

Abstract
Objective
Oxidative stress (OS)-induced stress signaler p38 mitogen-activated protein kinase (p38MAPK) activation and fetal membrane senescence are associated with parturition. This study determined changes in glycogen synthase kinase 3 beta (GSK3β) and its regulation by p38MAPK in effecting senescence to further delineate the molecular mechanism involved in senescence.
Methods
Primary human amnion epithelial cells and amnion mesenchymal cells were treated with cigarette smoke extract (CSE, OS inducer). Expression of total and phosphorylated GSK3β and p38MAPK, and that of GSK3β’s downstream targets: beta-catenin (β-Cat) and nuclear factor erythroid 2-related factor 2 (Nrf2) (western blot analysis), cell cycle regulation and senescence (flow cytometry) were determined. The specificity of GSK3β and p38MAPK’s mechanistic role was tested by co-treating cells with their respective inhibitors, CHIR99021 and SB203580. Exosomal secretion of β-Cat from OS-induced cells was confirmed by immunofluorescence confocal microscopy and western blot.
Results
OS induced by CSE resulted in phosphorylation of GSK3β (inactivation) and p38MAPK (activation) that was associated with cell cycle arrest and senescence. Inhibitors to GSK3β and p38MAPK verified their roles. Glycogen synthase kinase 3 beta inactivation was associated with nuclear translocation of antioxidant Nrf2 and exosomal secretion of β-Cat.
Conclusions
OS-induced P-p38MAPK activation is associated with functional downregulation of GSK3β and arrest of cell cycle progression and senescence of amnion cells. Lack of nuclear translocation of β-Cat and its excretion via exosomes further supports the postulation that GSK3β down-regulation by p38MAPK may stop cell proliferation preceding cell senescence. A better understanding of molecular mechanisms of senescence will help develop therapeutic strategies to prevent preterm birth.
Keywords: GSK3β, preterm labor, fetal membranes, oxidative stress, p38 MAPK
OS causes activation of p38MAPK that can inactivate GSK3? to promote senescence of human amnion cells.
Introduction
Fetal membranes (amniochorion or placental membranes) are layers of extra-embryonic tissue that function as a protective barrier during pregnancy [1, 2]. Fetal membranes undergo senescence, an irreversible arrest of cell growth that occurs naturally contributing to aging [3, 4]. An increase in oxidative stress (OS), at term or at preterm within the intrauterine compartment has been experimentally shown to cause senescence and inflammation of the amniochorion tissue [5–7]. Oxidative stress-induced fetal membrane senescence has been established as a plausible factor associated with labor not only at term but can increase the risk of preterm birth (PTB) and preterm premature rupture of membranes (pPROM) as well [3, 8].
Oxidative stress-induced fetal membrane senescence is mediated through telomere reduction, activation of stress signaler p38 mitogen-activated protein kinase (p38MAPK), and development of sterile inflammation called senescence-associated secretory phenotype (SASP). Oxidative stress experienced by fetal membranes can result from a noninfectious (often seen at term prior to labor) or infectious (often seen at preterm) stimulant; however, the extent of senescence and senescence-associated inflammation may vary based on the type of endogenous or exogenous stimulant. SASP from senescent fetal membranes are predominantly pro-parturient biochemical mediators that can cause activation of maternal tissues such as the uterus and decidua and initiate labor [4]. Mechanistically, one of the initiators of this process in fetal membranes is OS-induced increased expression and accumulation of transforming growth factor β (TGFβ). TGFβ through TGFβ receptor-TGFβ activated kinase 1 binding protein 1 (TAB) complex triggers a noncanonical pathway leading to activation of p38MAPK by autophosphorylation [9]. A constitutive and modest level of p38MAPK expression is likely during gestation, and this is expected to be a tightly regulated process to avoid premature senescence activation. The mechanism of balanced p38MAPK function during gestation and its increased activity contributing to senescence at term is unclear. This knowledge is critical to understand fetal membrane homeostasis during pregnancy and its imbalance contributing to pathologies associated with adverse pregnancies.
In an attempt to further elucidate mechanisms of senescence in the amnion layer of the fetal membranes, this study examined the role of glycogen synthase kinase 3 beta (GSK3β). p38 Mitogen-activated protein kinase has been demonstrated to regulate the function of GSK3β in other fields [10, 11]. Glycogen synthase kinase 3 beta is a serine/threonine kinase that is important for maintaining cellular homeostasis by regulating a number of important biological processes [12–14]. Unlike other kinases, GSK3β is constitutively active and is inactivated upon phosphorylation at the Ser9 site and has maximal activity upon phosphorylation at Tyr216 site [12, 15]. Multiple upstream regulators like the Wnt pathway, phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT), and p38MAPK dictate GSK3β’s downstream function [10, 16, 17]. Of the multitude of GSK3β’s downstream targets, β-catenin (β-Cat) is an important one [14, 18]. GSK3β phosphorylates and causes ubiquitin-mediated degradation of β-Cat along with Axin-1, adenomatous polyposis coli, and casein kinase-1 as part of the “β-catenin destruction complex” [19]. Phosphorylation of GSK3β at Ser9 phosphorylation site by upstream regulators causes inactivation of the kinase. Glycogen synthase kinase 3 beta is then unable to phosphorylate β-Cat, which becomes free to translocate into the nucleus to function as a potent transcription factor for up-regulating pathways determining cell fate [20]. Nuclear factor erythroid 2-related factor 2 (Nrf2), which is a transcription factor that plays a role in the antioxidant response of a cell, is another important downstream target of GSK3β [18, 21, 22]. Glycogen synthase kinase 3 beta is part of the noncanonical pathway of regulation of Nrf2 where inactivation of GSK3β can lead to activation and nuclear translocation of Nrf2 to cause its downstream effect [23].
In reproductive tissues, GSK3β has been reported to play an important role in various biological processes right from implantation of the blastocyst and development of the fetus and placenta, up to delivery of the baby [14, 16, 24–26]. Lim et al. recently reported that an increase in activity of GSK3 in fetal membranes causes an increase in cytokine expression (IL-6, IL-8, TNF α) leading to labor [27]. However, the possible contribution of GSK3β to OS-mediated p38MAPK-mediated senescence of fetal membranes has not been studied.
The present study aimed to understand the role of GSK3β in the OS-associated fetal membrane senescence in general and amnion cell senescence in particular and the labor pathways. The regulation of GSK3β’s function by p38MAPK and possible downstream targets of GSK3β including β-Cat and Nrf2 were analyzed. We demonstrate that p38MAPK can lead to the inhibition of GSK3β by phosphorylating at the Ser9 site. Further, GSK3β inhibition caused senescence of amnion cells—a function that can potentially be mediated by Nrf2. Further mechanistic studies are needed to determine the exact downstream target of GSK3β that mediates senescence in fetal membranes.
Materials and methods
Institutional review board approval
Placentas for this study were collected after term vaginal (term labor; TL) and scheduled cesarean deliveries (term not in labor; TNIL) from John Sealy Hospital (University of Texas Medical Branch (UTMB)) at Galveston, Texas according to the inclusion and exclusion criteria defined by our laboratory [6, 28]. As discarded placentas were used after delivery for the study, subject recruitment or consenting was not done. The Institutional Review Board at UTMB approved the study protocol, and placentas were collected according to the regulations of the IRB as an exempt protocol that allowed utilization of the discarded placentas (UTMB 11-251).
Clinical samples
Fetal membranes (amniochorion) were obtained from TNIL scheduled cesarean deliveries (n = 12) and TL vaginal deliveries (n = 12) as described previously by our laboratory [28]. Briefly, fetal membranes were dissected from the placenta and any adherent blood clots were removed by washing the membranes in normal saline followed by thorough cleaning using sterile cotton gauze. 6 mm biopsies (explants) were collected from the midzone of the fetal membranes. Intact fetal membrane explants (amniochorion), as well as explants with amnion and chorion layer separated, were collected and processed to perform immunohistochemistry (IHC) and western blotting (WB).
Isolation and culture of human amnion epithelial cells and human amnion mesenchymal cells
Human amnion epithelial cells (AECs) were isolated and cultured as previously described by our laboratory [6, 9, 29]. A total of 12 placentae were collected for preparing primary cells used for this study. Briefly, fetal membranes obtained from term scheduled cesarean deliveries (TNIL) were received by our laboratory and approximately 10 g of the amnion layer was separated from the chorion layer. The amnion was rinsed in saline and cut into small pieces of approximately 2 cm × 2 cm in size. The amnion was digested using 0.125% collagenase and 1.2% trypsin (Sigma-Aldrich, St. Louis, MO, USA) in Hanks’ balanced salt solution (HBSS; Mediatech Inc., Manassas, VA, USA) for 35 min. After the digestion step, the tissue was filtered through a 70-μm cell strainer (Thermo Fisher Scientific, Waltham, MA, USA), and trypsin was inactivated with AEC culture media. Complete AEC media contain Dulbecco’s modified Eagle’s medium: Nutrient Mixture F-12 media (DMEM/F12; Mediatech Inc.) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich), 10% penicillin/streptomycin (Mediatech Inc.), and 100 μg/mL epidermal growth factor (EGF; Sigma-Aldrich). The above digestion step was repeated once more and the final filtrate was centrifuged at 3000 rpm for 10 min. The pellet was resuspended in complete AEC media and the AECs were cultured in T75 flasks at approximately 3–5 million cells/flask [9]. The flasks were incubated at 37 °C, 5% CO2, and 95% air humidity till they reached 70–80% confluence and were ready to be passaged and treated.
Human amnion mesenchymal cells (AMCs) were isolated from the fetal membranes as per the protocol described previously by Kendal-Wright et al. [30, 31] with slight modifications. Briefly, fetal membranes obtained from term scheduled cesarean deliveries (TNIL) were received by our laboratory and approximately 10 g of the amnion layer was separated from the chorion layer. The blood clots adherent to the amnion were removed by rinsing the membrane 3–4 times in HBSS. Incubation of the amnion with 0.05% trypsin/EDTA (Corning, Corning, NY, USA) for 1 h in a 37 °C water bath helped remove AECs. The membrane was then washed 3–4 times in cold HBSS and incubated for 1 h in a rotator in a digestion buffer consisting of Minimum Essential Medium Eagle (Corning), 1 mg/mL collagenase type IV, and 25 μg/mL DNase I. Following complete digestion of the membrane, the solution was neutralized with complete AMC culture media. Complete AMC media consist of DMEM/F12 media supplemented with 5% FBS, 100 U/ml penicillin G, and 10% penicillin/streptomycin. After filtering through a 70 μm cell strainer, the solution was then centrifuged for 10 min at 3000 rpm. The cell pellet was resuspended in complete AMC media and the AMCs were seeded into T75 flasks at a density of 3–5 million cells per flask. The flasks were incubated at 37 °C, 5% CO2, and 95% air humidity till they reached 70–80% confluence and were ready to be passaged and treated.
Cell culture treatments
Cells isolated previously were passaged into T25 flasks once they reached 70–80% confluence. All experiments were conducted utilizing AECs and AMCs at passage 1. Approximately 1 million AECs/T25 and 800,000 AMCs/T25 were plated for 6 h treatments and million AECs/T25 and 800,000 AMCs/T25 were plated for 48 h treatments. Prior to each treatment, the cells were incubated in serum-free media for 1 h. The following treatments were performed in this study: media (control), oxidative stress inducer cigarette smoke extract (CSE) (6 h 1:10; 48 h 1:50), CSE+ functional p38 MAPK inhibitor SB203580 (30 μmol/L; Sigma Aldrich #S8307), GSK3β inhibitor CHIR99021 (5 μM; Sigma Aldrich #SML-1046),and as a positive control lithium chloride (20 mmol; Sigma Aldrich #62476). Glycogen synthase kinase 3 beta inhibitor concentration for treatments was determined based on cell viability titration studies conducted for this study as well as previously published reports [32]. The cells were incubated after treatment at 37 °C, 5% CO2, and 95% air humidity for 6 or 48 h based on the experiment requirements.
Cigarette smoke extract treatment
Cigarette smoke extract was utilized to induce OS in AECs and AMCs as previously described by our laboratory [6, 9, 33]. Cigarette smoke extract is used as an OS inducing laboratory reagent and not used to test the effect of cigarette smoke as a risk factor associated with pregnancy complications. To produce CSE, smoke from a single commercial cigarette (unfiltered Camel, R.J. Reynolds Tobacco Co, Winston Salem, NC, USA) was bubbled through 25 mL of AEC or AMC media. A 0.25 mm Steri-flip fifillter unit (Millipore, Billerica, MA, USA) was used to sterilize the stock CSE. The prepared stock CSE was diluted to 1:10 in complete DMEM/F12 media and utilized for 6-h treatments and diluted to 1:50 in complete DMEM/F12 media and utilized for 48-h treatments of the cells.
Protein extraction and western blotting
AECs, AMCs, and explant tissue were lysed with RIPA lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% Triton X-100, and 1.0 mM EDTA pH 8.0, 0.1% SDS) supplemented with protease and phosphatase inhibitor cocktail and phenylmethylsulfonyl fluoride as described previously by our laboratory [9]. The nuclear and cytoplasmic extraction reagents (NE-PER; Thermofisher #78835) were used, according to the manufacturer’s instructions, to extract the protein in the nuclear and cytoplasmic compartments of the control and CSE-treated AECs and AMCs. Determinations of protein concentrations were done using Pierce BCA Protein Assay Kit BCA (Thermo Scientific, Waltham, MA, USA). The protein concentrations were maintained the same for each sample that was loaded (28 μg of cell lysate/45 μg of tissue lysate). Western blotting was performed as previously described by our laboratory [6, 9]. Briefly, samples were run on a gradient (4–15%) SDS–PAGE Mini-PROTEAN TGX Precast Gels (Bio-Rad, Hercules, CA, USA) and transferred to the membrane using Biorad Gel Transfer Device (Biorad). Membranes were blocked in 5% nonfat milk in 1× Tris buffered saline-Tween 20 (TBS-T) for a minimum of 1 h at room temperature before adding primary antibody and incubating overnight at 4 °C. Appropriate secondary antibody conjugated with horseradish peroxidase was used to incubate the membranes after primary antibody incubation and immunoreactive proteins were visualized using chemiluminescence reagents ECL WB detection system (Amersham Piscataway, NJ, USA). The Restore Western Blot Stripping Buffer (Thermo Fisher) protocol was used before re-probing the blots and blots were not used more than three times. The antihuman antibodies used for WBs were as follows: total GSK3β (1:1000, Cell Signaling, Danvers, MA, USA), phospho-GSK3β (Ser 9) (1:1000, Cell Signaling), total β-Cat (1:1000, Cell Signaling), Nrf2 (1:500, Abcam, Cambridge, United Kingdom), P-p38MAPK (T180/Y182) (1:300, Cell Signaling), p38MAPK (1:1000, Cell Signaling), Heat Shock Protein 90 (HSP-90, 1:1000, Thermo Fischer Scientific, Rockford, IL, USA), CD-81 (1:400, Abnova, Taipei City, Taiwan), and β-actin (1:15,000, Sigma-Aldrich, St. Louis, MO, USA). The relative levels of the proteins in the specific bands were normalized densitometrically, using associated β-actin levels in the samples, using the Biorad-Image Lab 6.0 software.
Flow cytometry analysis
Senescence analysis
AECs and AMCs were treated and incubated for 48 h prior to analysis for cell senescence using flow cytometry. Senescence-associated β-galactosidase (SA-β-Gal), a biomarker for senescence, was detected by the flow cytometric SA–β-Gal assay as described previously by our laboratory [6, 33]. Briefly, cells were incubated for 1 h each in a medium with 100 nM bafilomycin A1 (Baf A1) and 6 μmol/L of 5-dodecanoylaminofluorescein di-β-d-galactopyranoside (C12FDG, eBioscience/Thermo Fischer Scientific, Waltham, MA, USA). Selection of viable cells was made possible by utilizing DNA Prep Stain which contains propidium iodide (Beckman Coulter). CytoFlex flow cytometer (Beckman Coulter, Indianapolis, IN, USA) was used for the assay and the corresponding CytExpert software (Beckman Coulter) was used for data analysis.
Cell cycle analysis
To determine the pattern of the cell cycle in control and treated cells, flow cytometric cell cycle analysis was performed as described previously [34] with modifications. The Coulter cell cycle kit (Beckman Coulter) was used for this experiment. Briefly, the cells were collected after treatment and spun at 3000 rpm for 10 min. The cell pellet was fixed using 70% ethanol and then centrifuged for 5 min at 300g. The cell pellet was resuspended in 250 μL of the cell cycle kit and incubated for 15 min at room temperature. The samples were then run on the Cytoflex flow cytometer and the corresponding Cytexpert software was used for analysis. Cycle analysis by measuring DNA content was used to distinguish between different phases of the cell cycles (sub-G0G1, G0G1, S, and G2).
Immunohistochemistry
IHC was performed on TL and TNIL fetal membrane samples using methods previously described by our laboratory [28, 35]. Briefly, fetal membrane sections were fixed for 48 h in 4% paraformaldehyde (PFA) and embedded in paraffin. Tissue sections of 5-μm thickness were cut and adhered to a positively charged slide. Xylene was used to deparaffinize the tissues and 100% alcohol, 95% alcohol, and normal saline (pH 7.4) was used for rehydration before proceeding to staining. Five images for each specimen were taken at 20x and 40x magnification. The following anti-human antibodies were used for IHC: total GSK3β (1:800, Cell Signaling, Danvers, MA, USA), P-GSK3β (Ser 9) (1:50, Cell Signaling), total β-Cat (1:200, Cell Signaling).
Isolation and characterization of exosomes
Exosomes were isolated from the collected culture media according to the protocol established and published by our laboratory [33, 34, 36], with slight modifications. Briefly, the collected medium was centrifuged in a Sorvall Legend X1R and TX-400 swinging bucket rotor (Thermo Fisher Scientific) (2000g for 20 min at 4 °C), to remove dead cells and cellular debris. Supernatants from the centrifugations were concentrated in Amicon-Ultra 15 tubes (centrifugation at 4000×g for 30 min) and then filtered in a Nalgene prefilter plus filter. This was followed by centrifugation for 30 min at 10,000g and ultracentrifugation in a Beckman Optima LX-80 ultracentrifuge with a 70.1Ti rotor (Beckman Coulter) for 2 h at 100,000g. The pellet was re-suspended in 1 × PBS and subjected to size exclusion chromatography through an Exo-spin column to remove co-precipitants that may contaminate the sample. The exosomes were stored at −80 °C. The size and concentration of the collected exosomes were determined using ZetaView PMX 110 (Particle Metrix, Meerbusch, Germany), with the help of the corresponding software (ZetaView 8.02.28; Particle Metrix). WB analysis was utilized to determine the exosome marker CD81 (1:400, Abnova, Taipei City, Taiwan).
Immunofluorescence and confocal microscopy
To localize β-Cat within exosomes, immunofluorescence (IF) was performed for the exosomal marker CD81 and β-Cat. AECs were passaged and seeded into Millicell EZ slides 8-well glass slides (Millipore, Billerica, MA, USA) at a concentration of 70,000 AECs/well. After 24 h, the cells were treated with either the control medium or medium with CSE (1:50 dilution) for 48 h. IF staining and colocalization experiments were performed as described earlier by our laboratory [33, 34]. Briefly, 4% PFA was used for fixation and 0.5% Triton X was used for permeabilization of the cells. After blocking for 30 min in 3% bovine serum albumin (BSA, Fisher Scientific, Waltham, MA, USA) in PBS, primary antibodies (β-Cat 1:1000 dilution in 3% BSA, Cell Signaling and CD81 1:250 dilution in 3% BSA, Abnova) were added and the slides were incubated at 4 °C overnight. Slides were incubated in secondary Alexa Fluor 488- or 594-conjugated antibodies (Life Technologies, Carlsbad, CA, USA) for 1 h at 1:1000 dilution in 3% BSA for β-Cat and 1:400 dilution in 3% BSA for CD81. NucBlue® Live ReadyProbes® Reagent (Life Technologies) for 3 min stained the nucleus for DAPI following washes with PBS. Confocal images (five images per condition) were obtained using a Zeiss LSM-880 confocal microscope with a 63 × 1.20 numerical aperture oil immersion objective. Z-stack acquisition was carried out with 0.41-μm z-steps. Image processing and analysis were performed with Fiji (open source). A linescan was traced over the areas in the membrane and cytoplasm where the highest intensity of the colocalized signal was noted (region of interest). Raw fluorescence intensity vs. calibrated distance (micrometers) along the linescan was plotted graphically using image J. Pearson correlation coefficient was obtained for the region of interest using coloc2 from Fiji for control and CSE-treated cells.
Statistical analysis
Statistical analyses for normally distributed data were performed using a Student t-test. Statistical values were calculated using GraphPad Prism 7 software (GraphPad Software, Inc., LaJolla, CA, USA). P values equal to or less than 0.05 were considered statistically significant. Data in graphs are represented as Mean ± SEM.
Results
Glycogen synthase kinase 3 beta and β-catenin are localized in fetal membranes
Fetal membranes are broadly divided into morphologically distinct layers, the amnion membrane and the chorion membrane each with its own well-defined microarchitecture [37]. In order to understand the localization of GSK3β and one of its major downstream targets, β-Cat, in the different layers of the fetal membranes, IHC was performed. Both P-GSK3β, the inactive form of GSK3β, and total GSK3β were localized in all layers of the fetal membranes in both TNIL as well as TL fetal membranes (Figure 1A, B, D, E). Similarly, β-Cat was also localized to all layers of the fetal membranes—the amnion epithelial and mesenchymal layers as well as the chorion mesenchymal and trophoblast layers in both TNIL and TL fetal membranes (Figure 1C and F). Prior studies from our laboratory had demonstrated that p38MAPK, an upstream regulator of GSK3β is expressed in all layers of the fetal membranes as well [38] and hence IHC of p38MAPK was not repeated in this study. This data provided evidence that both forms of GSK3βs and β-Cat are constitutively expressed in fetal membranes at term; however, did they not indicate their quantitative changes to determine any functional relevance associated with their localization.
Figure 1.
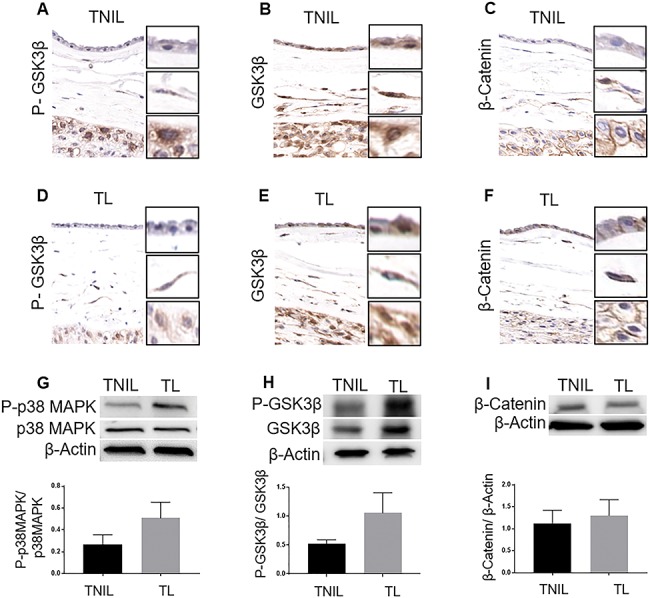
Localization and expression of GSK3β and β-Cat in fetal membranes. (A and D) Immunohistochemistry images of term not in labor (TNIL) and TL fetal membranes stained for P-GSK3β. (B and E) IHC images of TNIL and TL fetal membranes stained for total GSK3β. (C and F) IHC images of TNIL and TL fetal membranes stained for β-Cat. Representative images from a minimum n = 3 for each tissue type are shown. P-GSK3β, total GSK3β, and β-Cat were found localized to both amnion and chorion layers of the fetal membrane. (G) Western blots (WB) of P-p38MAPK in TL vs. TNIL fetal membranes. (H) WB of P-GSK3β in TL vs. TNIL fetal membranes. (I) WB of β-Cat in TL vs. TNIL fetal membranes. The relative levels of P-p38MAPK and P-GSK3β increased two-fold in TL compared to TNIL samples. Total p38MAPK, total GSK3β, and total cellular β-actin were used for normalization (in G, H and I, respectively). Representative blots from n = 12 for each tissue type are shown. Data are presented as mean ± SEM.
Term labor is associated with increased expression of inactive glycogen synthase kinase 3 beta in fetal membranes
In order to understand if the relative expression levels of GSK3β, β-Cat and p38MAPK change in TL compared to TNIL fetal membranes, WB was performed. Confirming our prior data, P-p38MAPK was two-fold higher in TL compared to TNIL fetal membranes (P = 0.09) (Figure 1G) [38]. The activation of p38MAPK corresponded to inactivation of GSK3β where we noted a two-fold increase in P-GSK3β in TL fetal membranes when compared to TNIL fetal membranes and this was marginally significant (P = 0.07) (Figure 1H). The relative levels of β-Cat, however, did not change between TL and TNIL fetal membranes (P = 0.3) (Figure 1I). These data suggest that the p38MAPK/GSK3β signaling pathway is possibly activated during the process of labor with no change in the levels of β-Cat.
Localization of P-GSK3β and total GSK3β to all layers of the fetal membranes and their increased expression at TL compared to TNIL membranes prompted us to determine whether their differential expression is limited specifically to the amnion or chorion layers. WBs were done to assess the differential expression of P-p38MAPK, P-GSK3β, and β-Cat in the amnion, chorion, and intact fetal membranes. We found that the expressions of P-p38MAPK, P-GSK3β, and β-Cat were higher in amnion than chorion and intact fetal membrane in both TL and TNIL tissues (data not shown). The data suggest that the p38MAPK/GSK3β signaling pathway is possibly more active in cells in the amnion layer of the fetal membranes. Therefore, further characterization of GSK3β mechanistic studies were conducted using amnion membrane cells (AECs and AMCs).
Oxidative stress induced by cigarette smoke extract increases p38 mitogen-activated protein kinase activation and glycogen synthase kinase 3 beta inactivation in amnion epithelial cells and amnion mesenchymal cells
Next, we performed primary cell culture experiments to confirm our tissue findings. AEC and AMC markers and morphological characterization using regular microscopy confirmed the specificity of these cells as reported previously [29, 39, 40]. In order to mimic OS that builds up prior to labor in utero, CSE has been utilized in vitro to treat AECs and AMCs [7].
Activation of p38MAPK by its increased phosphorylation was determined by WB in cell culture experiments. Similar to the data reported in the intact fetal membranes above, we were able to replicate data reported earlier where CSE treatment caused an increased expression of P-p38MAP in both AECs (Figure 2A)(P = 0.005) as well as AMCs (Figure 2B) (P = 0.009) compared to cells grown under normal cell culture conditions (untreated control cultures) [6, 29]. Activation of p38MAPK corresponded to inactivation of GSK3β as determined by the increased phosphorylation at the Ser 9 site in response to CSE. Increased phosphorylation of GSK3β (Ser 9) was noted in both AECs (Figure 2C) (P = 0.05) as well as AMCs (Figure 2D) (P = 0.04).
Figure 2.
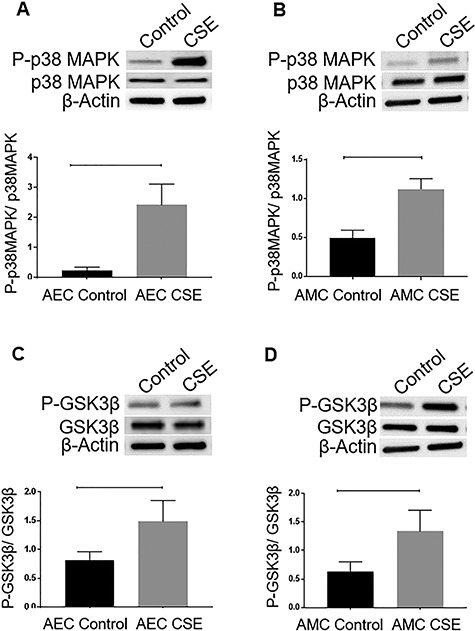
Effect of CSE on p38MAPK and GSK3β expression in the amnion membrane. (A and B) WBs of AECs and AMCs for P-p38MAPK in response to CSE treatment. (C and D) WBs of P-GSK3β in AECs and AMCs in response to CSE treatment. The relative levels of P-p38MAPK and P-GSK3β significantly increased in response to CSE treatment compared to controls in both AECs (P = 0.005 and P = 0.05, respectively) and AMCs (P = 0.009 and P = 0.04 respectively). Total p38MAPK and total GSK3β were used for normalization. Representative blots from a minimum n = 3 for each cell type are shown. Data are presented as mean ± SEM.
Interestingly, despite the inactivation of GSK3β, no change in the total β-Cat levels was noted in both AECs (Figure 3A) (P = 0.4) and AMCs (Figure 3B) (P = 0.4). β-catenin, being one of the most reported downstream targets of GSK3β, was expected to increase with increased phosphorylation of GSK3β as noted in other systems [14]. Nrf2, an antioxidant marker, is another downstream target of GSK3β which was also analyzed. An increase in the relative levels of Nrf2 was noted in response to CSE in both AECs (Figure 3C) (P = 0.01) and AMCs (Figure 3D) (P = 0.005). Since both β-Cat and Nrf2 need to be translocated into the nucleus in order to perform their functions, we next determined the relative level of both molecules in the nuclear fraction of the cells in response to CSE treatment. The nuclear fraction of β-Cat was unchanged in response to CSE treatment confirming lack of its increase in response to CSE. However, nuclear translocation of Nrf2 was increased in response to the same treatment in AECs (Figure 3E) (n = 3) and AMCs (Figure 3F) (n = 3). To note, nuclear localization of Nrf2 was visibly higher in AMCs (Figure 3F, middle panel) than AECs (Figure 3E, middle panel). This is expected as AMCs are much more vulnerable and more pronounced in their response to OS than AECs. Han et al. had also recently reported a similar observation [41] where immortalized human amniotic mesenchymal cellswere more sensitive to oxidative damage compared to immortalized human amniotic epithelial cells.
Figure 3.
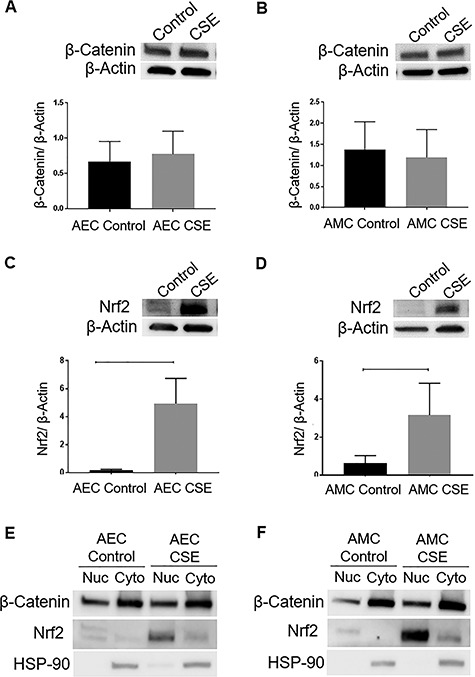
Effect of CSE on possible GSK3β downstream targets in the amnion membrane. (A and B) WBs of AECs and AMCs for β-Cat in response to treatment with CSE. (C and D) WBs of AECs and AMCs for Nrf2 in response to treatment with CSE. The relative levels of Nrf2 significantly increased in response to CSE treatment compared to controls in both AECs and AMCs (P = 0.01 and P = 0.005, respectively). Total cellular β-actin was used for normalization. Representative blots from a minimum n = 3 for each cell type are shown. Data are presented as mean ± SEM. (E) Western blot analysis of nuclear (Nuc) and cytoplasmic (Cyto) lysates demonstrates increased nuclear translocation of Nrf2 in response to CSE with no change in the level of β-Cat in AECs. (F) Western blot analysis of Nuc and Cyto lysates demonstrates increased nuclear translocation of Nrf2 in response to CSE with no change in the level of β-Cat in AMCs. HSP-90 was used to confirm the purity of extraction of nuclear and cytoplasmic proteins. Representative blots from a minimum n = 3 for each cell type are shown.
Thus, CSE-induced inactivation of GSK3β increased the nuclear translocation of Nrf2 with no change in β-Cat levels in amnion cells. To verify p38MAPK-GSK3β axis in determining cell fate, we utilized p38MAPK inhibitor (SB203580) and GSK3 inhibitor (CHIR 99021) in order to confirm the upstream regulator and downstream target of GSK3β.
p38 Mitogen-activated protein kinase-dependent inactivation of P-GSK3β in amnion epithelial cells and amnion mesenchymal cells
Previous experiments correlated the phosphorylation of p38MAPK (activation) as well as GSK3β (inactivation) in response to CSE treatment in both AECs and AMCs. In order to determine if p38MAPK is, in fact, a regulator of GSK3β’s function, a functional inhibitor to p38MAPK-SB203580 was tested. WB was performed and the relative expression of the proteins of interest was normalized densitometrically using associated β-actin levels in the samples (Supplemental Figure 1). Co-treatment of both AECs (Figure 4A) (P = 0.05) and AMCs (Figure 4B) (P = 0.03) with CSE and SB203580 caused a significant reduction in the Ser 9 phosphorylation of GSK3β. This suggests that p38MAPK is able to regulate the phosphorylation of GSK3β. Confirming our prior data, the relative level of β-Cat did not change in response to SB203580 treatment in AECs (Figure 4A) (P = 0.4) and AMCs (Figure 4B) (P = 0.2). However, the expression of Nrf2 was blunted in response to SB203580 in AMCs (Figure 4B) (P = 0.03) and remained unchanged in AECs (Figure 4A) (P = 0.1).
Figure 5.
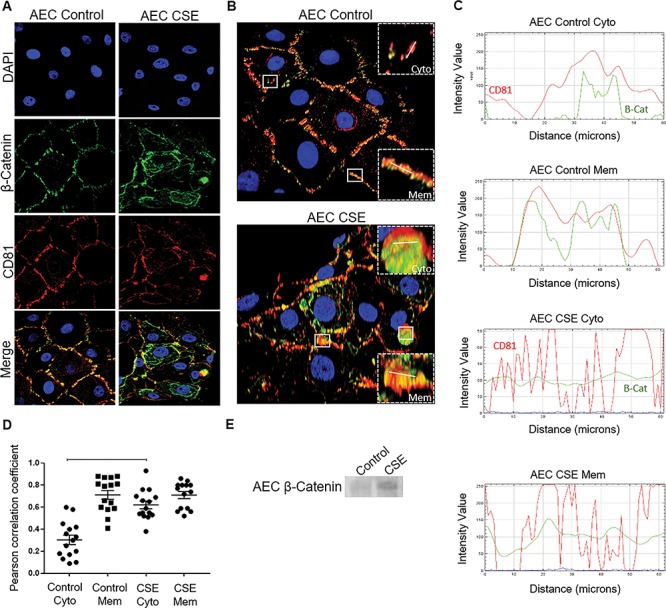
Fate of β-Cat in AECs. (A) Confocal microscopy of control and CSE-treated AECs showing β-Cat (green) and CD81 (red) localization. CSE treatment induced cytoplasmic β-Cat and CD81 compared to controls. (B) Three dimensional confocal images highlighting membrane (Mem) and cytoplasmic (Cyto) region of interest (crops; n = 15) selected for co-localization analysis. Control AEC (top) and CSE-treated AEC (bottom). White lines represent 60 μM line graph region of interest. DAPI—nuclear stain (blue); β-Cat (green); CD81—exosome marker (red). (C) The line graphs demonstrate overlap between CD81 and β-Cat signal at the region of interest at the cytoplasm (Cyto) and membranes (Mem) of both control and CSE-treated AECs. CSE exosomes contained more β-Cat-CD81 overlap than control exosomes. (D) The Pearson’s correlation coefficient showed significantly higher co-localization at the regions of interest in the cytoplasm of CSE-treated AECS compared to controls (P < 0.0001). Data are presented as mean ± SEM. (E) WB analysis of relative levels of β-Cat within AEC exosomes. Representative blot from a minimum of n = 3 is shown. Oxidative stress increased β-Cat in exosomes compared to exosomes from cells grown under normal conditions.
Figure 4.
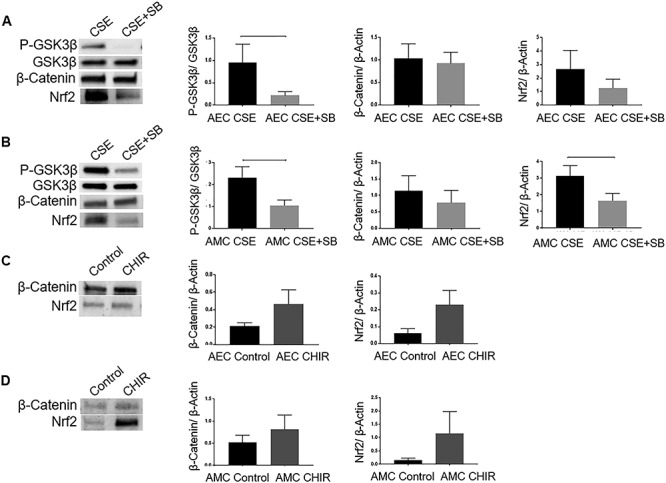
p38 Mitogen-activated protein kinase-dependent inactivation of P-GSK3β in AECs and AMCs. (A) WBs of CSE and CSE + SB203580 (SB)-treated AECs for P-GSK3β, total GSK3β, β-Cat, and Nrf2. (B) WBs of CSE and CSE + SB-treated AMCs for P-GSK3β, total GSK3β, β-Cat, and Nrf2. The relative levels of β-Cat were unchanged (AECs and AMCs) while that of Nrf2 was attenuated (AMCs) in response to treatment with SB. Total GSK3β was used for normalization. (C) WBs for β-Cat and Nrf2 in response to GSK3β inhibitor CHIR99021 (CHIR) in AECs. (D) WBs for β-Cat and Nrf2 in response to GSK3β inhibitor CHIR in AMCs. Total cellular β-actin was used for normalization. The relative levels of β-Cat or Nrf2 remained unchanged in AMCs. In AECs, however, the expression of both β-Cat (P = 0.08) and Nrf2 (P = 0.08) showed an increasing trend in response to treatment with CHIR. Representative blots from a minimum n = 4 for each cell type are shown. Data are presented as mean ± SEM.
In order to identify possible downstream targets of GSK3β, we utilized a potent pharmacological inhibitor of GSK3β-CHIR99021. In AMCs (Figure 4D), CHIR99021 did not change the expression of either β-Cat (P = 0.2) or Nrf2 (P = 0.1). In AECs (Figure 4C), however, the expression of both β-Cat (P = 0.08) and Nrf2 (P = 0.08) showed an increasing trend in response to treatment with CHIR99021. This is suggestive of independent effector mechanisms in AEC and AMC by GSK3β.
Cellular fate of β-catenin in amnion cells
Contrary to expected increase to β-Cat when GSK3β is downregulated, we did not observe any increase either in the cytoplasm or in the nuclear translocation of β-Cat in response to CSE in amnion cells. Therefore, we hypothesized that β-Cat is exported out of the cells by packaging them in the exosomes. To test this, we isolated exosomes from media collected from AECs grown under normal conditions and AECs exposed to CSE. Isolated exosomes were confirmed using exosome marker CD81 and ZetaView PMX 110 (Supplemental Figure 2).
Interestingly, IF and confocal microscopy (Figure 5A and B) demonstrated increased colocalization of the exosome marker CD81 and β-Cat within the cytoplasm of CSE-treated AECs compared to controls. This was demonstrable by the line graph of the intensity of signal vs. distance (Figure 5C) and Pearson’s correlation coefficient (Figure 5D) (P < 0.0001) at the regions of interest within the cytoplasm of CSE-treated AECs compared to control. The regions of interest at the membranes of control and CSE-treated AECs showed similar colocalization of CD81 and β-Cat as both proteins are present at the membranes [20, 42] (Figure 5C and D). The relative levels of β-Cat in exosomes from OS-induced cells were also higher than the control exosomes as demonstrated by WB (Figure 5E). Thus, β-Cat is possibly exported out of the cell in response to CSE preventing its nuclear translocation and its downstream function within the cell.
Cell fate in response to glycogen synthase kinase 3 beta inactivation
In order to understand the cell fate in response to inactivation of GSK3β, we used flow cytometry to identify changes in the cell cycle in AECs and AMCs. An increasing trend in the sub-G0G1 phase and a decrease in G2 phase were noted in response to the GSK3β inhibitor CHIR99021 in AECs (Figure 6A) suggesting that inhibition of GSK3β can possibly cause cell cycle arrest in AECs. This was similar to the cell cycle arrest in response to CSE previously documented by our laboratory [34]. However, no changes in cell cycle were noted in AMCs (Figure 6A), suggesting distinct mechanistic roles for GSK3β in AECs and AMCs.
Figure 6.
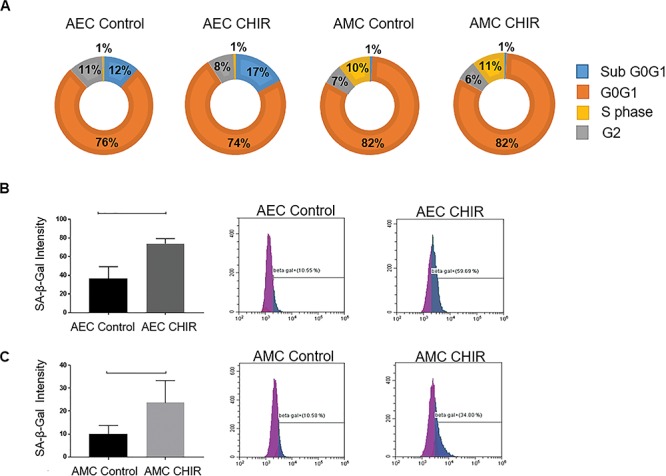
Cell fate in response to GSK3β inactivation. (A) Flow cytometric cell cycle analysis of AECs and AMCs in response to treatment with control media and CHIR. CHIR caused an increase in percent of cells in sub-G0G1 phase and reduced those in G2 phase in AECs compared to control. (B and C) Flow cytometric analysis of senescence (SA-β-Gal assay) of AECs and AMCs in response to treatment with CHIR compared to control. A significant increase in percent of senescence cells was noted following treatment in both AECs (P = 0.01) and AMCs (P = 0.04) compared to controls. Representative data from a minimum n = 3 for each cell type are shown. Data are presented as mean ± SEM.
Next, we performed flow cytometric SA–β-Gal assay to determine senescence induced by the GSK3β inhibitor CHIR99021. A significant increase in senescent cells compared to control was detected by C12FDG treatment in response to CHIR99021 in AECs (Figure 6B) (P = 0.01) and AMCs (Figure 6C) (P = 0.04). This finding was similar to what had been demonstrated earlier by our laboratory in response to CSE [6, 29]. This data suggests that inhibition of GSK3β can ultimately lead to senescence of amnion cells, and overall, this coincides with p38MAPK activation.
Discussion
The primary goal of our study was to understand how OS and p38MAPK regulate GSK3β’s function in the fetal membranes, particularly the amnion layer of the fetal membranes, and its possible contribution to the senescence phenotype. The effect of inducing OS on possible downstream targets of GSK3β that can determine cell fate, including β-Cat and Nrf2, was also analyzed. The key findings of our study are (1) TL is associated with activation of p38MAPK and inactivation of GSK3β. (2) In vitro, we recapitulated these data where OS conditions often seen at TL, induced active p38MAPK and inactive GSK3β in amnion membrane cells. Reduction of functional p38MAPK inhibited the phosphorylation of GSK3β, suggesting that p38MAPK can inactivate GSK3β. (3) Induction of OS on amnion cells was associated with an increased nuclear translocation of antioxidant Nrf2 with no change in pro-cell cycle mediator β-Cat but an increased secretion of β-Cat via exosomes. (4) Inhibition of GSK3β induced cell cycle arrest and senescence in amnion cells.
Glycogen synthase kinase 3 beta is a constitutively expressed pro-cell cycle protein during gestation and parturition. In reproductive tissues, GSK3β has been shown to play a role in survival, differentiation, and migration of placental trophoblast cells [43–45]. Interestingly, evidence that points to the contribution of GSK3β in promoting a seemingly opposing function of cell senescence also exists [46, 47]. Seo et al. linked GSK3 inhibition to enhanced glycogenesis and cell senescence [48]. Similarly, Kim et al. demonstrated that inactivation of both isoforms of GSK3 can cause an increase in lipogenesis and subsequent senescence of cells [49]. The dual role of GSK3β is possibly determined by the cell type, the stimulus, and phosphorylation of a specific site by its upstream regulator. Thus, understanding the exact functional and mechanistic role of GSK3β in the tissue of interest is a challenge. A systematic review conducted by our laboratory identified important knowledge gaps in studying mechanisms and functions involving GSK3β in reproductive tissues [14]. Although biological processes that are important during gestation and labor have been studied, the role of GSK3β in critical processes like cervical and fetal membrane remodeling, activation of the myometrium, cervical ripening, etc., has not been examined [14]. As reported previously discussed above, OS-induced p38MAPK activation has been demonstrated to contribute to senescence and inflammation of fetal membranes [7, 38]. Results from studies in other fields indicate that p38MAPK acts as an upstream regulator of GSK3β [10, 11]. However, the role of p38MAPK in regulating GSK3β’s function in reproductive tissues in general, and during parturition in particular, has not been studied. As far as we know, this is the first study in fetal membranes to establish the link between p38MAPK andGSK3β.
The role of p38MAPK in regulating GSK3β has been reported previously by Thornton TM et al. [10]. In thymocytes, authors reported a p38MAPK-dependent inactivation of GSK3β leading to an accumulation of β-Cat [10]. In these cells, p38MAPK mediated phosphorylation of GSK3β was at Thr390 [10]. This was confirmed in GSK3β Thr390 mutant cells where p38MAPK-mediated phosphorylation was partially abrogated [10]. Our experimental conditions and Thr390 specific antibody did not demonstrate GSK3β phosphorylation in either AECs or AMCs (data not shown). A similar phenomenon has been observed in AECs and AMCs with p38MAPK phosphorylation where these cells tend to have distinct mechanisms of activation where they do not exhibit canonical apoptosis signal-regulating kinase (ASK)-1-mediated activation of p38MAPK and are often activated by TGFβ-TAB1-mediated autophosphorylation [9]. Gene silencing of TAB1 prevented p38MAPK activation whereas ASK1 inhibition did not impact p38MAPK [9]. Lack of GSK3β phosphorylation at Thr390 but at Ser9 is also unique in these cell types under OS conditions induced by CSE. The uniqueness of activation and inactivation of various molecules related to cell growth and senescence is assuring timed events taking place during gestation and parturition. Canonical activation of p38MAPK can be detrimental to GSK3β-mediated feto-placental growth and survival during pregnancy. However, OS-mediated increase of TGFβ in the amniotic fluid and amniochorion assures p38MAPK activation that will promote GSK3β inhibition at a definite site to ensure senescence and inflammation required for timely delivery of the fetus. We postulate that unlike other cells, human fetal membrane cells exhibit unique pathways to regulate signaling molecules’ functions to ensure pregnancy survival as well as determine longevity of fetal membranes. This is a natural and physiologic response from the membranes as their functional role as protector of uterine cavity is over. On the contrary, redox balance during pregnancy prevents p38MAPK activation and maintains active GSK3β to perform proliferative functions in amnion membrane cells to help them remodel during pregnancy to maintain their integrity and function. Senescence and sterile inflammation from senescent cells, a nonreversible process, will eventually promote parturition by muting GSK3β function and transporting β-Cat outside the cell.
Our study analyzed a possible downstream target of p38MAPK-GSK3β. Like p38MAPK, GSK3β is a multifunctional kinase that regulates multiple biological processes within the cell [12, 14, 50, 51]. As an integral component of the Wnt pathway or the PI3K/AKT pathways, GSK3β and its downstream target β-Cat have played an important role in cellular proliferation, cell survival, and embryonic development [52–54]. β-catenin is one of the most studied downstream targets of GSK3β [14]. Amongst other functions, β-Cat is a pro-growth factor that helps maintain the pluripotency of stem cells [55, 56]. An increase in the nuclear translocation of β-Cat by abnormal activation of the Wnt pathway has been noted to play a major role in the development and progression of tumors [57]. Interestingly, some studies highlight the specific role of β-Cat in bypassing cellular senescence [58, 59]. In our study, the expression of β-Cat was unchanged in TL fetal membranes compared to TNIL samples. Further, treatment of AECs and AMCs with either CSE or CHIR99021 did not change the relative levels of β-Cat. Also, no nuclear translocation of β-Cat was noted in response to CSE. These findings suggest that OS that causes inactivation of GSK3β in amnion cells possibly does not involve the Wnt/β-Cat pathways. Further, a higher relative level of β-Cat was packaged within exosomes derived from OS-induced AECs compared to AECs grown under normal conditions and exported out of the cells. Exosomes carrying specific cargo can function as either “garbage bags” to remove unwanted molecules from the cell or can utilize the cargo as signaling molecules and a means of intercellular communication [36, 60]. We speculate that OS in AECs possibly suppresses the cell proliferative actions of the Wnt/β-Cat signaling pathway by removing any β-Cat that has been freed from the β-Cat destruction complex and packaging it within exosomes. Fetal membranes perform important biological functions during gestation; however, as discussed above, their need is limited to this specific period of time. It is thus possible that fetal membrane cells promote senescence by removing excess β-Cat from the cells under conditions of OS, as noted towards the end of the gestational period. Hoffmeyer et al. have demonstrated that β-Cat-deficient mouse embryonic stem cells have reduced telomerase activity and shortened telomeres [61]. Oxidative stress-induced telomere length loss has been noted in fetal membranes [4]. Cell-free fetal telomere fragments arising from fetal membranes are shown to get packaged in AEC-derived exosomes as well as accumulate in amniotic fluid prior to delivery at term [33]. Removal of β-Cat within exosomes under OS conditions to indirectly promote reduction of telomere length of amnion cells along with the senescence phenotype is a novel possibility. Future mechanistic studies planned by our laboratory will help determine the role of β-Cat present within exosomes derived from OS-induced AECs.
Although CSE is able to induce OS and p38 MAPK activation in amnion cells in vitro, the response of amnion cells to CSE may not be extrapolated to infection during pregnancy associated with preterm birth. We have reported an increase in the number of senescent amnion cells without p38 MAPK activation in response to treatment with lipopolysaccharide (LPS) and TNF-α to mimic changes associated with infection and inflammation [6]. Although both LPS and TNF-α induce p38 MAPK-mediated senescence, the extent of senescence was different than CSE. Further, preliminary studies conducted by our laboratory (data not shown) did not show any change in the phosphorylation of GSK3β in AECs in response to LPS treatments. It is likely that the pathways associated with LPS/infection-induced amnion cell senescence may bypass p38 MAPK/GSK3β-associated signaling. Distinct mechanisms of activation of p38MAPK and inflammatory pathways by infectious and noninfectious mediators have also been reported previously by us supporting this postulation [62].
Another downstream target of GSK3β is Nrf2 [18]. Nrf2 is a basic region leucine zipper (bZip) transcription factor which is a major regulator of the antioxidant response of a cell [22]. Nrf2 knockout mice were noted to be highly susceptible to oxidative stress-induced damage [63]. In fetal membranes, Nrf2 has been demonstrated to have anti-inflammatory properties [64]. Canonically, Nrf2 is regulated by Kelch-like ECH-associated protein 1 (Keap1) that is able to sequester Nrf2 in the cytoplasm and cause its ubiquitination and degradation [63, 65]. Oxidative stress and inducers of Nrf2 like sulforaphane can cause a conformational change in the structure of Keap1 and prevent the ubiquitination of Nrf2 [66]. Nrf2 can then translocate into the nucleus and bind to the antioxidant response element (ARE) of the DNA to induce a variety of genes, such as Heme-Oxygenease-1, to activate the antioxidant response in the cell [66–68]. A noncanonical or Keap1-independent regulation of Nrf2 includes the ubiquitination and degradation of Nrf2 by GSK3β [23]. The inactivation of GSK3β leading to the activation of Nrf2 has been demonstrated in several studies [23, 69–71]. In the present study, we noted that CSE was able to increase the relative levels of Nrf2 in amnion cells. The nuclear translocation of Nrf2 in response to CSE was also demonstrable. Pharmacological inhibition of p38 MAPK and GSK3β gave inconclusive data regarding the regulation of Nrf2 by p38MAPK or GSK3β. This could be due to insufficient power required for experiments using these inhibitors. Gene silencing and overexpression studies have been planned by our laboratory to better establish the possible role of Nrf2 in OS-induced amnion membrane senescence.
Treatment of amnion cells with CSE has been demonstrated to induce cell cycle arrest and senescence of the cells in previous studies conducted by our laboratory [6, 34]. In the present study, we noted that inhibition of GSK3β by CHIR99021 induced senescence as well which was comparable to that caused by CSE. Further, the treatments induced an arrest in cell proliferation in AECs. Thus, senescence of the amnion cells involves the activation of p38MAPK and the inactivation of GSK3β.
In conclusion, our present study demonstrates the novel regulation of GSK3β by p38MAPK in fetal membrane cells (Figure 7). Oxidative stress causes the activation of p38MAPK and the inactivation of GSK3β. This signaling mechanism can contribute to the senescence of amnion membrane cells. Further mechanistic studies are needed to determine if senescence of amnion cells can possibly be mediated by Nrf2, which has been recently demonstrated to occur in fibroblasts [72]. Nrf2 has also been shown to potentiate the actions of Peroxisome proliferator-activated receptor gamma (PPAR-γ) and cause the inhibition of the Wnt/β-Cat pathway [73–76]. Understanding the role of GSK3β in the oxidative stress response and senescence of fetal membranes will help determine if it is a potential target to develop therapeutic strategies to minimize damage associated with OS during pregnancy including PTB.
Figure 7.
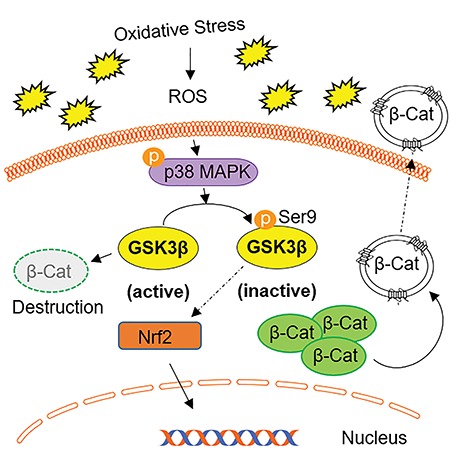
Summary figure representing the proposed pathway of OS-induced p38MAPK-GSK3β-mediated signaling in amnion cells. Oxidative stress-induced reactive oxygen species (ROS) activates p38MAPK and mediates the inactivation of GSK3β by phosphorylating Ser9. Inactivation of GSK3β effects downstream targets Nrf2 and β-Cat. In response to OS, there is increased translocation of Nrf2 into the nucleus likely as an antioxidant response. Active GSK3β degrades β-Cat, while OS-induced GSK3β inactivation allows for the accumulation of β-Cat in the cell. AEC β-Cat does not get translocated to the nucleus; instead, β-Cat is likely packaged within exosomes and cargoed out of the cell.
Conflict of interest
The authors declare no conflicts of interest.
Supplementary Material
{kind=link}
{kind=link}
References
- [1]. Martin L, Richardson L, Menon R. Characteristics, Properties and Functionality of Fetal Membranes: An Overlooked Area in the Field of Parturition, Encyclopedia of Reproduction. Amsterdam, the Netherlands: Elsevier; 2018:387–398. [Google Scholar]
- [2]. Bryant-Greenwood G.D., The extracellular matrix of the human fetal membranes: Structure and function, Placenta 1998; 19:1–11. [DOI] [PubMed] [Google Scholar]
- [3]. Behnia F., Taylor B.D., Woodson M., Kacerovsky M., Hawkins H., Fortunato S.J., Saade G.R., Menon R., Chorioamniotic membrane senescence: a signal for parturition?, Am J Obstet Gynecol 2015; 213:359.e1-16. [DOI] [PubMed] [Google Scholar]
- [4]. Menon R., Bonney E.A., Condon J., Mesiano S., Taylor R.N., Novel concepts on pregnancy clocks and alarms: Redundancy and synergy in human parturition, Hum Reprod Update 2016; 22:535–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5]. Menon R. Oxidative stress damage as a detrimental factor in preterm birth pathology. Front. Immunol. 2014; 5:567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6]. Dixon C.L., Richardson L., Sheller-Miller S., Saade G., Menon R., A distinct mechanism of senescence activation in amnion epithelial cells by infection, inflammation, and oxidative stress, Am J Reprod Immunol 2018; 79:e12790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7]. Menon R., Boldogh I., Urrabaz-Garza R., Polettini J., Syed T.A., Saade G.R., Papaconstantinou J., Taylor R.N., Senescence of primary amniotic cells via oxidative DNA damage, PLoS ONE 2013; 8:e83416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8]. Dutta E.H., Behnia F., Boldogh I., Saade G.R., Taylor B.D., Kacerovský M., Menon R., Oxidative stress damage-associated molecular signaling pathways differentiate spontaneous preterm birth and preterm premature rupture of the membranes, Mol Hum Reprod 2016; 22:143–157. [DOI] [PubMed] [Google Scholar]
- [9]. Richardson L., Dixon C.L., Aguilera-Aguirre L., Menon R., Oxidative stress-induced TGF-beta/TAB1-mediated p38MAPK activation in human amnion epithelial cells, Biol Reprod 2018; 99:1100–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10]. Thornton T.M., Pedraza-Alva G., Deng B., Wood C.D., Aronshtam A., Clements J.L., Sabio G., Davis R.J., Matthews D.E., Doble B., Rincon M., Phosphorylation by p38 MAPK as an alternative pathway for GSK3beta inactivation, Science 2008; 320:667–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11]. Rodriguez-Carballo E, Gamez B, Ventura F. p38 MAPK signaling in osteoblast differentiation. Front Cell Dev Biol 2016; 4:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12]. Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol Ther 2015; 148:114–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13]. Force T., Woodgett J.R., Unique and overlapping functions of GSK-3 isoforms in cell differentiation and proliferation and cardiovascular development, J Biol Chem 2009; 284:9643–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14]. Lavu N, Richardson L, Bonney E, Menon R. Glycogen synthase kinase (GSK) 3 in pregnancy and parturition: A systematic review of literature. J Matern Fetal Neonatal Med 2018; 1–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15]. Doble B., Woodgett J.R., GSK-3: Tricks of the trade for a multi-tasking kinase, J Cell Sci 2003; 116:1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16]. Rider V., Isuzugawa K., Twarog M., Jones S., Cameron B., Imakawa K., Fang J.W., Progesterone initiates Wnt-beta-catenin signaling but estradiol is required for nuclear activation and synchronous proliferation of rat uterine stromal cells, J Endocrinol 2006; 191:537–548. [DOI] [PubMed] [Google Scholar]
- [17]. Lassance L., Miedl H., Absenger M., Diaz-Perez F., Lang U., Desoye G., Hiden U., Hyperinsulinemia stimulates angiogenesis of human fetoplacental endothelial cells: A possible role of insulin in placental hypervascularization in diabetes mellitus, J Clin Endocrinol Metab 2013; 98:E1438–47. [DOI] [PubMed] [Google Scholar]
- [18]. Sutherland C. What are the bona fide GSK3 substrates? Int J Alzheimers Dis 2011; 2011:505607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19]. Voskas D, Ling LS, Woodgett JR. Does GSK-3 provide a shortcut for PI3K activation of Wnt signalling? F1000 Biol Rep 2010; 2:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20]. Valenta T., Hausmann G., Basler K., The many faces and functions of β-catenin, EMBO J 2012; 31:2714–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21]. Cheng X, Chapple SJ, Patel B, Puszyk W, Sugden D, Yin X, Mayr M, Siow RC, Mann GE. Gestational diabetes mellitus impairs Nrf2-mediated adaptive antioxidant defenses and redox signaling in fetal endothelial cells in utero. 2013; 62:4088–4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22]. Ray P.D., Huang B.-W., Tsuji Y., Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling, Cell Signal 2012; 24:981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23]. Cuadrado A, Kügler S, Lastres-Becker I. Pharmacological targeting of GSK-3 and NRF2 provides neuroprotection in a preclinical model of tauopathy. Redox Biol 2017; 14:522–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24]. Hou X.Y., Arvisais E.W., Davis J.S., Luteinizing hormone stimulates mammalian target of Rapamycin signaling in bovine luteal cells via pathways independent of AKT and mitogen-activated protein kinase: Modulation of glycogen synthase kinase 3 and AMP-activated protein kinase, Endocrinology 2010; 151:2846–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25]. Roseweir A.K., Katz A.A., Millar R.P., Kisspeptin-10 inhibits cell migration in vitro via a receptor-GSK3 beta-FAK feedback loop in HTR8SVneo cells, Placenta 2012; 33:408–15. [DOI] [PubMed] [Google Scholar]
- [26]. O'Connell B.A., Moritz K.M., Walker D.W., Dickinson H., Treatment of pregnant spiny mice at mid gestation with a synthetic glucocorticoid has sex-dependent effects on placental glycogen stores, Placenta 2013; 34:932–940. [DOI] [PubMed] [Google Scholar]
- [27]. Lim R., Lappas M., A novel role for GSK3 in the regulation of the processes of human labour, Reproduction 2015; 149:189–202. [DOI] [PubMed] [Google Scholar]
- [28]. Richardson L.S., Vargas G., Brown T., Ochoa L., Sheller-Miller S., Saade G.R., Taylor R.N., Menon R., Discovery and characterization of human amniochorionic membrane microfractures, Am J Pathol 2017; 187:2821–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29]. Jin J, Richardson L, Sheller-Miller S, Zhong N, Menon R. Oxidative stress induces p38MAPK-dependent senescence in the feto-maternal interface cells. Placenta 2018; 67:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30]. Casey M.L., MacDonald P.C., Interstitial collagen synthesis and processing in human amnion: A property of the mesenchymal cells, Biol Reprod 1996; 55:1253–60. [DOI] [PubMed] [Google Scholar]
- [31]. Sato BL, Collier ES, Vermudez SA, Junker AD, Kendal-Wright CE. Human amnion mesenchymal cells are pro-inflammatory when activated by the toll-like receptor 2/6 ligand, macrophage-activating lipoprotein-2. Placenta 2016; 44:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32]. Balamurugan K, Sharan S, Klarmann KD, Zhang Y, Coppola V, Summers GH, Roger T, Morrison DK, Keller JR, Sterneck E. FBXW7α attenuates inflammatory signalling by downregulating C/EBPδ and its target gene Tlr4. Nature Commun 2013; 4:1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33]. Sheller-Miller S, Urrabaz-Garza R, Saade G, Menon R. Damage-associated molecular pattern markers HMGB1 and cell-free fetal telomere fragments in oxidative-stressed amnion epithelial cell-derived exosomes. J Reprod Immunol 2017; 123:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34]. Sheller S., Papaconstantinou J., Urrabaz-Garza R., Richardson L., Saade G., Salomon C., Menon R., Amnion-epithelial-cell-derived exosomes demonstrate physiologic state of cell under oxidative stress, PLoS One 2016; 11:e0157614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35]. Polettini J, Richardson LS, Menon R. Oxidative stress induces senescence and sterile inflammation in murine amniotic cavity. Placenta 2018; 63:26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36]. Hadley EE, Sheller-Miller S, Saade G, Salomon C, Mesiano S, Taylor RN, Taylor BD, Menon R. Amnion epithelial cell derived exosomes induce inflammatory changes in uterine cells. Am J Obstet Gynecol 2018; 219:478.e1–478.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37]. Strauss J.F., 3rd, Extracellular matrix dynamics and fetal membrane rupture, Reprod Sci 2013; 20:140–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38]. Menon R., Boldogh I., Hawkins H.K., Woodson M., Polettini J., Syed T.A., Fortunato S.J., Saade G.R., Papaconstantinou J., Taylor R.N., Histological evidence of oxidative stress and premature senescence in preterm premature rupture of the human fetal membranes recapitulated in vitro, Am J Pathol 2014; 184:1740–51. [DOI] [PubMed] [Google Scholar]
- [39]. Martin LF, Richardson LS, Silva MG, Sheller-Miller S, Menon R. Dexamethasone induces primary amnion epithelial cell senescence through telomere-P21 associated pathway. Biol Reprod 2019; 100:1605–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40]. Richardson L., Menon R., Proliferative, migratory, and transition properties reveal metastate of human amnion cells, Am J Pathol 2018; 188:2004–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41]. Han LG, Zhao Q-L, Yoshida T, Okabe M, Soko C, Rehman MU, Kondo T, Nikaido T. Differential response of immortalized human amnion mesenchymal and epithelial cells against oxidative stress. Free Radi Biol Med 2019; 135:79–86. [DOI] [PubMed] [Google Scholar]
- [42]. Quast T., Eppler F., Semmling V., Schild C., Homsi Y., Levy S., Lang T., Kurts C., Kolanus W., CD81 is essential for the formation of membrane protrusions and regulates Rac1-activation in adhesion-dependent immune cell migration, Blood 2011; 118:1818–27. [DOI] [PubMed] [Google Scholar]
- [43]. Astuti Y., Nakabayashi K., Deguchi M., Ebina Y., Yamada H., Human recombinant H2 relaxin induces AKT and GSK3beta phosphorylation and HTR-8/SVneo cell proliferation, Kobe J Med Sci 2015; 61:E1–8. [PubMed] [Google Scholar]
- [44]. Sonderegger S., Haslinger P., Sabri A., Leisser C., Otten J.V., Fiala C., Knofler M., Wingless (Wnt)-3A induces Trophoblast migration and matrix Metalloproteinase-2 secretion through canonical Wnt signaling and protein kinase B/AKT activation, Endocrinology 2010; 151: 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45]. Fischer I., Weber M., Kuhn C., Fitzgerald J.S., Schulze S., Friese K., Walzel H., Markert U.R., Jeschke U., Is galectin-1 a trigger for trophoblast cell fusion?: The MAP-kinase pathway and syncytium formation in trophoblast tumour cells BeWo, Mol Hum Reprod 2011; 17:747–757. [DOI] [PubMed] [Google Scholar]
- [46]. Mancinelli R, Carpino G, Petrungaro S, Mammola CL, Tomaipitinca L, Filippini A, Facchiano A, Ziparo E, Giampietri C. Multifaceted roles of GSK-3 in cancer and autophagy-related diseases. Oxid Med Cell Longev 2017; 2017:4629495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47]. Kim YM, Seo YH, Park CB, Yoon SH, Yoon G. Roles of GSK3 in metabolic shift toward abnormal anabolism in cell senescence. Ann NY Acad Sci 2010; 1201:65–71. [DOI] [PubMed] [Google Scholar]
- [48]. Seo Y.H., Jung H.J., Shin H.T., Kim Y.M., Yim H., Chung H.Y., Lim I.K., Yoon G., Enhanced glycogenesis is involved in cellular senescence via GSK3/GS modulation, Aging Cell 2008; 7:894–907. [DOI] [PubMed] [Google Scholar]
- [49]. Kim Y.-M., Song I., Seo Y.-H., Yoon G., Glycogen synthase kinase 3 inactivation induces cell senescence through sterol regulatory element binding protein 1-mediated lipogenesis in chang cells, Endocrinol Metab (Seoul, Korea) 2013; 28:297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50]. Menon R., Papaconstantinou J., p38 mitogen activated protein kinase (MAPK): A new therapeutic target for reducing the risk of adverse pregnancy outcomes, Expert Opin Ther Targets 2016; 20:1397–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51]. Vijay G.V., Zhao N., Den Hollander P., Toneff M.J., Joseph R., Pietila M., Taube J.H., Sarkar T.R., Ramirez-Pena E., Werden S.J., Shariati M., Gao R., et al. . GSK3β regulates epithelial-mesenchymal transition and cancer stem cell properties in triple-negative breast cancer, Breast Cancer Res 2019; 21:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52]. McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Abrams SL, Montalto G, D'Assoro AB, Libra M, Nicoletti F, Maestro R, Basecke J, Cocco L et al. . Multifaceted roles of GSK-3 and Wnt/β-catenin in hematopoiesis and leukemogenesis: Opportunities for therapeutic intervention. Leukemia 2013; 28:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53]. Luo J., The role of GSK3beta in the development of the central nervous system, Front Biol 2012; 7:212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54]. Patel P, Woodgett JR. Chapter Eight—Glycogen synthase kinase 3: A kinase for all pathways? In: Jenny A. (ed.), Current Topics in Developmental Biology. Academic Press; 2017:277–302. [DOI] [PubMed] [Google Scholar]
- [55]. Anton R., Kestler H.A., Kuhl M., Beta-catenin signaling contributes to stemness and regulates early differentiation in murine embryonic stem cells, FEBS Lett 2007; 581:5247–54. [DOI] [PubMed] [Google Scholar]
- [56]. Pai V.C., Hsu C.-C., Chan T.-S., Liao W.-Y., Chuu C.-P., Chen W.-Y., Li C.-R., Lin C.-Y., Huang S.-P., Chen L.-T., Tsai K.K., Correction: ASPM promotes prostate cancer stemness and progression by augmenting Wnt–Dvl-3–β-catenin signaling, Oncogene 2019; 38:1354–1354. [DOI] [PubMed] [Google Scholar]
- [57]. Shang S., Hua F., Hu Z.-W., The regulation of β-catenin activity and function in cancer: Therapeutic opportunities, Oncotarget 2017; 8:33972–33989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58]. Larue L., Luciani F., Kumasaka M., Champeval D., Demirkan N., Bonaventure J., Delmas V., Bypassing melanocyte senescence by β-catenin: A novel way to promote melanoma, Pathol Biol 2009; 57:543–547. [DOI] [PubMed] [Google Scholar]
- [59]. Tian L.M., Xie H.F., Xiao X., Yang T., Hu Y.H., Wang W.Z., Liu L.S., Chen X., Li J., Study on the roles of beta-catenin in hydrogen peroxide-induced senescence in human skin fibroblasts, Exp Dermatol 2011; 20:836–8. [DOI] [PubMed] [Google Scholar]
- [60]. Rashed M. H., Bayraktar E., Helal G. K., Abd-Ellah M.F., Amero P., Chavez-Reyes A., Rodriguez-Aguayo C., Exosomes: From garbage bins to promising therapeutic targets, Int J Mol Sci 2017; 18: 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61]. Hoffmeyer K., Raggioli A., Rudloff S., Anton R., Hierholzer A., Del Valle I., Hein K., Vogt R., Kemler R., Wnt/beta-catenin signaling regulates telomerase in stem cells and cancer cells, Science 2012; 336:1549–54. [DOI] [PubMed] [Google Scholar]
- [62]. Behnia F., Sheller S., Menon R., Mechanistic differences leading to infectious and sterile inflammation, Am J Reprod Immunol 2016; 75:505–18. [DOI] [PubMed] [Google Scholar]
- [63]. Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 2013; 53:401–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64]. Lim R., Barker G., Lappas M., The transcription factor Nrf2 is decreased after spontaneous term labour in human fetal membranes where it exerts anti-inflammatory properties, Placenta 2015; 36:7–17. [DOI] [PubMed] [Google Scholar]
- [65]. Nguyen T., Nioi P., Pickett C.B., The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress, J Biol Chem 2009; 284:13291–13295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66]. Silva-Islas CA, Maldonado PD. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol Res 2018; 134:92–99. [DOI] [PubMed] [Google Scholar]
- [67]. Bryan H.K., Olayanju A., Goldring C.E., Park B.K., The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation, Biochem Pharmacol 2013; 85:705–717. [DOI] [PubMed] [Google Scholar]
- [68]. Loboda A., Damulewicz M., Pyza E., Jozkowicz A., Dulak J., Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism, Cell Mol Life Sci 2016; 73:3221–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69]. Culbreth M, Aschner M. GSK-3β, a double-edged sword in Nrf2 regulation: Implications for neurological dysfunction and disease. F1000 Res 2018; 7:1043–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70]. Salazar M., Rojo A.I., Velasco D., de Sagarra R.M., Cuadrado A., Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2, J Biol Chem 2006; 281: 14841–51. [DOI] [PubMed] [Google Scholar]
- [71]. Jiang Y., Bao H., Ge Y., Tang W., Cheng D., Luo K., Gong G., Gong R., Therapeutic targeting of GSK3β enhances the Nrf2 antioxidant response and confers hepatic cytoprotection in hepatitis C, Gut 2015; 64:168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72]. Hiebert P., Wietecha M.S., Cangkrama M., Haertel E., Mavrogonatou E., Stumpe M., Steenbock H., Grossi S., Beer H.D., Angel P., Brinckmann J., Kletsas D., et al. . Nrf2-Mediated Fibroblast Reprogramming Drives Cellular Senescence by Targeting the Matrisome, Dev Cell 2018; 46: 145–161.e10. [DOI] [PubMed] [Google Scholar]
- [73]. Polvani S, Tarocchi M, Galli A. PPARγ and oxidative stress: Con(β) Catenating NRF2 and FOXO. PPAR Res 2012; 2012:641087–641087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74]. Vallée A, Lecarpentier Y. Crosstalk between peroxisome proliferator-activated receptor gamma and the canonical WNT/β-catenin pathway in chronic inflammation and oxidative stress during carcinogenesis. Front Immunol 2018; 9:745–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75]. Lecarpentier Y, Vallée A. Opposite interplay between PPAR gamma and canonical Wnt/Beta-catenin pathway in amyotrophic lateral sclerosis. Front Neurol 2016; 7:100–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76]. Lecarpentier Y, Claes V, Vallée A, Hébert J-L. Interactions between PPAR gamma and the canonical Wnt/Beta-catenin pathway in type 2 diabetes and colon cancer. PPAR Res 2017; 2017:5879090–5879090. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.