

Introduction
 Access the complete reference list online at http://www.expertconsult.com
Access the complete reference list online at http://www.expertconsult.com
The relationship between two living organisms can be classified as parasitic, symbiotic, or commensal.1, 2, 3 This same classification scheme can be used to describe relationships between microorganisms and more complex living organisms that act as hosts. The term parasite is used here in its broad sense to mean a microorganism interacting with another organism (either vertebrate or invertebrate) in the same ecologic niche.
The following definitions are used in this chapter:
Parasitism: Association between two different organisms wherein one benefits at the expense of the other. All infectious agents causing illness belong to this category.
Commensalism: Association between two organisms in which one derives benefit from the other without causing it any harm. This intermediate category is not uniformly accepted. Often, upon detailed analysis, the relationship turns out to be either parasitic or symbiotic.2
Symbiosis or mutualism: Both organisms benefit from the relationship. The type of relationship also depends on host factors. For example, bacteria normally inhabiting the bowel live in an apparent commensal or (by inhibiting potential pathogens) symbiotic relationship with humans. However, in cases of cirrhosis with consequent hepatic insufficiency, bacteria can become a dangerous source of ammonia that leads to hepatic encephalopathy. A commensal relationship can be transformed into a potentially harmful one.
Microbial Factors
Principles of Microbial Evolution and Classification
The Earth is approximately 4.5 to 5 billion years old. There is good fossil evidence of microbial life approximately 3.5 billion years ago. Microbial life (stromatolites) was mostly photosynthetic, unicellular, and anaerobic.1, 4 Eukaryotes, bacteria, and archaea evolved from a still hypothetical universal common ancestor.5, 6, 7 Eukaryotes then evolved into protozoans, metazoans, plants, and animals, as we know them today. Moreover, there is strong evidence that primitive eukaryotic cells established relationships with bacterial organisms that later evolved into cytoplasmic organelles, such as chloroplasts in plants and mitochondria in animals.8
To put things into perspective, approximately five-sixths of the history of life on Earth has been exclusively microbial. Human beings appeared on the planet only 2 million years ago as very late newcomers to the biosphere. Life was initially anaerobic, but with the appearance of photosynthetic organisms and chloroplasts, oxygen was released into the atmosphere for the first time.9 Radiation in the upper atmosphere created the ozone layer from molecular oxygen, which then shielded the Earth's surface from dangerous radiation. Nucleic acids were therefore protected from harmful mutations. Organisms had to evolve to survive in the presence of oxygen. A few of the ancient anaerobes were able to survive in the highly oxidant atmosphere, and they represent the anaerobes as we know them today.
The phylogeny of living organisms is based on molecular approaches, particularly analysis of ribosomal RNA.5, 6, 10 Because of the antiquity of the protein synthesis machinery, these molecules are excellent evolutionary clocks. For prokaryotes, the 16S subunit of ribosomes is the most useful for classification purposes.
Viruses deserve special comment because of their molecular simplicity and at the same time their importance as human pathogens and as possible agents of hereditary changes and cancer.11, 12 A virus is a genetic element with either DNA or RNA coated by protein of viral origin and sometimes enveloped by lipid material of host origin. Some viruses have enzymes that are necessary for their replication. The only criterion that these organisms fulfill to be considered living organisms is that of reproduction. They are inert particles when outside of the host cell, and once they have access into a cell they become active and the cell is subverted to produce more viral particles. Sometimes the cell dies in the process, and sometimes the relationship is stable. Viral hosts include bacteria, protozoa, animals, and plants.
The classification of viruses is based on different criteria than the ones used for other organisms. The major criteria are type of nucleic acid, presence or absence of an envelope, manner of replication, and morphologic characteristics.12, 13
Simpler forms of self-replicating organisms include virusoids and viroids.14, 15 The former are satellite RNAs that are found encapsidated in the proteins encoded by their helper virus (e.g., hepatitis caused by the hepatitis D virus delta agent in conjunction with hepatitis B virus). The viroids are mostly plant pathogens that consist of single-stranded circular RNA molecules.
The concept of “infectious agent” was revolutionized by the discovery of proteinaceous infectious agents known as prions. These proteins are responsible for neurodegenerative diseases in animals and humans. The protein particles lack nucleic acids but still are able to reproduce and trigger conformational changes in host proteins, leading to cell death.16, 17
In contrast, protozoa are nucleated, single-cell organisms that, depending on the species, replicate by means as simple as binary fission (e.g., Trichomonas) or as complex as involving multiple sexual and asexual stages in both animal and invertebrate hosts (e.g., plasmodia). Protozoa include amebae (e.g., Entamoeba histolytica), flagellates (e.g., Giardia lamblia), ciliates (e.g., Balantidium coli), and sporozoa (e.g., Cryptosporidium). Even more complex are helminths, which are multicellular metazoan organisms with highly developed internal organs, including alimentary and reproductive tracts. The helminths include nematodes (roundworms), cestodes (tapeworms), and trematodes (flukes). Many helminths have complex life cycles with multiple developmental stages both in the animal host and in intermediate invertebrate or vertebrate hosts. Because of their size, helminths, the macroparasites, are solely extracellular pathogens; because of their prolonged life cycles and generation times, their capacity for genetic alteration is diminished compared to smaller, simpler microbes (the microparasites).
Development of Microbial Virulence
Evolution of Virulence
The traditional view assumes that natural selection would favor evolution toward a benign coexistence between host and parasite.18, 19 A modern view of evolution of virulence focuses on the tradeoff between the benefits that pathogens accrue through increased exploitation of hosts and the costs that result from any effects of disease that reduce transmission to susceptible hosts.19, 20 From this point of view, virulence could be the evolved as well as the primitive stage of the association between host and parasite, depending on the development of enhanced rather than reduced transmission.
According to Levin19 and Levin and Svanborg-Eden,20 there are three alternative models to explain evolution of a microparasite's virulence: direct selection, coincidental evolution, and short-sighted within-host selection. The direct selection model states that there is a direct relationship between the parasite's virulence and its rate of infectious transmission. The best documented and often cited example is that of the dramatic changes in virulence that the myxoma virus underwent after being released into the wild in Australia to “control” the population of wild rabbits. In the beginning, rabbit mortality and viral transmission rates were high. As the population of rabbits was decimated, the virulence of the virus decreased and its rate of transmission actually increased. This outcome is explained by the longer survival and duration of the period of shedding of the virus. At the same time, more resistant rabbits increased in number due to the selection process.21
According to the coincidental evolution model, the factors responsible for the virulence of a microparasite evolved for some purpose other than to provide the parasite with some advantage within a host or for its transmission to other hosts. Clostridial toxins are good examples in this category. There is no beneficial reason to kill a human host who became infected by Clostridium tetani spores from soil in order for the parasite to survive. They are mostly soil bacteria and do not need humans for their survival.19
Short-sighted within-host evolution posits that the parasites responsible for the morbidity and mortality of the host are selected for as a consequence of within-host evolution since that produces a local advantage for their survival within the host. The host dies and the rate of transmission would decrease. This is an example of evolutionary myopia in which the long-term consequences of killing a host would not matter to the parasite.22, 23 Natural selection is a local phenomenon that happens at a given time and place and goes perfectly with this model. Bacteria such as Neisseria meningitidis that normally live attached to human pharyngeal epithelial cells sometimes invade the central nervous system (CNS) and kill the host. Their replication in the CNS is favored since competition is low and defenses are not as abundant as in the tonsillar areas.19
It follows from the previous paragraphs that evolutionary theories addressing virulence have predicted that virulence of microbes evolves in response to changes in conditions or tradeoffs. One of the most commonly considered “tradeoffs” is between the benefit of a high within-host microbial density (allowing efficient transmission to the next host) and the cost of a reduced longevity of the infection in the host due to high parasite density that tends to kill the host.
Other approaches have been proposed such as the role of how pathogenic mechanisms affect virulence evolution. According to this model, pathogenic mechanisms that manipulate host immunity or escape from the host immune response (longer survival in the host) dominate as causes of virulence compared to mechanisms that incrementally alter transmission (higher microbial density and increased mortality).24, 25 Furthermore, natural selection would act more strongly on mutations influencing clearance than on mutations influencing transmission.26
Another cited model to explain dynamics of virulence evolution is the so-called “source–sink” model in which evolution of bacterial pathogens is evaluated from ecological points of view that microbes switch from permanent (source) and transient (sink) habitats.27
The generation times of mammalian hosts are much longer than those of microorganisms. Therefore, genetic mutations in these hosts, on which natural selection acts, take longer to become part of a large population. Nevertheless, there is evidence that specific microorganisms can exert selective pressure on the gene pool of human hosts. The evidence is strongest for the potentially lethal infections caused by falciparum malaria. In regions of the world where falciparum malaria is endemic, including Africa, there is a high prevalence of genetic mutations that alter hemoglobin structure or synthesis, decreasing or abolishing falciparum malaria parasites’ survival.28 The selective pressure of malaria on human gene expression is not confined solely to affecting erythrocytes but also likely involves the immune system, cytokines, and other systems.29
Other Modes of Altering Virulence and Pathogenicity
Although the selective pressures of evolution generally exert changes over a multitude of centuries, there are other mechanisms that may more rapidly alter microbial pathogenicity, virulence, and drug susceptibility. The expression of mutated genes in microorganisms is heightened when there are greater numbers of organisms and their generation times are brief. Hence, altered gene expression in helminths will be slow to be expressed, whereas in microparasites genetic alterations will be likely to develop. For mycobacterial infections, large numbers of bacilli that persist for a long time facilitate the genetic emergence of drug resistance to a single agent, and this likelihood underlies the principle of using more than a single drug to treat tuberculosis. Even more rapidly dividing microparasites can develop genetic alterations, and this is especially true when the fidelity of genetic replication is poor. This is prominent in human immunodeficiency virus type 1 (HIV-1), whose reverse transcriptase lacks a 3′ exonuclease proofreading activity.30 Alterations in cell tropism, pathogenicity, and drug sensitivity are frequent in HIV-1 infections. Again, several antiviral agents must be employed concomitantly to circumvent the highly frequent mutations that alter drug susceptibility in HIV-1 strains.
In addition to their own genetic material, many classes of microparasites either contain or are capable of acquiring transferable genetic elements in the form of plasmids, transposons, or bacteriophages. These transferable genetic elements also provide a means for the spread of resistance to antibacterial drugs, an increasing problem in all regions of the world.31
Causes of Acute or Chronic Infections in Individuals
One obvious impact of an infectious disease is on the individual infected. Hence, in any region of the world independent of other infectious diseases or malnutrition, the acute infection will cause morbidity and potential mortality in the infected human host. Among otherwise healthy people, the immediate impact of the infection is the symptomatic acute illness. For some infections that have prolonged courses, their impact may also continue over many years. Chronic infections include most of those caused by helminthic parasites, which characteristically live for years; persisting mycobacterial infections; and retroviral infections (HIV-1, HIV-2, and human T-cell lymphotropic virus type 1). Finally, the sequelae of some infections can include the development of neoplasms. Examples include hepatocellular carcinomas associated with chronic hepatitis B and C viral infections, bladder tumors with urinary schistosomiasis, cholangiocarcinomas with biliary fluke infections, and gastric adenocarcinomas and lymphomas associated with Helicobacter pylori infections.
Causes of Widespread Infections in Populations
Infectious diseases may affect not only individuals but also large groups of people or entire populations due to epidemic or highly endemic transmission. Throughout human history, a few microorganisms have been responsible for great epidemics and massive numbers of dead or crippled people as a result of infections spreading locally or throughout the world.32, 33, 34, 35 Typhus has been associated almost always with situations that involve overcrowding, famine, war, natural disasters, and poverty. The outcomes of several European wars were affected by the morbidity and mortality inflicted by typhus or other diseases on the military. Typhus epidemics were common during the world wars of the twentieth century and in the concentration camps where the ecological conditions were ideal for such a disease to spread.30 Today, typhus and other rickettsioses are still public health problems in some countries, but overall the disease was brought under control after its life cycle was described and antibiotics, insecticides, and public health measures became available.34
Bubonic plague, caused by Yersinia pestis, is another disease that has shaped history, especially in Europe during the Middle Ages.35 Millions of people were affected by pandemics that spread throughout the continent. Tuberculosis, smallpox, and measles had a tremendous effect on the native populations of the Americas after Columbus's voyages to the New World. It has been estimated that 90% of the population in Mexico was killed by these pathogens, which were novel to the native residents.
Acquired immunodeficiency syndrome (AIDS) represents the modern pandemic that will continue to affect human history for at least decades. Other examples are cholera and influenza, which are capable of causing pandemics.36
In addition to widespread diseases caused by epidemic spread of infections, some infectious diseases, because of their highly endemic prevalence in populations, continue to affect large segments of the world's population. These include enteric and respiratory infections, measles, malaria, tuberculosis and schistosomiasis. Furthermore, even the staggering mortality and morbidity of these tropical infectious diseases do not control populations but are associated with population overgrowth. This is true not only across the different countries of the world but also throughout the history of developed countries. Thus, the impact of these infections is not solely on the individual but, because of their highly endemic or epidemic occurrence, on populations. This has consequences on economic, political, and social functioning of entire societies.37
Polyparasitism and Effects on Nutrition and Growth
In an otherwise healthy and fully nourished person, a new infection is likely to be the only active infection in that person. In contrast, in regions where enteric and other infections are highly prevalent because of inadequate sanitation and poor socioeconomic conditions, adults and especially children may harbor several infections or be subject to repeated episodes of new enteric pathogens. Thus, the polyparasitism of multiple concurrent or recurrent infections adds a new dimension to the impact of acute infections, not often encountered in developed countries.
Moreover, the subclinical impact of a number of tropical infectious diseases is beginning to become apparent. Increasing data suggest that even “asymptomatic” giardial,38 cryptosporidial,39 and enteroaggregative Escherichia coli 40 infections may be very important in predisposing to malnutrition, thus reflecting a clinically important impact, even in the absence of overt clinical disease such as diarrhea. Likewise, chronic intestinal helminth infections also have a major impact on nutrition in those with already marginal nutrition. Anthelminthic therapy in these children, who lack symptomatic infections, has led to increases in growth, exercise tolerance, and scholastic performance.41, 42
Microbial Interactions with Human Hosts
Just as microorganisms have evolved over centuries or longer, mammalian hosts have evolved to contain and limit the deleterious consequences of infections with diverse microbes. The human immune system is composed of multiple elements, including those of innate immunity and those of adaptive immunity. Many of the elements of innate immunity are more primitive and found in invertebrate organisms, whereas the adaptive immune responses have evolved further in vertebrate hosts. Microorganisms that successfully infect human hosts must, at least in the short term, overcome elements of the host immune system, which then may react further to attempt to control these infections.
The study of microbial pathogenesis has been revolutionized with the advent of comparative genomics and the multitude of genomic tools that have become available in the last two decades or so. The first bacterium whose genome was fully sequenced was that of a laboratory strain of Haemophilus influenzae in 1995, followed by Mycoplasma genitalium the same year. Since then over 250 bacterial genome sequences have become available. Discovery of virulence genes has therefore expanded rapidly and has benefited from other strategies such as powerful computational methods, genetic signatures, analysis of physical linkage to accessory genetic elements, and biochemical and genetic approaches that depend on comprehensive genome sequence information.43 Furthermore, proteomics-based methods have also been used in combination with genome-sequence analysis to define new virulence factors.43 For obligate intracellular pathogens, whose genetic manipulation is far more difficult than for other microbes, the use of genomic and post-genomic tools (genomic-microarray methods and proteomics) has been virtually the only path to discovery of virulence factors. In general, these tools have advanced the understanding of virulence factors in pathogenic microorganisms exponentially.
Microorganisms that infect humans are exogenous to the host and must colonize or penetrate epithelial barriers to gain access to the host. Except for infections acquired during the intrauterine period, infectious agents must bridge host epithelial surfaces, the keratinized epithelium of the skin, or the mucosal epithelium of the respiratory, gastrointestinal, or genitourinary tracts. Ultimately, there are four types of microbial localization in the host (Fig. 1.1 ). Some microbes will enter intracellular sites either within the cytoplasm or within vesicular or vacuolar compartments in cells. Other microbes remain extracellular, either at epithelial surfaces or within the host in the blood, lymph, or tissues.
Figure 1.1.
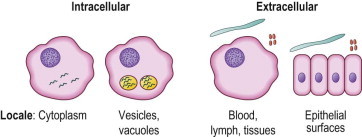
Microbial localization.
Interactions at Epithelial Barrier Surfaces
The barrier functions occurring at epithelial surfaces are part of the innate host defenses and are important in determining the outcome of interactions of potential pathogens with the host. Interactions at epithelial barriers involved in defense against external microbes include not only the physical properties of the epithelial surfaces but also the overlying mucous phase, the ciliated or other propulsive activities facilitating microbe clearance, and the normal microbial flora.
Normal Flora
Vertebrate warm-blooded organisms, such as humans, are an ideal site for the survival of many microbes and provide a rich source of organic material and a constant temperature and pH. Microbes coexist with us in and on our bodies, especially on epithelial surfaces where there is contact with the outside world, such as the bowel, upper respiratory tract, mouth, skin, and distal portions of the genitourinary tract.1, 2, 43 Most of these microorganisms are highly adapted to live with us and do not cause any harm. The presence of the same type of microorganisms at a particular site in the absence of disease is called colonization. Normal colonizing microbial flora help to limit access by potentially pathogenic microorganisms. One condition predisposing to infection is the alteration of the normal epithelial flora, as occurs with antibiotic therapy, since this may allow for the proliferation of pathogenic organisms normally held in balance by the endogenous normal microbial flora. Examples include Candida vaginitis or the development of pseudomembranous colitis due to toxigenic Clostridium difficile, which may complicate antibiotic therapy.
Adhesion to the Epithelium
Microorganisms maintain themselves in or on their host by adhesion to cells or the extracellular matrix. Adhesins are encoded by chromosomal genes, plasmids, or phages.44 They are usually divided into fimbrial and afimbrial adhesins.45 Fimbrial adhesins are present in organisms such as Neisseria gonorrhoeae and are in part responsible for the attachment to genitourinary tract epithelium, preventing the bacteria from being washed out by the urine stream.46, 47 An example of an afimbrial adhesin is the filamentous hemagglutinin of Bordetella pertussis, which is responsible for the attachment of B. pertussis to epithelial cells in the respiratory tract.48 Adhesins attach to receptors in the host. These receptors include proteins, glycolipids, and carbohydrates exposed on the surface of cells or in the extracellular matrix.44 Integrins are one class of proteins present on eukaryotic cell surfaces that can serve as bacterial receptors.44 Helicobacter pylori binds to Lewis blood group antigen present in the gastric epithelium.49 Neisseria has a ligand that binds to CD66 molecules on epithelial cells.
Some pathogens have even more evolved interactions with the host and activate signal transduction mechanisms in the host cell, which in turn upregulate other molecules that aid in the adhesion process.2, 44 Certain strains of enteropathogenic E. coli possess type III secretion or contact-mediated systems.50 In such cases, the secretion and synthesis of virulence factors is modulated by contact with host surfaces. The systems are complex (more than 20 genes are involved) and have not been elucidated completely at the molecular level.51, 52
Penetration of the Epithelial Barriers
Some microbes do not have the means to penetrate skin barriers and are only able to gain access through bites produced by arthropods (e.g., rickettsiae, arboviruses, plasmodia, and filariae).53, 54 In such cases, microbes may be introduced by direct inoculation (e.g., rickettsiae, arboviruses, and plasmodia) or may gain access by migrating through the puncture site (filariae). Other microbes (e.g., skin bacteria and fungi) depend on mechanical disruption of the skin (e.g., due to burns, trauma, or intravenous catheters) to invade deeper structures.55 Still others invade when defenses on mucosal surfaces are lowered due to combined local or generalized immunosuppression and altered mucosal integrity (mucositis) due to chemotherapy or malnutrition (e.g., Candida spp. and anaerobic and other enteric bacteria in the bowel). Some microbes do not invade tissues at all and affect the host locally and systemically by liberating toxins at the site of colonization (e.g., diphtheria exotoxin).44
For enteric pathogens, some, including poliovirus, Salmonella enterica serovar Typhimurium, Salmonella enterica serovar Typhi, Campylobacter jejuni, Yersinia enterocolitica, and Yersinia pseudotuberculosis, gain access to the host across the intestinal epithelium by utilizing uptake in specialized epithelial M cells.56 Internalization of some microorganisms is also achieved through other mechanisms, such as sequential “zipper-like” encircling of the organisms triggered by bacterial ligands and cellular receptors, as occurs in infections caused by Listeria monocytogenes.44 The trigger mechanism of the bacteria induces massive rearrangements of cytoskeletal proteins such as actin, which results in membrane ruffles, as occur with shigellosis and salmonellosis.44
In the genitourinary tract, invasion of some agents (e.g., HIV-1) is facilitated by mucosal erosions caused by other infectious agents.57
Spread from the Portal of Entry
Once the organisms gain access to the body after overcoming the first lines of defense, they either spread to other sites of the body or reproduce locally and often invade surrounding tissues. Local spread is facilitated by a number of factors, including collagenases, hyaluronidases, fibrinolysis, and other enzymes. They are produced by a wide range of organisms, and the role of these enzymes in invasion is, in some cases, controversial.2
Lymphatic spread occurs in most cases once the organisms gain access to subepithelial tissues or serosal surfaces. Lymphatic vessels are distributed in most tissues of the body, with few exceptions such as the brain. Lymph is carried by lymphatic vessels to regional lymph nodes, where it circulates through the node and eventually returns to the systemic circulation through the thoracic duct and the great lymphatic vein. One to three liters of lymph is returned to the systemic circulation every day. Most pathogens are filtered in lymph nodes before reaching the systemic circulation, but some actually reproduce either in the endothelium of lymphatic vessels (e.g., Mycobacterium leprae)2, 58 or in tissue macrophages present in the lymph nodes (e.g., Brucella spp.) or lymphocytes (HIV and herpesviruses, including Epstein–Barr virus).59 Some organisms reach the systemic circulation after overwhelming the defenses in the lymph nodes (e.g., Bacillus anthracis and Y. pestis).
Microorganisms carried in the blood are transported either extracellularly (e.g., most of those causing bacteremia) or intracellularly. Intracellular pathogens are carried by red blood cells (e.g., Plasmodium, Babesia, Colorado tick fever virus, and Bartonella), monocytes (e.g., measles virus, cytomegalovirus, and Toxoplasma), or neutrophils (e.g., Anaplasma phagocytophilum, Ehrlichia ewingii, and some pyogenic bacteria).2, 60
Once in the blood, by initial lymphatic or hematogenous spread, the microorganisms have access to virtually any site in the body. However, some pathogens exhibit tropism for certain tissues. This tropism depends on multiple factors, including the anatomy of the microcirculation in a given tissue (fenestrated capillaries versus continuous endothelial lining), receptors present on certain endothelial cells, and the presence of mononuclear phagocytic cells in organs such as bone marrow, liver, and spleen.2 Other less common routes of spread include peripheral nerves (e.g., rabies and varicella zoster virus), cerebrospinal fluid (after the organisms traverse the blood–brain barrier), and serosal cavities.
Localization in the Host
Microbes that have gained access to the host at or through epithelial barriers then, depending on the properties and size of the pathogens, have the capacity either to seek intracellular sites or remain extracellular (see Fig. 1.1). Mechanisms of host immune responses to the microorganisms vary depending on their sites of localization.
Intracellular Localization
Specific microorganisms use highly developed processes to gain access to and survive within host cells. The microorganisms may reside either in the cytoplasm or within vesicular or vacuolar compartments of targeted cells.
Targeting and penetration of cells is governed by the interactions of microbial surface proteins that may engage host cell molecules that function as receptors for the microbial ligands. The entry of malarial parasites into erythrocytes is a good example, and the nature of the erythrocyte receptors used by different malarial parasite species governs which red blood cells are infected. Plasmodium vivax binds to the Duffy blood group antigens present on some people's red blood cell membranes. The expression of the Duffy blood group antigen is genetically determined, and this antigen is present mostly in whites and Asians and largely absent in blacks of sub-Saharan African ancestry.61, 62, 63, 64 This genetic absence of a receptor on red blood cells required for vivax malaria's survival explains why vivax malaria is rare in regions of Africa. Plasmodium vivax also exhibits a characteristic restriction in the age of erythrocytes it infects. Only young red blood cells and reticulocytes are susceptible to infection, even though the Duffy blood group antigen is present on red blood cells of all ages. The basis for this restriction to younger red blood cells also rests with receptor-mediated limitations. Plasmodium vivax parasites contain reticulocyte-binding proteins, which recognize and bind to reticulocyte-specific antigens on the red blood cell surface.65, 66 Thus, host cell receptor–microbial ligand interactions have an impact on the geographic range of infections based on host genetic differences in requisite receptor expression and on the specific cells that a microbe may enter.
Another example of the intricacies of microbe–receptor interactions has been recognized with HIV-1. Although CD4 is the primary cellular receptor for HIV entry, binding to CD4 alone is not sufficient for entry of HIV-1 into cells. Cellular coreceptors that are members of the chemokine receptor family of seven-transmembrane G protein-coupled molecules are also important. T-cell tropic strains use the CXCR-4 chemokine receptor, and macrophage tropic HIV-1 strains use the CCR-3 and CCR-5 chemokine receptors as coreceptors in concert with CD4. The differences among strains of HIV-1 in their capacities to bind to different chemokine receptor–coreceptors may help explain differences in cell tropism and pathogenicity, the lack of infectability of nonprimate cells, and, for those with genetically altered coreceptors, the apparent resistance to HIV-1 infection of some individuals.67, 68, 69, 70
Typical of those etiologic agents that have an intracellular localization are viruses. The entry of these agents into cells is increasingly recognized to be dependent on their interactions with specific host cell proteins that act as their “receptors.” For instance, host cell molecules that function as viral receptors include multiple isoforms of membrane cofactor protein (CD46), a complement regulatory protein, for measles; the integrin, intracellular adhesion molecule-1 (ICAM-1), for rhinovirus; erythrocyte P antigen for parvovirus B19; and the C3d complement receptor (CR2) for Epstein–Barr virus.71, 72, 73, 74
Microbes that exist principally within the cytoplasm are sequestered from many immune response mechanisms active on extracellular pathogens, including antibody and phagocytic cells. Viral intracellular proteins will be processed and displayed with class I major histocompatibility complex (MHC) proteins, which enable CD8 cytotoxic T cells to recognize and kill the virally infected cell.
Other microbes are internalized within phagocytic cells, especially macrophages. Once internalized in host cells, organisms such as Salmonella, Mycobacterium, Chlamydia, and Legionella use an extraordinary assortment of mechanisms to prevent their phagocytic vacuole from fusing with the host cells’ acidifying lysosomes.75, 76, 77 For some parasites, the intracellular environment is an important determinant of parasitism. For example, Leishmania and Coxiella (unlike other pathogens) benefit from the acidic environment of the macrophage phagolysosome. Leishmania use the proton gradient across the lysosome to drive the energy-dependent uptake of two important substrates: glucose and proline.78 Thus, Leishmania amastigotes actually survive in the macrophage phagolysosome because they benefit from its proton gradient and because they avoid activating the processes that normally kill ingested microorganisms. Leishmanial lipophosphoglycan inhibits the action of β-galactosidase, chelates calcium, inhibits protein kinase C and the oxidative burst, and may scavenge toxic oxygen metabolites.79
Conversely, other intracellular pathogens such as Toxoplasma gondii survive within the macrophage by using an alternative pathway of entry that avoids fusion of the parasitophorous vacuole with lysosomes.76, 80 In contrast, dead or antibody-coated T. gondii enter via the Fc receptor and are routed to a different intracellular compartment, which fuses with the lysosome, and are then killed in the phagolysosome.76, 80, 81
Other organisms, such as Shigella, Listeria, and Rickettsia, breach their vacuolar membrane to multiply freely in the cytoplasm and may also usurp host cellular actin to propel their further spread to neighboring cells, continuing to exploit their intracellular sanctuary.82, 83, 84
Immune responses against microbes within macrophages rely heavily on class II MHC-mediated presentation of host antigenic peptides to T-helper 1 (Th1) types of CD4+ T cells, which then augment the microbicidal activities of the macrophages.
Extracellular Localization
Some types of microbes that remain extracellular typically reside at epithelial surfaces, including bacteria such as N. gonorrhoeae, H. pylori, Vibrio cholerae, and E. coli, and helminths such as adult Ascaris lumbricoides, hookworms, and Trichuris trichiura. Mucosal immune responses, including IgA and leukocytes, participate in host immune reactions to these pathogens.
Other microbes that survive extracellularly are present within the blood, lymph, or tissues of the host, and these organisms include fungi, viruses, bacteria, protozoa, and notably the helminths. Multicellular helminths, due to their large size, remain forever extracellular and may be found in the blood (e.g., microfilariae), lymph (adult lymphatic filarial worms), tissues (migrating larvae and adult stages of some helminths), and cerebrospinal fluid. Host defense against extracellular pathogens uses antibodies, complement, phagocytic cells, and, for helminths, IgE, eosinophils, and mast cells.85
Tissue Damage
There are multiple mechanisms by which microbes inflict damage on host tissues.
Direct Damage or Alteration of Host Cell Function
Host cells can be killed directly by the infectious agent, as in some viral or bacterial infections that are highly cytopathic (e.g., yellow fever virus in hepatocytes and Salmonella in macrophages).86, 87 Some microorganisms multiply intracellularly until the cell bursts and dies (e.g., Rickettsia prowazekii).34 Some bacteria, viruses, and other parasites, such as Shigella, HIV-1, and Listeria, can induce apoptosis of host cells.59, 88, 89 Apoptosis is triggered by different mechanisms, such as activation of the interleukin-converting enzyme pathway.90, 91 This form of programmed cell death is probably more widespread as a mechanism of cell death in infectious diseases than previously thought.
Damage is sometimes caused by toxins secreted by bacterial cells (exotoxins). In this case, bacteria can either invade host tissues or colonize mucosal sites and then release toxins at the mucosal site that are absorbed systemically and cause distant damage.92 Exotoxins can act through different pathways that damage the components of the cell membranes such as phospholipids88, 93 or affect signaling pathways (e.g., V. cholerae).44, 94 Other exotoxins, such as streptolysins and listeriolysins, alter membrane permeability. Still others, such as exfoliatin (e.g., Staphylococcus aureus) and elastase (e.g., Pseudomonas spp.), are capable of degrading extracellular elements.2 Some toxins are translocated to the intracellular environment, where they affect multiple enzymatic systems. These toxins are classified according to their enzymatic activity, such as adenosine diphosphate (ADP) ribosyl transferase (e.g., diphtheria toxin, P. aeruginosa exotoxin A, and pertussis toxin), depurinase (e.g., Shiga toxin), adenylate cyclase (e.g., pertussis hemolysin and anthrax edema factor), and zinc protease (e.g., tetanus).94 The end result ranges from blockade of protein synthesis and cell death or blockade of exocytosis (especially CNS neurotransmitters at the synaptic cleft)95, 96 to increases of cyclic adenosine monophosphate (AMP) or cyclic guanosine monophosphate (GMP) and changes in cell permeability.94 Still other organisms, such as C. difficile, produce toxins that change basic cell signaling transducers such as Rho to alter cell function or affect their spread. Finally, organisms can interact with host cell or microbial transcriptional regulation of genes (such as iron-binding proteins for uropathogenic E. coli 92, 97) or cytokine release (such as H. pylori or enteroaggregative E. coli 40, 98, 99, 100) to enhance their survival or elicit pathogenic responses. The evolutionary advantages to a microbe of its remarkable array of traits that we call “virulence” hold many of the clues to their control, if we can but truly understand them.
Endotoxins are a subset of lipopolysaccharides present in the outer membrane of Gram-negative bacteria that can trigger a wide variety of responses in the host, including massive cytokine release leading to hypotension and shock.101, 102 These deleterious effects occur with high-grade invasion of the blood by Gram-negative bacteria, including enteric Gram-negative bacteremias and meningococcemia.
Indirect Damage
Damage to the host may also develop as a consequence of immune reactions to the infectious agents. One scheme for classifying immunopathologic responses divides the reactions into four types based on the elements of the immune response involved.103
Type I reactions involve elements of strong Th2 responses that lead to increased IgE, eosinophilia, and eosinophil and mast cell activation. Adverse reactions of this type include the development of urticaria (with several helminthic parasites), the occurrence of potentially life-threatening anaphylactic shock in IgE-mediated mast cell degranulation (e.g., triggered by systemic release of antigens from echinococcal cysts104), and exuberant eosinophilic infiltration of tissues due to migrating helminth larvae (e.g., Löffler's pneumonia with the pulmonary migration of Ascaris larvae).
Type II reactions are also dependent on elements of Th2 cell responses that lead to increased IgM and then IgG antibodies directed toward the infectious agents. These antibodies, if cross-reactive with host antigens, may lead to complement-mediated cytotoxicity or antibody-dependent cell-mediated cytotoxicity by natural killer cells, which have Fc receptors. An example of this type of immunopathologic response is the uncommon hemolytic anemia associated with Mycoplasma pneumoniae infection that is mediated by complement-induced hemolysis triggered by IgM (cold agglutinin) antibodies against erythrocyte I antigen.
Type III reactions are caused by the deposition of immune complexes. When neither antibody nor antigen is present in excess of the other, the complexing of antibodies with soluble antigen results in the formation of immune complexes that may cause disease. This may develop acutely as antibody titers rise in the presence of microbial antigens, causing the syndrome of serum sickness. In addition, when soluble antigen is persistently abundant, sustained formation of immune complexes develops, leading to chronic immune complex-mediated tissue damage (especially glomerulonephritis), as found in subacute bacterial endocarditis, chronic hepatitis B antigenemia, and chronic Plasmodium malariae infections.105
Type IV reactions include adverse reactions mediated by macrophages and cytotoxic T cells. Examples are damage caused by granulomas in leprosy, tuberculosis, tertiary syphilis, and fungal infections. Likewise, granulomas developing around schistosomal eggs, depending on their location, may cause ureteral obstruction or hepatic presinusoidal lesions. Other deleterious inflammatory reactions in this category are mediated by parasite-elicited host cytokines, such as the hepatic fibrosis elicited by schistosomal eggs.
Immune Interactions
Immune Evasion
The human immune system has evolved in concert with microbes and is very sophisticated, especially with regard to host defenses against microbes, but the system is not perfect. Interactions of the immune system with microbes are an ongoing affair. Microbes have a high mutation rate compared to human beings. Microbes have evolved a diversity of mechanisms that can enable microorganisms to subvert immediate immunologically mediated elimination (Table 1.1 ). Persistence within the host is necessary for the propagation of some parasites.
Table 1.1.
Mechanisms of Immune Evasion Used by Pathogenic Microbes
| Type of Immune Response | Immune Evasion Mechanisms |
|---|---|
| Innate immunity | Block natural killer cells |
| Recognition avoidance |
|
| Complement inactivation or blockage |
|
| Avoidance of phagocytosis by macrophages and polymorphonuclear neutrophils |
|
| Manipulation of cell surface | |
| Early induced response | Modulation of inflammatory response |
| Interference with signaling pathways |
|
| Interference with RNAi |
|
| Antimicrobial peptide degradation |
|
| Modulation of endosomal trafficking |
|
| Modulation of cytoskeleton | |
| Adaptive immunity | Induce apoptosis |
| Interfere with receptors and signaling |
|
| Modulation of antigen presentation and processing |
|
| Modulation of cell maturation |
|
| Superantigens | |
| Protective immunity | Antigenic variation |
| Phase shift |
|
| Escape mutants | |
There are multiple mechanisms by which microbes can persist in the body and evade the immune system. Tolerance is defined as specific reduction in the response of the immune system to a given antigen.106, 107 In the case of transplacental infection, the fetus develops a certain degree of tolerance to antigens to which it is exposed. The immune system of fetuses is rather incompletely developed in utero, and microorganisms survive easily. Cytomegalovirus infects the fetus transplacentally and produces extensive damage to multiple tissues. After delivery, infants continue shedding virions for weeks to months because they are unable to destroy the virus. Other mechanisms include the production of superantigens that stimulate a large population of T cells, which then become deleted if the encounter occurs during early development. Exposure to massive amounts of antigen in the circulation can also lead to tolerance.2, 103 Immunosuppression is a well-demonstrated phenomenon that occurs during certain infections caused by viruses, bacteria, protozoa, and helminths. These infections usually involve the lymphoid tissues and macrophages and hamper the immune response.
Intracellular pathogens that are able to spread from cell to cell without exposure to the extracellular compartment can avoid exposure to some elements of the immune system. In other cases, pathogens reside in sites relatively inaccessible to the immune system, such as glandular luminal spaces or kidney tubules. In many infections, antibodies are produced but do not effect microbial killing. Sometimes, antibody avidity is low, the epitopes against which the antibody is directed are not critical to the microorganism's survival, or the mechanism of immune elimination is not antibody dependent.2
Other microorganisms have developed means of counteracting specific elements of immune responses, such as production of an IgA-degrading enzyme, IgAase, by certain strains of N. gonorrhoeae.108 Some strains of amebae also produce proteases that destroy complement.2 Reactivation of infections in old age due to waning immunity has been well demonstrated in cases of tuberculosis and varicella zoster virus, allowing transmission to new hosts.
One well-studied mechanism of immune evasion is the capability of changing the antigenic structure by genetic mutation or by programmed sequential expression of genes encoding different surface antigens.109Antigenic drift and recombination between influenzaviruses affecting humans and animals are well documented. Borrelia recurrentis and Trypanosoma gambiense are also capable of changing their surface antigens after antibodies control the initial bloodstream infection.110, 111 The new antigens are not recognized by the antibodies, allowing relapse of the infection. Parasites in which sexual reproduction is possible benefit enormously.112 Genetic variability introduced by crossing over during meiotic divisions is much greater than the variability introduced by asexual reproduction. As many as four crossovers on a single pair of chromosomes have been demonstrated in P. falciparum.113
Microparasites also have multiple mechanisms by which they can evade the initial line of defense provided by phagocytes. These strategies include killing of the phagocyte (e.g., Streptococcus pyogenes and Entamoeba histolytica), inhibition of chemotaxis (e.g., Clostridium perfringens), decreased internalization of microbes by phagocytic cells (e.g., T. gondii), inhibition of opsonins (e.g., Treponema pallidum), inhibition of phagolysosome fusion (e.g., M. leprae and Mycobacterium tuberculosis), and escape from the phagosome into the cytoplasm (e.g., Rickettsia spp., Trypanosoma cruzi, and Listeria).2, 44, 75, 92 With cell-to-cell spread, microorganisms may be minimally exposed to complement, antibodies, or phagocytes in the extracellular or intravascular spaces.77, 78 Rickettsial infections spread from cell to cell throughout the infected foci in the endothelial layer of the microvasculature.82, 83, 94
Macroparasites, the helminths, have evolved diverse mechanisms that enable them to survive in vivo.85 Characteristically, helminths live for months to years in infected hosts within the lumen of the bowel, within tissues, or in the blood or lymphatic vessels. Many helminths are in intimate and recurring contact with all elements of the immune system. As a consequence of their size, helminthic worms do not use intracellular mechanisms to evade immune responses but have evolved a number of capabilities that permit their survival. For instance, interference with antigen processing has been well documented in animal models and patients infected with the filarial nematodes Brugia malayi and Onchocerca volvulus. These helminths produce a family of proteins called the cystatins that are capable of inhibiting proteases responsible for antigen degradation and subsequent presentation through MHC class II pathways in antigen-presenting cells. These proteins are also capable of modulating T-cell proliferation and elicit upregulation of interleukin-10 (IL-10) expression. Other modulators include helminthic derivatives of arachidonic acid such as lipoxin A4, which is capable of blocking production of IL-12 in dendritic cells. Helminthic prostaglandins can also inhibit IL-12 production by dendritic cells. Since helminths have very complex genomes (~21 000 protein-encoding genes in some of them), they are capable of producing a large variety of proteins. Some of them are cytokines and related proteins also capable of modulating the host immune response to their advantage. For example, B. malayi has been shown to express transforming growth factor (TGF)-β-like proteins capable of binding TGF-β human receptors. Other cytokines include macrophage-migration inhibition factors produced by several nematodes including B. malayi. Blockade of effector mechanisms has also been demonstrated in some helminth infections, including proteases that target effector molecules such as eotaxin. Neutrophil proteases can also be inhibited by serpins.
Emerging Infectious Diseases
The concept of emerging infectious diseases is not new but has been the focus of attention due to the resurgence of old infectious diseases that were thought to be controlled and the recognition of new pathogens as humans increase their interaction with the biosphere. By definition, an emerging infectious disease is one that has newly appeared in the population or has existed but is rapidly increasing in incidence or geographic range.114 Others define emerging infections as “new emerging or drug resistant infections whose incidence in humans has increased within the last two decades, or whose incidence threatens to increase in the near future.” Emerging infections are classified by some as newly emerging, re-emerging/resurging, and deliberately emerging. Since 1967, after a widely publicized statement from the US Surgeon General declaring victory in the war against infectious diseases, more than 85 new pathogens have been described. These include viruses, bacteria, protozoans, and helminths. These pathogens can cause a wide diversity of syndromes including acute respiratory infections (influenza A H1N5, H1N1, SARS-CoV, Sin Nombre virus, human metapneumovirus), systemic diseases caused by viral hemorrhagic fever viruses (Lassa, Ebola, dengue), encephalitic syndromes (Nipah virus, West Nile virus), arthropod-borne agents (Borrelia burgdorferi, Rickettsia spp., Ehrlichia spp., Anaplasma), enteric pathogens (Cryptosporidium, microsporidia), chronic viral diseases (HIV-1 and 2, human T-cell lymphotropic virus types 1–3, human herpesviruses 6–8), hepatotropic viruses (hepatitis C and E), and other infectious agents.
The factors involved in the emergence or reemergence of infectious diseases are complex and include ecological changes (deforestation, reforestation, flooding, and climatic changes), changes in human demographics and behavior (sexual, cultural, and war), increased international travel, technological advances (organ transplantation and antibiotics), microbial evolution with the appearance of antibiotic-resistant or antigenically distinct strains, and deficiencies in surveillance and public health policy.113, 115, 116, 117 The classic triad of microbe, host, and environment is again exemplified.
Tropical Infectious Diseases
Globally, as assessed in terms of disability-adjusted life years (DALYs), which measure morbidity and mortality,118 infectious diseases in 1990 accounted for 36.4% of total DALYs. Infectious disease DALYs were considerably in excess of those attributable to cancer (5.9%), heart disease (3.1%), cerebrovascular disease (3.2%), or chronic lung disease (3.5%).119
However, these calculations admittedly miss the disproportionate impact of tropical infectious diseases on the still rapidly increasing populations living in impoverished, tropical areas, and they grossly underestimate the major developmental impact of common childhood enteric, helminthic, and other infections.38, 120, 121, 122 For those caring for individual patients with infectious diseases, appropriate diagnosis and treatment are important considerations for the individual. Even more important is the consideration of approaches that will lead to diminished acquisition of infectious diseases. For some infectious agents, immunization holds promise, as witnessed by the successful global eradication of smallpox and the potential eradication of poliomyelitis. Greater progress in the control of infectious diseases, however, rests with improvements related to socioeconomic conditions of the population at risk. In developed countries, tuberculosis was diminished well before the introduction of the first antimicrobial agents active against M. tuberculosis, and the reduction was attributable to improved socioeconomic conditions. For the major infectious diseases of the tropics, improvements in sanitation, living conditions, and general public health will be critical in helping control the impact of the diverse infectious agents that currently contribute to human morbidity and mortality. The impact of these infections is related not only to their effect on the health of the infected individual but also to their contribution to the morbidity associated with malnutrition and to their larger societal impact as an impediment to the full development of the political, economic, and social potential of entire populations.
References
- 1.Madigan MT, Martinko JM, Dunlap PV. 12th ed. Benjamin Cummings; San Francisco, CA: 2008. Brock Biology of Microorganisms. [Google Scholar]
- 2.Mims C, Nash A, Stephen J. 5th ed. Academic Press; London: 2001. Mims’ Pathogenesis of Infectious Disease. [Google Scholar]
- 3.Nelson KE, Masters Williams C. 2nd ed. Jones and Bartlett; Boston, MA: 2007. Infectious Disease Epidemiology: Theory and Practice. [Google Scholar]
- 4.Schopf JW. Princeton University Press; Princeton, NJ: 1983. Earth's Earliest Biosphere. Its Origin and Evolution. [Google Scholar]
- 5.Lepp PW, Brinig MM, Ouverney CC. Methanogenic Archaea and human periodontal disease. Proc Natl Acad Sci. 2004;101:6176. doi: 10.1073/pnas.0308766101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woese CR. Bacterial evolution. Microbiol Rev. 1987;51:221. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pace NR. A molecular view of microbial diversity and the biosphere. Science. 1997;276:734. doi: 10.1126/science.276.5313.734. [DOI] [PubMed] [Google Scholar]
- 8.Margulis L. 2nd ed. WH Freeman; New York: 1992. Symbiosis in Cell Evolution. [Google Scholar]
- 9.Kasting JF. Earth's early atmosphere. Science. 1993;259:921. doi: 10.1126/science.11536547. [DOI] [PubMed] [Google Scholar]
- 10.Olsen GJ, Lane DL, Giovannoni SJ. Microbial ecology and evolution: A ribosomal RNA approach. Annu Rev Microbiol. 1986;40:337. doi: 10.1146/annurev.mi.40.100186.002005. [DOI] [PubMed] [Google Scholar]
- 11.Bishop JM. The molecular genetics of cancer. Science. 1987;235:305. doi: 10.1126/science.3541204. [DOI] [PubMed] [Google Scholar]
- 12.Benjamin T, Vogt PK. Cell transformation by viruses. In: Knipe DM, Howley PM, Griffin DE, editors. Fields Virology. Lippincott Williams & Wilkins; New York: 2001. [Google Scholar]
- 13.Strauss JH, Strauss EG. 2nd ed. Academic Press, Elsevier; New York: 2008. Viruses and Human Disease. [Google Scholar]
- 14.Francki RB. Encapsidated viroid-like RNA. In: Diener TO, editor. The Viroids. Plenum Press; New York: 1987. p. 205. [Google Scholar]
- 15.Keese P, Synons RH. The structure of viroids and virusoids. In: Semanicik JS, editor. Viroids and Viroid-like Pathogens. CRC Press; Boca Raton, FL: 1986. p. 1. [Google Scholar]
- 16.Prusiner SB. Shattuck lecture – Neurodegenerative diseases and prions. N Engl J Med. 2001;344:1516. doi: 10.1056/NEJM200105173442006. [DOI] [PubMed] [Google Scholar]
- 17.DeArmond SJ, Prusiner SB. Perspectives on prion biology, prion disease pathogenesis, and pharmacologic approaches to treatment. Clin Lab Med. 2003;23:1. doi: 10.1016/s0272-2712(02)00041-0. [DOI] [PubMed] [Google Scholar]
- 18.Ewald PW. Guarding against the most dangerous emerging pathogens: Insights from evolutionary biology. Emerg Infect Dis. 1996;2:245. doi: 10.3201/eid0204.960401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levin BR. The evolution and maintenance of virulence in microparasites. Emerg Infect Dis. 1996;2:93. doi: 10.3201/eid0202.960203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levin BR, Svanborg-Eden C. Selection and the evolution of virulence in bacteria: An economical excursion and modest suggestion. Parasitology. 1990;100:5103. doi: 10.1017/s0031182000073054. [DOI] [PubMed] [Google Scholar]
- 21.Flint SJ, Enquist LW, Racaniello VR. ASM Press; Washington, DC: 2004. Principles of Virology: Molecular Biology, Pathogenesis, and Control of Animal Viruses. [Google Scholar]
- 22.Anita R, Levin BR, May RM. Within host population dynamics, and the evolution and maintenance of microparasite virulence. Am Naturalist. 1994;144:457. [Google Scholar]
- 23.Frank SA, Schmid-Hempel P. Mechanisms of pathogenesis and the evolution of parasite virulence. J Evol Biol. 2008;21:396. doi: 10.1111/j.1420-9101.2007.01480.x. [DOI] [PubMed] [Google Scholar]
- 24.Schmid-Hempel P. Parasite immune evasion: a momentous molecular war. Trends Ecol Evol. 2008;23:318. doi: 10.1016/j.tree.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 25.Andre JB, Godelle B. J Theor Biol. 2006;241:402. doi: 10.1016/j.jtbi.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 26.Sokurenko EV, Gomulkiewicz R, Dykhuizen DE. Source-sink dynamics of virulence evolution. Nature Rev Microbiol. 2006;4:548. doi: 10.1038/nrmicro1446. [DOI] [PubMed] [Google Scholar]
- 27.Bonhoeffer S, Nowak MA. Intrahost versus interhost selection: Viral strategies of immune function impairment. Proc Natl Acad Sci USA. 1994;91:8062. doi: 10.1073/pnas.91.17.8062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flint J, Harding RM, Boyce AJ. The population genetics of the haemoglobinopathies. Baillieres Clin Haematol. 1993;6:215. doi: 10.1016/s0950-3536(05)80071-x. [DOI] [PubMed] [Google Scholar]
- 29.Weatherall DJ, Clegg JB. Genetic variability in response to infection: Malaria and after. Genes Immun. 2002;3:331. doi: 10.1038/sj.gene.6363878. [DOI] [PubMed] [Google Scholar]
- 30.Wain-Hobson S. Running the gamut of retroviral variation. Trends Microbiol. 1996;4:135. doi: 10.1016/0966-842x(96)10023-8. [DOI] [PubMed] [Google Scholar]
- 31.Amyes SG, Gemmell CG. Antibiotic resistance in bacteria. J Med Microbiol. 1992;36:4. doi: 10.1099/00222615-36-1-4. [DOI] [PubMed] [Google Scholar]
- 32.Burnet M, White DO. 4th ed. Cambridge University Press; Cambridge, UK: 1972. Natural History of Infectious Disease. [Google Scholar]
- 33.McNeill WH. Anchor Press; Garden City, NY: 1976. Plagues and Peoples. [Google Scholar]
- 34.Walker DH, editor. Biology of Rickettsial Diseases. CRC Press; Boca Raton, FL: 1988. [Google Scholar]
- 35.Wills C. Addison-Wesley; Reading, MA: 1996. Yellow Fever and Black Goddess. The Coevolution of Peoples and Plagues. [Google Scholar]
- 36.Colwell RR. Global climate and infectious disease: the cholera paradigm. Science. 1997;274:2025. doi: 10.1126/science.274.5295.2025. [DOI] [PubMed] [Google Scholar]
- 37.Guerrant RL. Why America must care about tropical medicine: threats of global health and security from tropical infectious diseases. Am J Trop Med Hyg. 1998;59:3. doi: 10.4269/ajtmh.1998.59.3. [DOI] [PubMed] [Google Scholar]
- 38.Simsek Z, Zeyrek FY, Kurcer MA. Effect of Giardia infection on growth and psychomotor development of children aged 0–5 years. J Trop Pediatr. 2004;50:90. doi: 10.1093/tropej/50.2.90. [DOI] [PubMed] [Google Scholar]
- 39.Checkley W, Gilman RH, Epstein LD. Asymptomatic and symptomatic cryptosporidiosis: their acute effect on weight gain in Peruvian children. Am J Epidemiol. 1997;145:156. doi: 10.1093/oxfordjournals.aje.a009086. [DOI] [PubMed] [Google Scholar]
- 40.Steiner TS, Lima AA, Nataro JP. Enteroaggregative Escherichia coli produce intestinal inflammation and growth impairment and cause interleukin-8 release from intestinal epithelial cells. J Infect Dis. 1998;177:88. doi: 10.1086/513809. [DOI] [PubMed] [Google Scholar]
- 41.Crompton DWT, Montresor A, Nesheim MC, editors. Controlling Disease Due to Helminth Infections. World Health Organization; Geneva: 2003. [Google Scholar]
- 42.Stephenson LS. Helminth parasites, a major factor in malnutrition. World Health Forum. 1994;15:169. [PubMed] [Google Scholar]
- 43.Raskin DM, Seshadri R, Pukatzki SU. Bacterial genomics and pathogen evolution. Cell. 2006;124:703. doi: 10.1016/j.cell.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 44.Isenberg LT, D’Amato RF. Indigenous and pathogenic microorganisms of humans. In: Murray PR, Baron EJ, Jorgensen JH, editors. Manual of Clinical Microbiology. 8th ed. ASM Press; Washington, DC: 2003. [Google Scholar]
- 45.Finlay B, Falkow S. Common themes in microbial pathogenicity revisited. Microbiol Mol Biol Rev. 1997;61:136. doi: 10.1128/mmbr.61.2.136-169.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hultgren SJ, Abraham S, Caparon M. Pilus and nonpilus bacterial adhesins: Assembly and function in cell recognition. Cell. 1993;73:887. doi: 10.1016/0092-8674(93)90269-v. [DOI] [PubMed] [Google Scholar]
- 47.Blum G, Falbo V, Caprioli A. Gene clusters encoding the cytotoxic necrotizing factor type 1. Prs-fimbriae and alpha-hemolysin form the pathogenicity island II of the uropathogenic Escherichia coli strain J96. FEMS Microbiol Lett. 1995;126:189. doi: 10.1111/j.1574-6968.1995.tb07415.x. [DOI] [PubMed] [Google Scholar]
- 48.Sandros J, Tuomanen E. Attachment factors of Bordetella pertussis: Mimicry of eukaryotic cell recognition molecules. Trends Microbiol. 1993;96:884. doi: 10.1016/0966-842x(93)90090-e. [DOI] [PubMed] [Google Scholar]
- 49.Boren T, Falk P, Roth KA. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science. 1993;262:1892. doi: 10.1126/science.8018146. [DOI] [PubMed] [Google Scholar]
- 50.Jarvis KG, Giron JA, Jerse AE. Enteropathogenic Escherichia coli contains a putative type III secretion system necessary for the export of proteins involved in attaching and effacing lesion formation. Proc Natl Acad Sci USA. 1995;92:7996. doi: 10.1073/pnas.92.17.7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.von Gijsegem F, Genia S, Boucher C. Conservation of secretion pathways for pathogenicity determinants of plants and animal bacteria. Trends Microbiol. 1993;1:175. doi: 10.1016/0966-842x(93)90087-8. [DOI] [PubMed] [Google Scholar]
- 52.Coburn B, Sekirov I, Finlay BB. Type III secretion systems and disease. Clin Microbiol Rev. 2007;20:535. doi: 10.1128/CMR.00013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garnham PC. Malaria parasites of man: Life cycles and morphology. In: Wernsdorfer WH, McGregor I, editors. vol 1. Churchill Livingstone; London: 1988. p. 61. (Malaria. Principles and Practice of Malariology). [Google Scholar]
- 54.Manz J, Herbert J. Arboviral encephalitis. In: Connor DH, Chandler FW, editors. Pathology of Infectious Diseases. Appleton & Lange; Stamford, CT: 1997. [Google Scholar]
- 55.Schell WA, Salkin IF, McGinnis MR. Bipolaris, Exophiala, Scedosporium, Sporopthrix and other dematiaceous fungi. In: Murray PR, Baron EJ, Jorgensen JH, editors. Manual of Clinical Microbiology. 8th ed. ASM Press; Washington, DC: 2003. [Google Scholar]
- 56.Neutra MR, Pringault E, Kraehenbuhl JP. Antigen sampling across epithelial barriers and induction of mucosal immune responses. Annu Rev Immunol. 1996;14:275. doi: 10.1146/annurev.immunol.14.1.275. [DOI] [PubMed] [Google Scholar]
- 57.Sewankambo N, Gray RH, Wawer MJ. HIV-1 infection associated with abnormal vaginal flora morphology and bacterial vaginosis. Lancet. 1997;350:546. doi: 10.1016/s0140-6736(97)01063-5. [DOI] [PubMed] [Google Scholar]
- 58.Cockerell CJ. Leprosy. In: Connor DH, Chandler FW, editors. Pathology of Infectious Diseases. Appleton & Lange; Stamford, CT: 1997. [Google Scholar]
- 59.Douek DC. Disrupting T-cell homeostasis: how HIV-1 infection causes disease. AIDS Rev. 2003;5:172. [PubMed] [Google Scholar]
- 60.Dumler JS, Walker DH. Ehrlichial infections. In: Connor DH, Chandler FW, editors. Pathology of Infectious Diseases. Appleton & Lange; Stamford, CT: 1997. [Google Scholar]
- 61.Chitnis CE, Chaudhuri A, Horuk R. The domain on the Duffy blood group antigen for binding Plasmodium vivax and P. knowlesi malarial parasites to erythrocytes. J Exp Med. 1996;184:1531. doi: 10.1084/jem.184.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miller LH, Mason SJ, Clyde DF. The resistance factor to Plasmodium vivax in blacks. N Engl J Med. 1976;295:302. doi: 10.1056/NEJM197608052950602. [DOI] [PubMed] [Google Scholar]
- 63.Hadley TJ, Peiper SC. From malaria to chemokine receptor: the emerging physiologic role of the Duffy blood group antigen. Blood. 1997;87:3077. [PubMed] [Google Scholar]
- 64.Horuk R, Chitnis CE, Darbonne WC. A receptor for the malarial parasite Plasmodium vivax: the erythrocyte chemokine receptor. Science. 1993;261:1182. doi: 10.1126/science.7689250. [DOI] [PubMed] [Google Scholar]
- 65.Galinski MR, Barnwell JW. A reticulocyte-binding complex of Plasmodium vivax merozoites. Cell. 1992;69:1213. doi: 10.1016/0092-8674(92)90642-p. [DOI] [PubMed] [Google Scholar]
- 66.Barnwell JR, Galinski MR. Plasmodium vivax: A glimpse into the unique and shared biology of the merozoite. Ann Trop Med Parasitol. 1995;89:113. doi: 10.1080/00034983.1995.11812941. [DOI] [PubMed] [Google Scholar]
- 67.Choe H, Farzan M, Su NY. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 1996;85:1135. doi: 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- 68.Moore JP. Coreceptors: Implications for HIV pathogenesis and therapy. Science. 1997;276:51. doi: 10.1126/science.276.5309.51. [DOI] [PubMed] [Google Scholar]
- 69.Doranz BJ, Berson JF, Rucker J. Chemokine receptors as fusion cofactors for human immunodeficiency virus type 1 (HIV-1) Immunol Res. 1997;16:15. doi: 10.1007/BF02786321. [DOI] [PubMed] [Google Scholar]
- 70.Garred P. Chemokine-receptor polymorphisms: clarity or confusion for HIV-1 prognosis. Lancet. 1998;351:2. doi: 10.1016/S0140-6736(98)22001-0. [DOI] [PubMed] [Google Scholar]
- 71.Manchester M, Liszewski MK, Atkinson JP. Multiple isoforms of CD46 (membrane cofactor protein) serve as receptors for measles virus. Proc Natl Acad Sci USA. 1994;91:2161. doi: 10.1073/pnas.91.6.2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Greve JM, Davis G, Meyer AM. The major human rhinovirus receptor is ICAM-1. Cell. 1989;56:839. doi: 10.1016/0092-8674(89)90688-0. [DOI] [PubMed] [Google Scholar]
- 73.Fingeroth JD, Weis JJ, Tedder TF. Epstein–Barr virus receptor of human B lymphocytes is the C3d receptor CR2. Proc Natl Acad Sci USA. 1984;81:4510. doi: 10.1073/pnas.81.14.4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brown KE, Hibbs JR, Gallinella G. Resistance to parvovirus B19 infection due to lack of virus receptor (erythrocyte P antigen) N Engl J Med. 1994;330:1192. doi: 10.1056/NEJM199404283301704. [DOI] [PubMed] [Google Scholar]
- 75.Buchmeier NA, Heffron F. Inhibition of macrophage phagosome-lysosome fusion by Salmonella typhimurium. Infect Immun. 1991;59:2232. doi: 10.1128/iai.59.7.2232-2238.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Walker DH, Valbuena GA, Olano JP. Pathogenic mechanisms of diseases caused by Rickettsia. Ann NY Acad Sci. 2003;990:1. doi: 10.1111/j.1749-6632.2003.tb07331.x. [DOI] [PubMed] [Google Scholar]
- 77.Clemens DL, Horwitz MA. Characterization of the Mycobacterium tuberculosis phagosome and evidence that phagosomal maturation is inhibited. J Exp Med. 1995;181:257. doi: 10.1084/jem.181.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zilberstein D, Dwyer DM. Proton motive force-driven active transport of d-glucose and l-proline in the protozoan parasite Leishmania donovani. Proc Natl Acad Sci USA. 1990;82:1716. doi: 10.1073/pnas.82.6.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Turco SJ, Descoteaux A. The lipophosphoglycan of Leishmania parasites. Annu Rev Microbiol. 1992;46:65. doi: 10.1146/annurev.mi.46.100192.000433. [DOI] [PubMed] [Google Scholar]
- 80.Sibley LD, Weidner E, Krahenbuhl JL. Phagosome acidification blocked by intracellular Toxoplasma gondii. Nature. 1985;315:416. doi: 10.1038/315416a0. [DOI] [PubMed] [Google Scholar]
- 81.Joiner KA, Fuhrman SA, Miettinen HM. Toxoplasma gondii: fusion competence of parasitophorous vacuoles in Fc receptor-transfected fibroblasts. Science. 1990;249:641. doi: 10.1126/science.2200126. [DOI] [PubMed] [Google Scholar]
- 82.Cossart P. Subversion of the mammalian cytoskeleton by invasive bacteria. J Clin Invest. 1997;99:2307. doi: 10.1172/JCI119409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Heinzen RA. Rickettsial actin-based motility: behavior and involvement of cytoskeletal regulators. Ann NY Acad Sci. 2003;990:535. doi: 10.1111/j.1749-6632.2003.tb07424.x. [DOI] [PubMed] [Google Scholar]
- 84.Portnoy DA, Chakraborty T, Goebel W. Molecular determinants of Listeria monocytogenes pathogenesis. Infect Immun. 1992;60:1263. doi: 10.1128/iai.60.4.1263-1267.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Maizels RM, Yazdanbakhsh M. Immune regulation by helminth parasites: cellular and molecular mechanisms. Nature Rev Immunol. 2003;3:733. doi: 10.1038/nri1183. [DOI] [PubMed] [Google Scholar]
- 86.Chen LM, Kaniga K, Galan JE. Salmonella spp. are cytotoxic for cultured macrophages. Mol Microbiol. 1996;21:1101. doi: 10.1046/j.1365-2958.1996.471410.x. [DOI] [PubMed] [Google Scholar]
- 87.Strano A. Yellow fever. In: Connor DH, Chandler FW, editors. Pathology of Infectious Diseases. Appleton & Lange; Stamford, CT: 1997. [Google Scholar]
- 88.Zychlinsky A, Kenny B, Menard R. IpaB mediates macrophage apoptosis induced by Shigella flexneri. Mol Microbiol. 1994;11:619. doi: 10.1111/j.1365-2958.1994.tb00341.x. [DOI] [PubMed] [Google Scholar]
- 89.Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992;358:167. doi: 10.1038/358167a0. [DOI] [PubMed] [Google Scholar]
- 90.Che NYJ, Smith MR, Thirumalai K. A bacterial invasin induces macrophage apoptosis by binding directly to ICE. EMBO J. 1996;15:3853. [PMC free article] [PubMed] [Google Scholar]
- 91.Fraser A, Evan G. A license to kill. Cell. 1996;85:781. doi: 10.1016/s0092-8674(00)81005-3. [DOI] [PubMed] [Google Scholar]
- 92.Bobak DA, Guerrant RL. New developments in enteric bacterial toxins. Adv Pharmacol. 1992;23:85. doi: 10.1016/s1054-3589(08)60963-1. [DOI] [PubMed] [Google Scholar]
- 93.Walker DH, Firth WT, Ballard JG. Role of phospholipase-associated penetration mechanism in cell injury by Rickettsia rickettsii. Infect Immun. 1983;40:840. doi: 10.1128/iai.40.2.840-842.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shames SR, Auweter SD, Finlay BB. Co-evolution and exploitation of host cell signalling pathways by bacterial pathogens. Int J Biochem Cell Biol. 2009;41:380. doi: 10.1016/j.biocel.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 95.Montecucco C, Schiavo G. Mechanism of action of tetanus and botulinum neurotoxins. Mol Microbiol. 1994;13:1. doi: 10.1111/j.1365-2958.1994.tb00396.x. [DOI] [PubMed] [Google Scholar]
- 96.Schiavo G, Benfenati F, Poulain B. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature. 1992;359:832. doi: 10.1038/359832a0. [DOI] [PubMed] [Google Scholar]
- 97.Zhang JP, Normark S. Induction of gene expression in Escherichia coli after pilus-mediated adherence. Science. 1996;273:1234. doi: 10.1126/science.273.5279.1234. [DOI] [PubMed] [Google Scholar]
- 98.Keates S, Hitti YS, Upton M. Helicobacter pylori infection activates NF-κB in gastric epithelial cells. Gastroenterology. 1997;113:1099. doi: 10.1053/gast.1997.v113.pm9322504. [DOI] [PubMed] [Google Scholar]
- 99.Rieder G, Hatz RA, Moran AP. Role of adherence in interleukin-8 induction in Helicobacter pylori-associated gastritis. Infect Immun. 1997;65:3622. doi: 10.1128/iai.65.9.3622-3630.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guerrant RL, Steiner TS, Lima AA. How intestinal bacteria cause disease. J Infect Dis. 1999;179(suppl 2):S331. doi: 10.1086/513845. [DOI] [PubMed] [Google Scholar]
- 101.Holst O, Ulmer AJ, Brade H. Biochemistry and cell biology of bacterial endotoxins. FEMS Immunol Med Microbiol. 1996;16:83. doi: 10.1111/j.1574-695X.1996.tb00126.x. [DOI] [PubMed] [Google Scholar]
- 102.Sweet MJ, Hume DA. Endotoxin signal transduction in macrophages. J Leukoc Biol. 1996;60:8. doi: 10.1002/jlb.60.1.8. [DOI] [PubMed] [Google Scholar]
- 103.Abbas AK, Lichtman AH, Pillai S. 6th ed. Saunders/Elsevier; Philadelphia: 2007. Cellular and Molecular Immunology. [Google Scholar]
- 104.Diaz de Durana MD, Lopez A, Fraj J. Anaphylaxis and cerebral hydatidic disease. Ann Intern Med. 1997;126:745. doi: 10.7326/0003-4819-126-9-199705010-00032. [DOI] [PubMed] [Google Scholar]
- 105.Abbas AK. Diseases of immunity. In: Kumar V, Abbas AK, Fausto N, Aster JC, editors. Robbins and Cotran Pathologic Basis of Disease. 8th ed. Saunders/Elsevier; Philadelphia: 2009. [Google Scholar]
- 106.Marrack P, Kappler J. Subversion of the immune system by pathogens. Cell. 1994;76:323. doi: 10.1016/0092-8674(94)90339-5. [DOI] [PubMed] [Google Scholar]
- 107.Janeway CA, Travers P, Walport M, editors. Immunobiology. 6th ed. Garland; New York: 2004. [Google Scholar]
- 108.Klauser T, Pohlner J, Meyer TF. The secretion pathway of IgA protease-type proteins in Gram-negative bacteria. Bioessays. 1993;15:799. doi: 10.1002/bies.950151205. [DOI] [PubMed] [Google Scholar]
- 109.Brunham RC, Plummer FA, Stephens RS. Bacterial antigenic variation, host immune response, and pathogen–host coevolution. Infect Immun. 1993;61:2273. doi: 10.1128/iai.61.6.2273-2276.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cross GAM, Holder AA, Allen G. An introduction to antigenic variation in trypanosomes. Am J Trop Med Hyg. 1980;29:1027. doi: 10.4269/ajtmh.1980.29.1027. [DOI] [PubMed] [Google Scholar]
- 111.Saint Girons I, Barbour AG. Antigenic variation in Borrelia. Res Microbiol. 1991;142:711. doi: 10.1016/0923-2508(91)90085-o. [DOI] [PubMed] [Google Scholar]
- 112.Howard RS, Lively CM. Parasitism, mutation accumulation and the maintenance of sex. Nature. 1994;367:554. doi: 10.1038/367554a0. [DOI] [PubMed] [Google Scholar]
- 113.Walker-Jonah A, Dolan SA, Gwadz RW. An RFLP map of the Plasmodium falciparum genome, recombination rates and favored linkage groups in a genetic cross. Mol Biochem Parasitol. 1992;51:313. doi: 10.1016/0166-6851(92)90081-t. [DOI] [PubMed] [Google Scholar]
- 114.Morse SS. Factors in the emergence of infectious diseases. Emerg Infect Dis. 1995;1:7. doi: 10.3201/eid0101.950102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Satcher D. Emerging infections: getting ahead of the curve. Emerg Infect Dis. 1995;1:1. doi: 10.3201/eid0101.950101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wilson ME. Travel and the emergence of infectious diseases. Emerg Infect Dis. 1995;1:39. doi: 10.3201/eid0102.950201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Peiris JS, Yuen KY, Osterhaus AD. The severe acute respiratory syndrome. N Engl J Med. 2003;349:2431. doi: 10.1056/NEJMra032498. [DOI] [PubMed] [Google Scholar]
- 118.Murray CJL. Quantifying the burden of disease: The technical basis for disability-adjusted life years. In: Murray CJL, Lopez AD, editors. Global Comparative Assessments in the Health Sector. World Health Organization; Geneva: 1994. [PMC free article] [PubMed] [Google Scholar]
- 119.Murray CJL, Lopez AD, editors. The Global Burden of Disease. Harvard University Press; Cambridge, MA: 1996. [Google Scholar]
- 120.Guerrant RL, Kosek M, Lima AAM. Updating the DALYs for diarrhoeal disease. Trends Parasitol. 2002;18:191. doi: 10.1016/s1471-4922(02)02253-5. [DOI] [PubMed] [Google Scholar]
- 121.Chan M-S. The global burden of intestinal nematode infections – fifty years on. Parasitol Today. 1997;13:438. doi: 10.1016/s0169-4758(97)01144-7. [DOI] [PubMed] [Google Scholar]
- 122.Bundy DAP, Chan M-S, Medley GF. Harvard University Press; Cambridge, MA: 1997. Health Priorities and Burden of Disease Analysis: Methods and Applications from Global, National and Subnational Studies. [Google Scholar]