regions of the genome with higher numbers of TATA boxes displayed lower H2A.Z occupancy (51).
We found that although H2A.Z localizes to the TSS of TATA-containing genes, its incorporation is less apparent and the signal more diffuse than the precise and strong signal found at TATA-less genes (Figure 7A, B and Supplementary Figure S14). The average profile reveals that at TATA-containing genes, less H2A.Z is incorporated into the +1 and −1 nucleosomes, and that it can be incorporated at low levels throughout the gene body and within the NDR (Figure 7B). This difference was also apparent when comparing enrichment at called +1 nucleosomes (Supplementary Figure S14). These results indicate that, when a TATA box is present, H2A.Z is less likely to be incorporated in nucleosomes surrounding the NDR, and that when it is incorporated, its localization at the +1 nucleosome is not as strictly maintained. The Swr1 and Ino80 binding patterns at TATA-containing and TATA-less genes revealed that in WT cells, both complexes were depleted from TATA-containing genes, whereas Ino80 accumulated at the TATA-containing genes in the absence of H2A.Z (Figure 7C). Similarly, at highly expressed genes in WT cells, Swr1 and Ino80 showed reduced binding at the NDR relative to other genes in the genome but in the absence of H2A.Z, Ino80 in particular accumulated at the NDR and upstream of these genes (Supplementary Figure S15). The reason for increased occupancy of TATA-containing genes by Ino80 in the absence of H2A.Z is not clear at present. One possibility could be that the low levels of H2A.Z at TATA-containing genes are actively maintained by the evicting actions of Ino80 which is somehow retained at these promoters in the absence of H2A.Z. This predicts an increase in H2A.Z accumulation at these promoters in the absence of Ino80. However, when we examined previously published data for H2A.Z occupancy in the absence of Ino80, there was no accumulation of H2A.Z at TATA-containing promoters (Supplementary Figure S16).
Figure 7.

H2A.Z localization at the TSS with respect to the TATA box and at ribosomal protein genes. (A) Heatmap of H2A.Z localization at genes with TATA-less promoters, ribosomal protein (RP) genes, and TATA box-containing genes. Six RP genes also have TATA boxes in their promoters, but their H2A.Z occupancy pattern is similar to the other 130 RP genes. (B) Average H2A.Z levels across TATA-containing and TATA-less genes. Genes with TATA boxes show lower +1 and −1 nucleosome incorporation of H2A.Z, but also display increased H2A.Z levels in the gene body. (C) Swr1 and Ino80 binding levels at TATA-containing and TATA-less genes in WT and htz1Δ background strains. (D) Average H2A.Z occupancy levels across RP and non-RP genes. RP genes are strongly depleted for H2A.Z. (E) Swr1 and Ino80 binding levels at RP and Non-RP genes in WT and htz1Δ background strains.
H2A.Z is depleted from ribosomal genes
The 78 proteins in the yeast ribosome are encoded for by 137 gene loci (57). These ribosomal protein (RP) genes, which tend to be highly expressed, were previously shown to be depleted for H2A.Z, in contrast to the enrichments seen at mitochondrial RP genes and ribosome biogenesis genes (48,51). We also found a pronounced depletion of H2A.Z at RP genes (Figure 7A, D). Swr1 was also extremely depleted from the NDRs of these genes (Figure 7E). However, similar to TATA-containing genes, Ino80 showed higher occupancy of the NDR of RP genes in the absence of H2A.Z (Figure 7E). Again, however, there was no increase of H2A.Z at RP gene promoters in the absence of Ino80, leaving open the question of why Ino80 preferentially binds these promoters in the absence of H2A.Z (Supplementary Figure S16) (58,59).
The presence of a spliced intron has been reported to increase the directionality associated with an NDR (60). Given the association of H2A.Z with bidirectional transcription, we examined the relationship of H2A.Z occupancy with intron presence. In yeast, intron-containing genes overlap the RP genes to a large degree; out of 307 genes containing introns in the yeast genome, 115 are RP genes. When we examined H2A.Z occupancy at these genes, we found that RP genes were strongly depleted for H2A.Z but intron-containing non-RP genes were not (Supplementary Figure S17A). On average, RP genes displayed higher levels of sense transcription and lower levels of upstream antisense transcription than intron-containing non-RP genes and intron-less non-RP genes (Supplementary Figure S17B, C). These results corroborate a strong bias in H2A.Z localization to sites of bidirectional transcription.
DISCUSSION
The prevailing notion of a core promoter is that it comprises an NDR bounded by two nucleosomes, labeled as the +1 nucleosome in the direction of transcription and the −1 nucleosome forming the upstream boundary. Our data reveal that the −1 nucleosome, which appears as the upstream barrier of the NDR, is in reality also a +1 nucleosome but for a divergently transcribed gene or short upstream antisense non-coding RNA. The apparent 3′ end incorporation of H2A.Z is also due to its presence at the start of a transcript that happens to overlap the 3′ end of a gene. Thus, H2A.Z is present almost exclusively in the single nucleosome at a transcription start site (Figure 8A, B). The finding that H2A.Z levels correlate with UAN RNAs but not steady-state transcript levels could be due to the fact that the non-coding UAN RNA promoters are weaker and these transcripts are short, with short half-lives that are likely similar. Therefore, the steady-state levels we measure in our RNA-seq experiments are likely to be proportional to initiation rate and H2A.Z incorporation. mRNA steady-state levels however are a function of both initiation rate and varying half-lives, complicating the relationship between H2A.Z occupancy and steady-state levels. Our results also shed light on the different patterns of H2A.Z occupancy at promoters in different organisms. In humans, peaks of H2A.Z enrichment are also found at both sides of the NDR, and correspondingly, bidirectional promoters producing bidirectional transcription are common (61,62). In contrast, the Drosophila NDR shows H2A.Z enrichment at the +1 nucleosome but not at the −1 (63). The lack of an upstream −1 nucleosome containing H2A.Z could reflect the fact that Drosophila gene promoters also display a pronounced lack of bidirectional transcription and a larger number of directional motifs (64).
Figure 8.
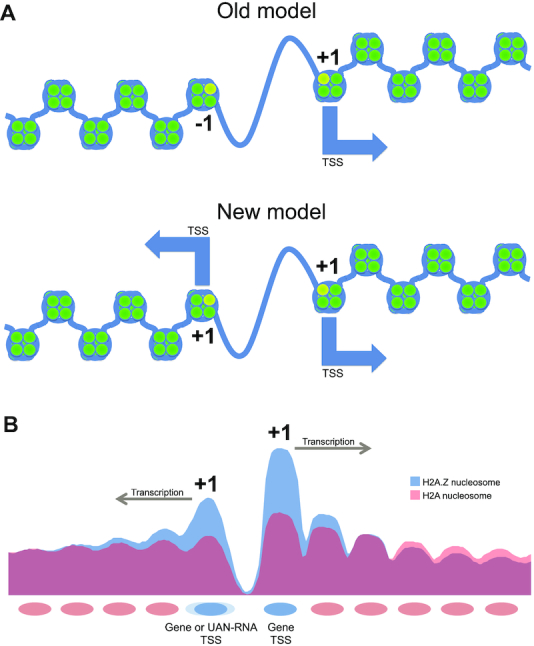
Models of H2A.Z incorporation at the NDR. (A) Earlier view showing H2A.Z incorporation at both the +1 and −1 nucleosomes flanking the NDR at a promoter. The new model supported by our data shows that incorporation of H2A.Z at both sides of an NDR is indicative of transcription in both directions. Hence, −1 nucleosomes that incorporate H2A.Z are +1 nucleosomes of divergent transcripts. (B) Average nucleosome occupancy levels for both H2A.Z-containing nucleosomes and background H2A containing nucleosomes around the TSS.
What determines the precise localization of H2A.Z to the +1 nucleosome? It has been proposed that Swr1 localization to the NDR explains it, but our data suggests that Swr1 is unlikely to be the exclusive determinant, for many reasons. First, Swr1 localization to the NDR is not as strong and specific as that of H2A.Z (Figures 1 and 3A). Second, the correlation between Swr1 signal and H2A.Z signal at the promoter is poor (Figure 2B), though this could be due to the fact that ChIP measures steady-state levels of occupancy rather than recruitment directly. Third, Swr1 localization to the NDR is itself strongly dependent on the presence of H2A.Z at the +1 nucleosome, inconsistent with a simple model where Swr1 localization to the NDR occurs independently of H2A.Z and directs H2A.Z incorporation. Finally, Swr1 occupancy of the promoters of divergent and tandem promoters is indistinguishable, but the extent of H2A.Z incorporation at the two flanking nucleosomes is markedly different. Thus, the sharp localization of H2A.Z to the single nucleosome at the start site of transcription suggests that additional factors such as promoter sequence elements, the transcription machinery and the initiation process, in conjunction with Swr1 occupancy at the NDR, serve to define the +1 nucleosome and determine the strong specificity of H2A.Z in the genome at least at genes that are not completely transcriptionally silent. A short 22 bp fragment containing a homopolymeric dA:dT sequence and a Reb1 binding site inserted into a silent gene was shown to be sufficient to specify the formation of an NDR and the incorporation of flanking H2A.Z nucleosomes (4). However, it is not clear that there was no low-level transcription from cryptic promoters in this situation. Swr1 has been reported to switch to a ‘promiscuous’ mode in the context of acetylation of H3 at lysine 56 (H3K56Ac) where it can exchange either H2A or H2A.Z-containing dimers (65). There is also some evidence for random incorporation of H2A.Z inside transcriptionally silent genes in human cells (58,59). Further highlighting the complexities of their relationship, genes that require H2A.Z for expression but not Swr1, as well as genes that require Swr1 but not H2A.Z have been reported, and swr1Δ and htz1Δ differ in drug sensitivity (66).
The chromatin remodeler Ino80 shares a number of subunits with Swr1 as well as with other chromatin remodelers and modifiers that are generally targeted to the NDR (67–69). Ino80 can evict H2A.Z nucleosomes from chromatin in vitro, and, in vivo, H2A.Z has been reported to become redistributed or accumulate at promoters in the absence of key Ino80 complex components (7,11). However, recent evidence suggesting that H2A.Z can be removed from genes by formation of the pre-initiation complex and transcription rather than by Ino80 calls into question the necessity of this chromatin remodeler for the maintenance of H2A.Z occupancy levels (8,9). Despite this, the severely affected phenotype of ino80Δ cells (70) demonstrates that Ino80 plays a vital role in proper cell function. Our data reveals that Ino80 is dramatically redistributed in the absence of H2A.Z, and specifically accumulates at regions that have low H2A.Z occupancy in WT cells. This behavior of Ino80 is not matched by Swr1. It is possible that Ino80 is rapidly recycled at loci where it is most active, such that in the absence of its target H2A.Z, it accumulates. Given the conservation of H2A.Z and its chaperones and the fact that it is affected in human diseases (71,72), similar experiments measuring H2A.Z incorporation in conjunction with precise transcript analysis are likely to shed light on the functions of H2A.Z in normal human cellular biology and in disease.
DATA AVAILABILITY
The genomic datasets from this study have been deposited in NCBI’s Gene Expression Omnibus (GEO) database under accession number GSE104147 (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE104147).
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Genomic Sequencing and Analysis Facility at The University of Texas at Austin and the NGS Facility at the MD Anderson Cancer Center, Science Park (which was supported by CPRIT Core Facility Support Grant RP170002) for Illumina sequencing and the Texas Advanced Computing Center (TACC) and Biomedical Research Computing Facility at UT Austin for the use of computational facilities.
Notes
Present address: Daechan Park, Department of Biological Sciences, College of Natural Sciences, Ajou University, Suwon 16499, Republic of Korea.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH [CA095548 and CA198648 to V.R.I.] (in part). Funding for open access charge: NIH [CA198648].
Conflict of interest statement. None declared.
REFERENCES
- 1. Yuan G.C., Liu Y.J., Dion M.F., Slack M.D., Wu L.F., Altschuler S.J., Rando O.J.. Genome-scale identification of nucleosome positions in S. cerevisiae. Science. 2005; 309:626–630. [DOI] [PubMed] [Google Scholar]
- 2. Albert I., Mavrich T.N., Tomsho L.P., Qi J., Zanton S.J., Schuster S.C., Pugh B.F.. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007; 446:572–576. [DOI] [PubMed] [Google Scholar]
- 3. Iouzalen N., Moreau J., Mechali M.. H2A.ZI, a new variant histone expressed during Xenopus early development exhibits several distinct features from the core histone H2A. Nucleic Acids Res. 1996; 24:3947–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Raisner R.M., Hartley P.D., Meneghini M.D., Bao M.Z., Liu C.L., Schreiber S.L., Rando O.J., Madhani H.D.. Histone variant H2A.Z marks the 5′ ends of both active and inactive genes in euchromatin. Cell. 2005; 123:233–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gu M., Naiyachit Y., Wood T.J., Millar C.B.. H2A.Z marks antisense promoters and has positive effects on antisense transcript levels in budding yeast. BMC Genomics. 2015; 16:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mizuguchi G., Shen X., Landry J., Wu W.H., Sen S., Wu C.. ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science. 2004; 303:343–348. [DOI] [PubMed] [Google Scholar]
- 7. Papamichos-Chronakis M., Watanabe S., Rando O.J., Peterson C.L.. Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell. 2011; 144:200–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jeronimo C., Watanabe S., Kaplan C.D., Peterson C.L., Robert F.. The histone chaperones FACT and Spt6 Restrict H2A.Z from intragenic locations. Mol. Cell. 2015; 58:1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tramantano M., Sun L., Au C., Labuz D., Liu Z., Chou M., Shen C., Luk E.. Constitutive turnover of histone H2A.Z at yeast promoters requires the preinitiation complex. Elife. 2016; 5:e14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ranjan A., Mizuguchi G., FitzGerald P.C., Wei D., Wang F., Huang Y., Luk E., Woodcock C.L., Wu C.. Nucleosome-free region dominates histone acetylation in targeting SWR1 to promoters for H2A.Z replacement. Cell. 2013; 154:1232–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yen K., Vinayachandran V., Pugh B.F.. SWR-C and INO80 chromatin remodelers recognize nucleosome-free regions near +1 nucleosomes. Cell. 2013; 154:1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Altaf M., Auger A., Monnet-Saksouk J., Brodeur J., Piquet S., Cramet M., Bouchard N., Lacoste N., Utley R.T., Gaudreau L. et al.. NuA4-dependent acetylation of nucleosomal histones H4 and H2A directly stimulates incorporation of H2A.Z by the SWR1 complex. J. Biol. Chem. 2010; 285:15966–15977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Neves L.T., Douglass S., Spreafico R., Venkataramanan S., Kress T.L., Johnson T.L.. The histone variant H2A.Z promotes efficient cotranscriptional splicing in S. cerevisiae. Genes Dev. 2017; 31:702–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gonzalez-Arranz S., Cavero S., Morillo-Huesca M., Andujar E., Perez-Alegre M., Prado F., San-Segundo P.. Functional impact of the H2A.Z histone variant during meiosis in saccharomyces cerevisiae. Genetics. 2018; 209:997–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brahma S., Henikoff S.. RSC-Associated subnucleosomes define MNase-Sensitive promoters in yeast. Mol. Cell. 2019; 73:238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Singh R.K., Fan J., Gioacchini N., Watanabe S., Bilsel O., Peterson C.L.. Transient kinetic analysis of SWR1C-Catalyzed H2A.Z deposition unravels the impact of nucleosome dynamics and the asymmetry of histone exchange. Cell Rep. 2019; 27:374–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cakiroglu A., Clapier C.R., Ehrensberger A.H., Darbo E., Cairns B.R., Luscombe N.M., Svejstrup J.Q.. Genome-wide reconstitution of chromatin transactions reveals that RSC preferentially disrupts H2AZ-containing nucleosomes. Genome Res. 2019; 29:988–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Santisteban M.S., Kalashnikova T., Smith M.M.. Histone H2A.Z regulates transcription and is partially redundant with nucleosome remodeling complexes. Cell. 2000; 103:411–422. [DOI] [PubMed] [Google Scholar]
- 19. Zlatanova J., Thakar A.. H2A.Z: view from the top. Structure. 2008; 16:166–179. [DOI] [PubMed] [Google Scholar]
- 20. Thakar A., Gupta P., Ishibashi T., Finn R., Silva-Moreno B., Uchiyama S., Fukui K., Tomschik M., Ausio J., Zlatanova J.. H2A.Z and H3.3 histone variants affect nucleosome structure: biochemical and biophysical studies. Biochemistry. 2009; 48:10852–10857. [DOI] [PubMed] [Google Scholar]
- 21. Bonisch C., Hake S.B.. Histone H2A variants in nucleosomes and chromatin: more or less stable. Nucleic Acids Res. 2012; 40:10719–10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Weber C.M., Ramachandran S., Henikoff S.. Nucleosomes are context-specific, H2A.Z-modulated barriers to RNA polymerase. Mol. Cell. 2014; 53:819–830. [DOI] [PubMed] [Google Scholar]
- 23. Dion M.F., Kaplan T., Kim M., Buratowski S., Friedman N., Rando O.J.. Dynamics of replication-independent histone turnover in budding yeast. Science. 2007; 315:1405–1408. [DOI] [PubMed] [Google Scholar]
- 24. Dhillon N., Oki M., Szyjka S.J., Aparicio O.M., Kamakaka R.T.. H2A.Z functions to regulate progression through the cell cycle. Mol. Cell Biol. 2006; 26:489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Adam M., Robert F., Larochelle M., Gaudreau L.. H2A.Z is required for global chromatin integrity and for recruitment of RNA polymerase II under specific conditions. Mol. Cell Biol. 2001; 21:6270–6279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lemieux K., Larochelle M., Gaudreau L.. Variant histone H2A.Z, but not the HMG proteins Nhp6a/b, is essential for the recruitment of Swi/Snf, Mediator, and SAGA to the yeast GAL1 UAS(G). Biochem. Biophys. Res. Commun. 2008; 369:1103–1107. [DOI] [PubMed] [Google Scholar]
- 27. Brunelle M., Nordell Markovits A., Rodrigue S., Lupien M., Jacques P.E., Gevry N.. The histone variant H2A.Z is an important regulator of enhancer activity. Nucleic Acids Res. 2015; 43:9742–9756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Day D.S., Zhang B., Stevens S.M., Ferrari F., Larschan E.N., Park P.J., Pu W.T.. Comprehensive analysis of promoter-proximal RNA polymerase II pausing across mammalian cell types. Genome Biol. 2016; 17:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Subramanian V., Mazumder A., Surface L.E., Butty V.L., Fields P.A., Alwan A., Torrey L., Thai K.K., Levine S.S., Bathe M. et al.. H2A.Z acidic patch couples chromatin dynamics to regulation of gene expression programs during ESC differentiation. PLos Genet. 2013; 9:e1003725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zovkic I.B., Paulukaitis B.S., Day J.J., Etikala D.M., Sweatt J.D.. Histone H2A.Z subunit exchange controls consolidation of recent and remote memory. Nature. 2014; 515:582–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zovkic I.B., Walters B.J.. H2A.Z helps genes remember their history so we can remember ours. Bioessays. 2015; 37:596–601. [DOI] [PubMed] [Google Scholar]
- 32. Yang Y., Yamada T., Hill K.K., Hemberg M., Reddy N.C., Cho H.Y., Guthrie A.N., Oldenborg A., Heiney S.A., Ohmae S. et al.. Chromatin remodeling inactivates activity genes and regulates neural coding. Science. 2016; 353:300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Winzeler E.A., Shoemaker D.D., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J.D., Bussey H. et al.. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999; 285:901–906. [DOI] [PubMed] [Google Scholar]
- 34. Ghaemmaghami S., Huh W.K., Bower K., Howson R.W., Belle A., Dephoure N., O'Shea E.K., Weissman J.S.. Global analysis of protein expression in yeast. Nature. 2003; 425:737–741. [DOI] [PubMed] [Google Scholar]
- 35. Knop M., Siegers K., Pereira G., Zachariae W., Winsor B., Nasmyth K., Schiebel E.. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast. 1999; 15:963–972. [DOI] [PubMed] [Google Scholar]
- 36. Shivaswamy S., Bhinge A., Zhao Y., Jones S., Hirst M., Iyer V.R.. Dynamic remodeling of individual nucleosomes across a eukaryotic genome in response to transcriptional perturbation. PLoS Biol. 2008; 6:e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wal M., Pugh B.F.. Genome-wide mapping of nucleosome positions in yeast using high-resolution MNase ChIP-Seq. Methods Enzymol. 2012; 513:233–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Park D., Morris A.R., Battenhouse A., Iyer V.R.. Simultaneous mapping of transcript ends at single-nucleotide resolution and identification of widespread promoter-associated non-coding RNA governed by TATA elements. Nucleic Acids Res. 2014; 42:3736–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Park D., Shivram H., Iyer V.R.. Chd1 co-localizes with early transcription elongation factors independently of H3K36 methylation and releases stalled RNA polymerase II at introns. Epigenet. Chromatin. 2014; 7:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee Y., Park D., Iyer V.R.. The ATP-dependent chromatin remodeler Chd1 is recruited by transcription elongation factors and maintains H3K4me3/H3K36me3 domains at actively transcribed and spliced genes. Nucleic Acids Res. 2017; 45:7180–7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xu Z., Wei W., Gagneur J., Perocchi F., Clauder-Munster S., Camblong J., Guffanti E., Stutz F., Huber W., Steinmetz L.M.. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009; 457:1033–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Dijk E.L., Chen C.L., d’Aubenton-Carafa Y., Gourvennec S., Kwapisz M., Roche V., Bertrand C., Silvain M., Legoix-Ne P., Loeillet S. et al.. XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature. 2011; 475:114–117. [DOI] [PubMed] [Google Scholar]
- 44. Harlen K.M., Trotta K.L., Smith E.E., Mosaheb M.M., Fuchs S.M., Churchman L.S.. Comprehensive RNA polymerase II interactomes reveal distinct and varied roles for each Phospho-CTD residue. Cell Rep. 2016; 15:2147–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rhee H.S., Pugh B.F.. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature. 2012; 483:295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nakao A., Yoshihama M., Kenmochi N.. RPG: The ribosomal protein gene database. Nucleic Acids Res. 2004; 32:D168–D170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Park D., Lee Y., Bhupindersingh G., Iyer V.R.. Widespread misinterpretable ChIP-seq Bias in Yeast. PLoS One. 2013; 8:e83506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rhee H.S., Bataille A.R., Zhang L., Pugh B.F.. Subnucleosomal structures and nucleosome asymmetry across a genome. Cell. 2014; 159:1377–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ramachandran S., Zentner G.E., Henikoff S.. Asymmetric nucleosomes flank promoters in the budding yeast genome. Genome Res. 2015; 25:381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vinayachandran V., Reja R., Rossi M.J., Park B., Rieber L., Mittal C., Mahony S., Pugh B.F.. Widespread and precise reprogramming of yeast protein-genome interactions in response to heat shock. Genome Res. 2018; 28:357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang H., Roberts D.N., Cairns B.R.. Genome-wide dynamics of Htz1, a histone H2A variant that poises repressed/basal promoters for activation through histone loss. Cell. 2005; 123:219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li B., Pattenden S.G., Lee D., Gutierrez J., Chen J., Seidel C., Gerton J., Workman J.L.. Preferential occupancy of histone variant H2AZ at inactive promoters influences local histone modifications and chromatin remodeling. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:18385–18390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Millar C.B., Xu F., Zhang K., Grunstein M.. Acetylation of H2AZ Lys 14 is associated with genome-wide gene activity in yeast. Genes Dev. 2006; 20:711–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Santisteban M.S., Hang M., Smith M.M.. Histone variant H2A.Z and RNA polymerase II transcription elongation. Mol. Cell Biol. 2011; 31:1848–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Neil H., Malabat C., d’Aubenton-Carafa Y., Xu Z., Steinmetz L.M., Jacquier A.. Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature. 2009; 457:1038–1042. [DOI] [PubMed] [Google Scholar]
- 56. Basehoar A.D., Zanton S.J., Pugh B.F.. Identification and distinct regulation of yeast TATA box-containing genes. Cell. 2004; 116:699–709. [DOI] [PubMed] [Google Scholar]
- 57. Warner J.R. The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 1999; 24:437–440. [DOI] [PubMed] [Google Scholar]
- 58. Hardy S., Jacques P.E., Gevry N., Forest A., Fortin M.E., Laflamme L., Gaudreau L., Robert F.. The euchromatic and heterochromatic landscapes are shaped by antagonizing effects of transcription on H2A.Z deposition. PLoS Genet. 2009; 5:e1000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hardy S., Robert F.. Random deposition of histone variants: A cellular mistake or a novel regulatory mechanism. Epigenetics. 2010; 5:368–372. [DOI] [PubMed] [Google Scholar]
- 60. Agarwal N., Ansari A.. Enhancement of transcription by a splicing-competent intron is dependent on promoter directionality. PLos Genet. 2016; 12:e1006047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Trinklein N.D., Aldred S.F., Hartman S.J., Schroeder D.I., Otillar R.P., Myers R.M.. An abundance of bidirectional promoters in the human genome. Genome Res. 2004; 14:62–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Preker P., Almvig K., Christensen M.S., Valen E., Mapendano C.K., Sandelin A., Jensen T.H.. PROMoter uPstream Transcripts share characteristics with mRNAs and are produced upstream of all three major types of mammalian promoters. Nucleic Acids Res. 2011; 39:7179–7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mavrich T.N., Jiang C., Ioshikhes I.P., Li X., Venters B.J., Zanton S.J., Tomsho L.P., Qi J., Glaser R.L., Schuster S.C. et al.. Nucleosome organization in the Drosophila genome. Nature. 2008; 453:358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Core L.J., Waterfall J.J., Gilchrist D.A., Fargo D.C., Kwak H., Adelman K., Lis J.T.. Defining the status of RNA polymerase at promoters. Cell Rep. 2012; 2:1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Watanabe S., Radman-Livaja M., Rando O.J., Peterson C.L.. A histone acetylation switch regulates H2A.Z deposition by the SWR-C remodeling enzyme. Science. 2013; 340:195–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Morillo-Huesca M., Clemente-Ruiz M., Andujar E., Prado F.. The SWR1 histone replacement complex causes genetic instability and genome-wide transcription misregulation in the absence of H2A.Z. PLoS One. 2010; 5:e12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Morrison A.J., Shen X.. Chromatin remodelling beyond transcription: the INO80 and SWR1 complexes. Nat. Rev. Mol. Cell Biol. 2009; 10:373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bao Y., Shen X.. SnapShot: chromatin remodeling: INO80 and SWR1. Cell. 2011; 144:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gerhold C.B., Hauer M.H., Gasser S.M.. INO80-C and SWR-C: guardians of the genome. J. Mol. Biol. 2015; 427:637–651. [DOI] [PubMed] [Google Scholar]
- 70. Papamichos-Chronakis M., Peterson C.L.. The Ino80 chromatin-remodeling enzyme regulates replisome function and stability. Nat. Struct. Mol. Biol. 2008; 15:338–345. [DOI] [PubMed] [Google Scholar]
- 71. Yang H.D., Kim P.J., Eun J.W., Shen Q., Kim H.S., Shin W.C., Ahn Y.M., Park W.S., Lee J.Y., Nam S.W.. Oncogenic potential of histone-variant H2A.Z.1 and its regulatory role in cell cycle and epithelial-mesenchymal transition in liver cancer. Oncotarget. 2016; 7:11412–11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Vardabasso C., Hake S.B., Bernstein E.. Histone variant H2A.Z.2: a novel driver of melanoma progression. Mol. Cell Oncol. 2016; 3:e1073417. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genomic datasets from this study have been deposited in NCBI’s Gene Expression Omnibus (GEO) database under accession number GSE104147 (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE104147).