

Abstract
Glycan-protein interactions occur at many physiological events, and the analyses are of considerable importance for understanding glycan-dependent mechanisms. Biophysical approaches including 3D structural analysis are essential for revealing glycan-protein interactions at the atomic level. The inherent diversity of glycans suits them to function as identification tags, e.g., distinguish self from the nonself components of pathogens. However, the complexity of glycans and poor affinities for interaction partners limit the usefulness of conventional analyses. To cope with such troublesome glycans, a logical sequence of biophysical analyses need to be developed. In this chapter, we introduce a workflow of glycan-protein interaction analysis consisting of six steps: preparation of lectin and glycan, screening of glycan ligand, determination of binding epitope, quantitative interaction analysis, 3D structural analysis, and molecular dynamics simulation. Our increasing knowledge and understanding of lectin-glycan interactions will hopefully lead to the design of glyco-based medicines and vaccines.
Keywords: Glycan microarray, Nuclear magnetic resonance, X-ray crystallography, Isothermal titration calorimetry, Surface plasmon resonance, Molecular dynamics simulation, Frontal affinity chromatography
Introduction
Glycan-protein interactions are crucial for numerous biological processes such as quality control of nascent proteins, cell-cell interaction, and pathogenic recognition. Of special interest is the breadth of glycan-mediated recognition events, which comes from the extremely high information-coding capacity of glycans. The diversity of oligosaccharides is inherent in their configurational variability, created by the broad range of monosaccharide species, anomeric configurations, multiple linkages, and branching. Glycan complexity is potentially suitable for coding biological information, and interest is growing to “decode” the sugar codes. To this end, investigating glycan-protein interaction at the atomic level is a necessity as only then will we understand the detailed mechanisms of these glycan-mediated biological activities. Recent techniques introduced include glycan microarray analysis and other high-throughput screening methods, and now we can begin to have a more systematic approach to analyzing glycan-protein interactions. We here introduce biophysical approaches to analyze the structure and function of glycan-protein interactions.
A Flowchart for Analyzing Glycan-Protein Interactions by Biophysical Methods
Biophysical investigation of glycan-protein interactions is typically divided into six steps in our laboratory:
Large-scale preparation of protein and glycan
Screening of glycan ligand
Determination of binding epitope (glycotope)
Quantitative glycan-protein interaction analysis
3D structural study on protein-glycan interaction
Molecular dynamics simulation of glycan-protein interaction
Crystallographic and nuclear magnetic resonance (NMR) analyses are direct methods for obtaining atomic details of glycan-protein interactions if the binding epitope is known. In those cases where the binding epitopes are unknown, biophysical approaches such as listed above are required for structural studies. A computational approach combined with experiments is especially effective to investigate the flexible nature of glycans.
Large-Scale Preparation of Protein and Glycan
Biophysical studies require large amounts of protein and glycan. In general, much effort needs to be paid for their preparation. Examples of our achievements are shown below.
Preparation of Protein
Carbohydrate recognition domain (lectin domain) or glyco-enzymes directly bind glycan, and it is convenient to use the isolated domain for biophysical interaction analysis. Normally, 5–10 mg of purified protein is required. Bacterial expression systems are frequently used to express lectin domains in recombinant form (Fig. 7.1a), but obtaining large amounts of pure lectin in a soluble form is often difficult using bacterial expression systems. For instance, C-type lectin and C-type lectin-like domains contain three conserved disulfide bridges, and the cysteine residues hamper soluble expression with correct folding. Refolding of protein from inclusion bodies may be needed to produce functional lectin domains. The protein is solubilized and refolded under controlled conditions. This refolding procedure has been effective for the purification of mammalian C-type lectins and C-type lectin-like domains (Nagae et al. 2013b, 2014a, b, 2016b).
Fig. 7.1.

Large-scale preparation of recombinant proteins
(a) A flowchart of large-scale preparation of lectins for 3D structure and interaction studies
(b) Small-scale expression of differently fused MLS128-scFv in BL21 CodonPlus (left) and Origami B (right) strains analyzed by SDS-PAGE. M: Molecular weight of marker proteins; Lanes 1–8, odd number lanes are composition of total cell lysates, and even number lanes are composition of soluble cell lysates. Lanes 1 and 2 for scFv without fusion tag, Lanes 3 and 4 for thioredoxin (Trx)-fused scFv, Lanes 5 and 6 for DsbC-fused scFv, and Lanes 7 and 8 for PDI-fused scFv. The figure was adapted from a previous paper (Subedi et al. 2012) with permission
(c) Comparison of expression yield of Dectin-1 with and without GB1 fusion tag before and after concentration (to 1 mL) of purified sample. Results are presented as mean ± SD (n = 3) for pET28a murine Dectin-1, pCold murine Dectin-1, pCold-human Dectin-1 (codon-optimized), pCold murine Dectin-1 (optimized), and pCold GB1-fused murine Dectin-1 (codon-optimized). The figure was adapted from the paper (Dulal et al. 2016) with permission
Fusion of a tag protein can improve the solubility of target proteins. In our laboratory, several fusion tags have been used, including protein disulfide isomerase (PDI) and B1 domain of streptococcal protein G (GB1). Single chain Fv (scFv) originating from anti-carbohydrate MLS128 antibody was successfully expressed as a soluble form with PDI fusion (Subedi et al. 2012), while the other fusion tags such as thioredoxin or disulfide isomerase DsbC were not sufficient to produce soluble scFv (Fig. 7.1b). The expressed MLS128-scFv retains full binding activity toward synthetic carbohydrate antigens. The lectin domain of a C-type lectin-like receptor Dectin-1 was expressed as a GB1-fused protein (Dulal et al. 2016). The domain is correctly folded, is a monomer, and specifically binds a β-glucan ligand. The addition of the GB1 tag to the lectin domain greatly increased both yield (fivefold) and solubility (14-fold) (Fig. 7.1c).
Bacterial expression systems are the first choice for protein expression but are not always successful for glycosylated proteins. In these cases, mammalian expression systems may be necessary to express correctly folded proteins with posttranslational modifications. HEK293 cells, CHO cells, and their variant cells are frequently used as hosts. For example, protein O-mannosyl kinase (POMK), which phosphorylates the mannose residue on α-dystroglycan, has three N-glycosylation sites within the catalytic domain, and two sites out of three are partially N-glycosylated. To abolish the heterogeneity originating from partial N-glycan occupancy, two asparagine residues were mutated to glutamine and the mutated POMK expressed in HEK293 variant cells lacking N-acetylglucosaminyl transferase I (GnT-I), to produce uniformly glycosylated protein (Man5GlcNAc2-glycan attached), which was successfully crystallized and the atomic structure elucidated (Nagae et al. 2017b).
Preparation of Glycan
It is another challenge to obtain sufficient amounts of glycan and glycoconjugates suitable for biophysical assays. Prior to interaction analysis, it is necessary to design and prepared a suitable glycan ligand. Ligand design is an extremely important step. The design may arise from information in the literature, or else the binding specificity needs to be ascertained experimentally (Steps 2 and 3). Several methods can be used to prepare glycan ligands: (1) purification of a free natural glycan or liberation of a specific glycan from natural glycoconjugates ( glycoprotein, glycolipid, etc.), (2) chemical synthesis, and (3) chemoenzymatic synthesis. Liberation of glycan from natural glycoprotein can yield large amounts of glycan provided that there is a plentiful supply of the natural glycoprotein. N-glycans attached onto glycoproteins can be liberated by chemical methods (e.g., hydrazinolysis) or by enzymatic methods (e.g., PNGase F) and then labeled with a fluorescent tag 2-aminopyridine for detection. Pyridylamino derivatives of oligosaccharides can be purified by HPLC, and their chemical structures confirmed by solution NMR spectroscopy and mass spectrometry. For example, a biantennary N-glycan bearing bisecting GlcNAc was successfully purified with a yield of 120 μg from 200 mg of IgG and used for the analysis of a plant lectin E4-PHA (Nagae et al. 2014c, 2016b).
Glycan ligands “designed” through synthetic approaches are of great importance in structural studies. N-glycan core units in particular cannot be prepared easily by enzymatic approaches, and chemical synthesis plays a significant role (Hanashima et al. 2014b). Synthetic bisected hexasaccharide has been used as ligand for the C-type lectin receptor murine DCIR2, the legume lectin PHA-E, and the plant lectin Calsepa. The use of designed synthetic ligands has been invaluable for revealing the ligand recognition mode of these lectins by X-ray crystallography and NMR.
Recombinant and/or chemoenzymatic methods have been developed to obtain sufficient amounts of glycopeptides for structural analysis. It is known that several lectins recognize not only the glycan part but also the aglycon part (Nagae and Yamaguchi 2015), and here it is best to use the appropriate glycoconjugate (e.g., glycopeptide, glycolipid, etc.) as the ligand molecule. A C-type lectin receptor CLEC-2 binds to O-glycosylated podoplanin, and binding requires both disialyl core 1 glycan (NeuAcα2-3Galβ1–3[NeuAcα2–6] GalNAc) and neighboring amino acid residues. The O-glycosylated podoplanin glycopeptide was prepared by overexpression using engineered yeast cells and in vitro sialylation (Kato et al. 2008). The substrate of POMK was chemoenzymatically synthetized, composed of core M3 trisaccharide unit ( GalNAcβ1–3GlcNAcβ1-4Man) and α-dystroglycan peptide. Mono-mannosylated peptide was chemically synthesized in advance, and then additional disaccharide units were successively introduced by two glycosyltransferases (Nagae et al. 2017b).
Screening of Glycan Ligand
It is very important to understand how glycans function in recognition and signaling systems within an organism and the responses with microbes and pathogens (Cummings and Pierce 2014). Heterogeneous glycans are potentially a vast source of information, and glycan microarrays embedding such diverse glycans are now becoming one of the key technologies to provide information on recognition determinants (glycotopes) of carbohydrate-binding proteins (Feizi and Chai 2004; Geissner and Seeberger 2016).
Glycan Microarray
When doing the glycan microarray analysis, it is essential to immobilize a series of glycans onto the plate. The immobilization is one of the critical issues in the development of a glycan microarray. There are several ways to immobilize carbohydrate probes onto a suitable surface with retention of function. The chemically modified or unmodified glycans can be non-covalently or covalently attached onto the surface (Fig. 7.2a). Neoglycolipid (NGL) is an artificial glycolipid used to construct glycan microarrays via non-covalent interaction. In NGL technology, lipid-conjugated oligosaccharides prepared from natural sources or chemically synthesized are immobilized onto nitrocellulose-coated microarray slides (Palma et al. 2014). In covalent immobilization, various chemical reactions have been developed (Hyun et al. 2017). Among them, the conjugation reaction using amine-linked glycans is most widely used, and the Consortium for Functional Glycomics uses this chemistry for a glycan microarray platform (Fig. 7.2b). Several different functional groups which react with amines are utilized such as cyanuric chloride, N-hydroxysuccinimide ester (NHS), and epoxide. Measuring lectin binding requires labeling of the lectin with a fluorescent probe or the use of an antibody. Even so carbohydrate-lectin interaction is often too weak to be detected. In such cases, oligomerization of the lectin is a strategy to enhance inherent weak binding. Interactions may be detected by introducing dimeric glutathione S-transferase (GST) into the lectin as a fusion tag, as in the case of a mammalian lectin ZG16p-binding immobilized glycans (Kanagawa et al. 2014; Hanashima et al. 2015). Alternatively, it is possible to enhance binding by increasing the amount of surface glycans (Hanashima et al. 2015). However, it should be noted that the density of immobilized glycans may affect the specificity of lectins (Horan et al. 1999). Instead of fluorescence detection, label-free glycan biosensors have been developed to push the limit of detection (Hushegyi and Tkac 2014).
Fig. 7.2.

Glycan microarray analysis for the lectin specificity
(a) Non-covalent and covalent glycan immobilization for glycan microarray analysis
(b) On-array inhibition assays for analyzing the binding of GST-fused human ZG16p to selected ligands which are non-covalently immobilized on the plate as neoglycolipids. Man-α-OMe, Gal-β-OMe (upper panel), oligosaccharide fractions (>20-mer) of heparin (Hep), and hyaluronic acid (HA) (lower panel) were used as inhibitors. This figure was adapted from a previous paper (Kanagawa et al. 2014) with permission
Glycan microarrays have various applications. Microarrays usually simply display relative binding affinity, but quantitative parameters (e.g., dissociation constant, K D) can be obtained by applying various concentrations (Liang et al. 2007). The estimated K D values are comparable to those obtained by other methods such as surface plasmon resonance (SPR). Moreover, effects of heterogeneous glycan mixing can be evaluated by immobilizing a mixture of glycans onto one spot (Liang et al. 2011). On-array inhibition assays can show if two different ligands bind competitively to the lectin (sharing a common binding site) or independently (each using a different binding site) (Fig. 7.2b) (Kanagawa et al. 2014). Glycan microarrays can also evaluate direct binding of mammalian cells and viruses (Nimrichter et al. 2004; Song et al. 2011), as well as biologically active glycans, if cell signaling can be linked to a fluorescent probe (Pai et al. 2016). This could result in rapid progress toward elucidating pathway-specific glycans.
Other Screening Methods to Evaluate Lectin-Glycan Interaction
Glycan microarray analysis often may overestimate the strength of weak binding, and weak binding is often seen in carbohydrate-lectin interactions. In addition, non-specific binding is usually present, which contributes a background signal. Therefore it is preferable to include other screening methods to validate the results. Several methods have already been developed to screen glycans which bind target lectins. The thermal shift assay is widely used for ligand screening. In this approach, melting temperature is measured in the absence and the presence of ligands and any shifts assessed (Vedadi et al. 2006). A temperature shift is indicative of increased stability acquired upon ligand binding. The purified protein is mixed with ligand in the presence of a fluorescent probe, such as SYPRO Orange, that binds to the denatured state. This method is simple and can be used for rapid ligand screening. Frontal affinity chromatography (FAC) can quantitatively evaluate weak interactions between lectins and glycans, as explained in the following section. The FAC method can also be used to screen for ligand binding. NMR is an excellent technique to determine the binding epitope of a lectin-glycan interaction. When the binding epitope is determined, a suitable ligand of minimum size is designed for use in X-ray crystallography. NMR can also be applied to high-throughput ligand screening (Meyer and Peters 2003) and be advantageously used with a mixture of binder and non-binders.
Determination of Binding Epitope
The binding epitope of a glycan (glycotope) can be deduced from the results of glycan microarray and FAC analysis using a series of glycans. More directly, NMR spectroscopy can provide the glycotope at atomic level. NMR spectroscopy has demonstrated its potential with a wide range of applications for studies of structure and function of biological molecules. The unique potential of NMR spectroscopy is its ability to generate high-resolution data not only on structure but also on dynamics and binding interactions. There are many types of NMR experiments that can be applied for very different experimental conditions both for the sample and the NMR parameters. NMR experiments provide various parameters such as chemical shifts, spin-spin couplings (J couplings), nuclear Overhauser effect (NOEs), and relaxation times. These parameters provide the atomic information on chemical environment, dihedral angles, distance, and mobility, respectively. Here two NMR techniques, saturation transfer difference NMR (STD-NMR) and deuterium-induced isotope shift (DIS), are described below.
Saturation Transfer Difference NMR in Lectin–Glycan Interactions
Saturation transfer difference NMR (STD-NMR) spectroscopy is a method for studying protein-ligand interactions by observing signals emanating from the ligand (Mayer and Meyer 2001; Haselhorst et al. 2009). When the target protein is saturated with selective pulses, the saturation is transferred to a bound ligand through intermolecular 1H-1H cross relaxation (Fig. 7.3a). Protons which are spatially close to the protein surface receive a higher degree of saturation compared with protons further away, and those on a non-binding ligand do not receive any saturation from the protein and are therefore not attenuated in the on-resonance spectrum. The subtraction of two spectra, one with protein resonance saturation (on-resonance) and the other without saturation of the protein resonance (off-resonance), results in a final STD-NMR spectrum showing only signals from bound ligands. For binding epitope analyses, the signal intensities of each peak are evaluated from an amplification factor (AF) = (I off – I on)/I off, where I on is the on-resonance signal intensity and I off is off-resonance signal intensity. Association constants can be estimated from a plot of AF against ligand concentration or initial growth rates of STD AF (Hanashima et al. 2010; Angulo et al. 2010). STD-NMR requires only a small amount of native protein, and no expensive protein labeling is necessary. STD-NMR spectroscopy can be utilized to detect binding of ligands with K D values in the 10−2–10−8 M range. Often screening for optimum STD-NMR conditions is required, such as probe temperature and optimal ligand-to-protein ratio (Fig. 7.3b). D2O solvent is preferred over H2O, to avoid direct and water-mediated saturation artifacts. High-affinity ligands typically reside longer within the protein binding site, undergo slow chemical exchange, and are thus not detectable by STD-NMR spectroscopy.
Fig. 7.3.

Saturation transfer difference NMR (STD-NMR) analysis for group epitope mapping
(a) Basic principle of saturation transfer difference NMR (STD-NMR). The ligand protons which receive saturation from the protein are highlighted in red. From the degree of ligand saturation, one can map the group epitope which is recognized by the protein
(b) STD-NMR spectra of (Neu5Ac)6 with an anti-polysialic acid antibody 12E3 at 10 °C (ii), 20 °C (iii), and 30 °C (iv) with the control 1H-NMR spectrum (off-resonance) at 10 °C (i). STD-NMR spectra were collected by irradiating iteratively at 7 ppm (on resonance)/ 40 ppm (off resonance). x; the signals from low-molecular-weight impurities, which are nearly null in STD-NMR spectra. This figure was adapted from a previous paper (Hanashima et al. 2013) with permission
(c) STD-NMR spectrum (lower) and reference 1H-NMR spectrum (upper) of the di-galactosylated biantennary glycan bearing bisecting GlcNAc and core fucose (top panel) in the presence of PHA-E (glycan: subunit molar ratio is 1:2). Normalized amplification factor (GlcNAc-2 signal is set to 100%) is shown in parenthesis. The signal indicated with an asterisk originates from PHA-E protein. This figure was adapted from a previous paper (Nagae et al. 2014c) with permission. Crystal structure of PHA-E in complex with bisected N-glycan (PDB code: 5AVA) is shown in right panel
1D STD-NMR analysis is often hampered by severe overlapping of non-anomeric signals. For instance, the ring proton signals from the two branches (α1-3 and α1-6 branches) of bisected biantennary N-glycan largely overlap in 1D STD-NMR analysis. In contrast, the methyl proton signals from GlcNAc and core fucose are sharp and well separated for certain glycans. Hence these signals can be used as a reliable probe for branch-specific epitope mapping as exemplified by an analysis of the interaction between biantennary bisected glycan and a legume lectin PHA-E (Nagae et al. 2014c). The methyl proton signals of GlcNAc from outer branches and inner chitobiose unit show almost comparable intensities, indicating that the entire glycan contributes equally to binding with PHA-E (Fig. 7.3c). Consistent with this result, the crystal structure of bisected glycan-PHA-E complex showed that most sugar residues of the glycan ligand are recognized by PHA-E (Fig. 7.3c, right panel). Another approach is the use of 2D 1H-13C STD-heteronuclear single quantum coherence ( HSQC) to avoid the severe signal overlap in 1D NMR spectra. 2D STD- spectra have been collected to investigate the interaction between oligosialic acid and anti-oligosialic acid antibodies, 12E3 and A2B5 (Hanashima et al. 2013). STD-NMR is also applicable to analyze multiple binding modes of a unique ligand in a single binding site (Angulo and Nieto 2011). The duplicate sugar-binding modes of C-type lectin, DC-SIGN (Angulo et al. 2008), and anti-HIV antibody, 2G12 (Enríquez-Navas et al. 2012), have been analyzed in this way.
Application of 13C-NMR Using Deuterium-Induced Isotope Shift
In solution NMR experiments, it is difficult to directly observe the hydroxyl protons of glycans because of their inherently fast exchange with water protons. Deuterium secondary isotope shifts on 13C-chemical shifts (deuterium-induced 13C isotope shifts, DIS) have been observed at the geminal 13C-signal of exchangeable hydroxyl protons (Pfeffer et al. 1979). In the presence of H2O and D2O, the line shape of the 13C-OH/D signal is highly dependent on the H/D exchange rate (Fig. 7.4a). In a very slow H/D exchange environment, a deuterium-induced 13C isotope shift (DIS) is observed in corresponding isotopomers, 13C-OH and 13C-OD, with a 0.09–0.15 ppm difference, whereas in the case of fast exchange, the equilibrium makes each isotopomer indistinguishable and gives a singlet signal at an averaged chemical shift.
Fig. 7.4.

Hydrogen exchange analysis using deuterium-induced isotope shift
(a) Deuterium-induced 13C isotope shifts for analyzing the proton exchange rate of sugar hydroxyl groups. k ex, exchange rates of hydroxyl protons with water. The chemical shift difference (isotope shift) is ~0.15 ppm, which is not dependent on the magnetic field
(b) 13C-NMR spectra of 40 mM LewisX tetrasaccharide with expanded secondary carbon area. LewisX at 5 °C (top), and LewisX with 1.0 M calcium chloride at 5 °C (bottom). The sample was dissolved in 10 mM sodium acetate buffer (pH 6.0) composed of H2O:D2O = 1:1
(c) 13C-NMR spectra of short β1–3-glucans DP7 (red) and laminarin (black) at 5, 10, 15, 20, and 25 °C. 13C-NMR signal focuses on the C4 position. All ligands were dissolved in the same buffer shown in (B). The figures were adapted from previous papers (Hanashima et al. 2011, 2014a) with modifications
This NMR technique has been applied in the weak carbohydrate-carbohydrate interaction of LewisX (Hanashima et al. 2011). LewisX, a trisaccharide of galactose, fucose and GlcNAc, is often found at the termini of glycolipids and glycoproteins and initiates cell-cell interactions by forming a LewisX-Ca2+ complex (Eggens et al. 1989). Calcium-dependent intermolecular interactions of the LewisX-Ca2+ complex are observed, via the proton exchange rates of each hydroxyl group in an equimolar H2O/D2O solution (Fig. 7.4b). The conformation of the glycan ligand can also be analyzed by this method, including that of β(1,3)-glucan dependent on the degree of polymerization (DP) (Hanashima et al. 2014a). With β-glucan, the exchange of 13C-OH/D is dramatically retarded as DP extends, suggesting changes in quaternary structure (Fig. 7.4c). Recently a new two-dimensional 1H-13C NMR method, named INTOXSY, was developed to estimate the H/D exchange rate constants of OH groups based on DIS (Battistel et al. 2017).
Quantitative Lectin-Glycan Interaction Analysis
Like other protein-ligand interactions, lectin-glycan interactions are characterized by several biophysical properties, such as the dissociation constant K d, thermodynamics parameters such as enthalpy ΔH and entropy ΔS, and kinetics (e.g., k on and k off rate constants). Many quantitative biophysical methods exist to analyze lectin-glycan interactions such as frontal affinity chromatography (FAC) analysis, isothermal titration calorimetry (ITC), and surface plasmon resonance (SPR). All these methods can provide K d; however, a suitable method needs to be selected for each purpose. Among them, ITC can be performed without labeling or immobilization of lectin or glycan. A lot of sample (typically 10 mmol for ligand and protein) is usually needed. In FAC analysis, immobilization of lectin is required, and the glycan must be labeled for detection. SPR also requires immobilization of lectin or glycan, but amount of sample is typically small. Combining complementary methods provides more insights than a single method.
Frontal Affinity Chromatography (FAC)
Frontal affinity chromatography (FAC) is a powerful method to assess lectin-glycan interactions as it is applicable to weak interactions (Tateno et al. 2007). In FAC analysis, glycans are applied to a lectin-immobilized column and the elution profile monitored. Dissociation constants for each glycan are calculated from the equation:
where V and V 0 are the elution volumes of the column and negative control, respectively. B t is the effective lectin content expressed in mole, and [A]0 is the initial glycan molar concentration. The amount of analyte needed depends on the affinity of analyte and ligand: higher affinities require smaller amounts.
An example of FAC analysis is that of the binding of various N-glycans to a mannose-binding Jacalin-related lectin Calsepa. The lectin was immobilized on the column, and a panel of pyridylaminated (PA) or p-nitrophenyl (pNP) glycans were successively passed through the column. In this way, Calsepa was found to specifically bind bisected N-glycans (Nagae et al. 2017a) (Fig. 7.5a). The specificity is explained by the crystal structure of bisected glycan-Calsepa complex (Fig. 7.5a, right). The ligand is in a flipped-back conformation, which bisected glycan prefers, and the unique ligand conformation enables the 2:1 sandwich binding mode.
Fig. 7.5.

Quantitative analyses of lectin-glycan interaction
(a) FAC analysis of a mannose-binding Jacalin-related lectin Calsepa toward N-glycans. Association constant (K A) of Calsepa lectin for each glycan is shown. Monosaccharide symbols follow the SNFG (symbol nomenclature for glycans) system (Varki et al. 2015). The FAC data is adapted from a paper (Nagae et al. 2017a) with permission. Crystal structure of Calsepa in complex with bisected glycan (corresponding to #104) is shown in right panel (PDB code: 5AV7)
(b) Isothermal titration calorimetry (ITC) analysis of scFv735 using polysialic acid (PolySia, DP 80–130) (left). Bar graph and table of thermodynamic parameters for DP4, DP5, DP6, and PolySia are shown in middle panel. Crystal structure of scFv735 in complex with octa- sialic acid (DP8) is shown in the right panel (PDB code: 3WBD)
(c) Surface plasmon resonance (SPR) analyses of the protein-protein interaction between p24β1 and p24δ1 GOLD domains. (i) Sensorgram showing the interaction between immobilized p24δ1 GOLD domain and analyte p24β1 GOLD domain. p24β1 GOLD domain aliquots (0, 5, 25, 50, 100, 200, and 400 μM) were injected for 60 s at a flow rate of 30 μl/min. Subtracted sensorgrams with blank lanes are shown. (ii) Sensorgram showing the interaction between immobilized p24β1 GOLD domain and analyte p24δ1 GOLD domain. The dilution series of p24δ1 GOLD domain are the same as those of p24β1 GOLD domain shown in (i). The SPR data was adapted from a paper (Nagae et al. 2016a) with permission
Isothermal Titration Calorimetry (ITC)
Isothermal titration calorimetry (ITC) is a technique that directly determines thermodynamic parameters of protein-ligand interactions (Dam et al. 2016). In a single experiment, ITC measures n (number of binding sites on the protein), ΔH ( enthalpy of binding), and K A ( association constant). From the K A value, the free energy of binding (ΔG) and the entropy of binding (ΔS) can be calculated from the following equation:
where ΔG, ΔH, and ΔS are the changes in free energy, enthalpy, and entropy of binding, respectively. T is the absolute temperature and R is the gas constant (8.314 J/mol/K).
An example is the ITC analysis of an anti-polysialic acid antibody 735. This antibody shows high affinity toward α2–8-linked polysialic acid as the degree of polymerization (DP) increases. To investigate the DP-dependent affinity enhancement mechanism, a series of oligo (DP4–6) and polysialic acids (DP 80–130) and single chain Fv originating from antibody 735 (scFv735) were evaluated by ITC (Nagae et al. 2013a) (Fig. 7.5b). As DP increases so does the affinity of scFv735. Thermodynamic parameters revealed that a large entropy loss upon interaction (TΔS = −72.7 kcal/mol for polysialic acid) is compensated for by a larger enthalpy gain (ΔH = −80.1 kcal/mol). Importantly, the crystal structure of scFv735-ligand complex indicates that antibody 735 can recognize internal residues of polysialic acids (Fig. 7.5b, right). Therefore, increasing DP may provide a rapid cycle of dissociation-association and thereby enhance the affinity (Nagae and Yamaguchi 2014).
Typical lectin-glycan interactions are exothermic, but occasionally an endothermic reaction is observed, e.g., interaction between heparan sulfate and heparin-binding hemagglutinin (Huang et al. 2017). Kinetic parameters can also be obtained from ITC experiments (Burnouf et al. 2012; Vander Meulen et al. 2016).
Surface Plasmon Resonance (SPR)
Surface plasmon resonance (SPR) is one of the most widely used analytical methods for detecting biomolecular interactions and kinetics. It monitors interactions between an immobilized ligand on a thin metal layer and a soluble-free analyte by detecting changes in the resonance angle due to an increase in concentration of analyte at the surface. SPR can monitor real-time interaction with biosensors by measuring the resonance units against time.
Sensorgrams can be utilized for the determination of kinetic parameters, association/dissociation constants (K A/K D), on/off rates (k on/k off), as well as thermodynamics parameters. There are two major methods to calculate kinetic parameters, first is by directly fitting the curve sensorgram to ideal binding models (more applicable to slow association/dissociation binding) and the second is steady-state analysis (for fast association/dissociation interactions). Maximum resonance values of a series of analyte concentrations (R eq) are plotted against analyte concentration. In simple 1:1 steady-state binding model, the plot is fitted to the following equation:
where C is concentration of analyte, R max is maximum binding response, and K D is the equilibrium dissociation constant. For example, the heterophilic interaction of p24 Golgi dynamics (GOLD) domains is shown between p24β1 and p24δ1. These GOLD domains interact with each other to form a hetero p24 family protein complex which is involved in the transport of glycosylphosphatidylinositol (GPI)-anchored proteins. The sensorgram shows fast association/dissociation binding and was analyzed by a 1:1 steady-state model (Nagae et al. 2016a) (Fig. 7.5c). To avoid immobilization artifacts, two experiments were performed: first p24β1 was immobilized and p24δ1 injected and then the vice versa experiment performed. The estimated K D values of the two runs are comparable (1.2 × 10−4 and 1.5 × 10−4 M). SPR is widely used in lectin-glycan interactions, either alone or combined with other methods such as the enzyme-linked immunosorbent assay (ELISA), ITC, and thermal shift assays (Wesener et al. 2015; Ribeiro et al. 2016; Machon et al. 2017). When the lectin is immobilized and glycan injected, the SPR detection limit needs to be confirmed in advance. To overcome the sensitivity problem, biotinylated carbohydrates are immobilized onto streptavidin or neutravidin-coated sensor chips. Diluted lectins are injected and affinities calculated .
3D Structural Studies on Lectin-Glycan Interactions
Biophysical data of lectin-glycan interaction is strengthened when combined with structural data obtained from X-ray crystallography and NMR spectroscopy. X-ray crystallography is the favored method to determine the 3D structures of protein-ligand complexes at atomic resolution. However, glycans are highly flexible in solution, and the electron density is sometimes missing even in the presence of lectin. In contrast, solution NMR analyses such as titration assays and transferred nuclear Overhauser effects (trNOE) can give a more realistic picture under physiological conditions. Titration assays provide affinities, while trNOEs give intramolecular distance constraints of the bound glycan. This chapter focuses on NMR analysis of lectin-glycan interaction.
NMR Titration Study
For any binding scheme , a ligand exchanges between free and bound states with rate constant k (expressed per second). In NMR spectra, resonance frequencies of bound and free ligand are different, with Δν representing the chemical shift difference (expressed in Hz). In a fast exchange regime, k >> Δν, the signal will appear in a population-weighted position between free and bound resonances. This allows one to monitor binding using the chemical shift change. In 1H-15N HSQC experiments, the dissociation constant K D can be obtained by plotting the weighted chemical shift change, Δδ, as a function of the carbohydrate/protein molar ratio. Δδ of each concentration is calculated with the following equation:
where Δδ H and Δδ N are the observed chemical shift changes (ppm) of 1H and 15N and α is a scaling factor (typically 0.1~0.2). Then the dissociation constants is estimated by fitting the curve:
where Δδ max is the maximum chemical shift change, [L]T the total ligand concentration, [P]T the total protein concentration, and K D the dissociation constant for a 1:1 binding equilibrium.
In a slow exchange regime, k < < Δν, two distinct resonances are observed, corresponding to the free and bound states. Increasing concentrations of carbohydrate increases the signal of the bound form and decreases that of the free. When the exchange rate is comparable to the frequency difference between the two states (intermediate exchange regime), k ~ Δν, extensive line broadening occurs, and sometimes the signals are beyond detection. Occasionally, it may be possible to escape this undesirable intermediate exchange situation by changing experimental conditions, aiming to either accelerate or slow the exchange.
A first example is the interaction of human soluble lectin ZG16p and phosphatidylinositol mannosides (PIMs) (Hanashima et al. 2015). PIM2 was titrated into a 15N-labeled ZG16p solution, and signal perturbations in the 1H-15N HSQC spectra were recorded (Fig. 7.6a). The binding is a fast exchange process, since each set of specific signals featured a gradual chemical shift change. The binding interface of PIM2 can be inferred from a map of the hot spots on the 3D structure of ZG16p, and the dissociation constant for PIM2 was 3.0 mM from a fit of the titration curve.
Fig. 7.6.


NMR titration studies and trNOE-based conformational analyses
(a) NMR titration study of the interaction between ZG16p and PIM2. (Left panel) 1H-15N HSQC spectra of uniformly 15N-labeled ZG16p in titration with PIM2 glycan (Black, 0 equiv.; red, 5 equiv.; and green, 20 equiv. of PIM2). Blue arrows indicate directions of the chemical shift changes. The figure was adapted from a paper (Hanashima et al. 2015) with permission.Fig. 7.6 (continued) (Right panel) Mapping surface residues in the PIM2 glycan interaction on the crystal structure of human ZG16p (PDB ID; 3APA). The signal from I149 (green) was broadened upon PIM2 binding
(b) NMR titration study of the interaction between a C-type lectin receptor mDCIR2 CRD and glycans. The pyridylaminated bisected and non-bisected N-glycans (glycans a and b) used in this study are shown in middle panel. A part of 1H-NMR spectra of 20 μM mDCIR2 CRD in the absence of glycan (top), in the presence of 40 μM glycan a (second), and 40–120 μM glycan b (third to the last). The figure was adapted and modified from a previous paper (Nagae et al. 2013b) with permission. Crystal structure of mDCIR2 CRD in complex with bisected glycan corresponding to glycan b is shown in right panel (PDB code: 3VYK)
(c) Basic principle of transferred NOE (trNOE). If the glycan ligand is bound to protein for a sufficiently short time (in fast exchange), one can observed the bound NOEs (reflecting the interproton distances in the bound state) by using the free ligand signals
(d) NOE build up curves of Man-3 H1 and Man-3 O-methyl proton signals in the presence (left) and absence (right) of Calsepa lectin. Man-3 H1 signal was selectively inverted using a 180° rectangular pulse with 40-ms duration. This figure was adapted from a paper (Nagae et al. 2016c) with permission
(e) 1D selective NOESY spectra of bisected glycan in the presence (upper) and absence (lower) of Calsepa. Strong intra-residue trNOE was observed from Man-3 H1 to Man-3 H2, and long-range Fig. 7.6 (continued) trNOEs were also observed from Man-3 H1 to GlcNAc-5′ (α1–6 branch) H1 and to Man-4’ H2 signals (Right panel). Proton-proton distances were indicated between Man-3 H1 and GlcNAc-5’ H1 and between Man-3 H1 and Man-4’ H2. Structures of back-fold conformations are derived from Calsepa complex (PDB code: 5AV7). Hydrogen atoms are generated with PyMOL. This figure was adapted and modified from a paper (Nagae et al. 2016c) with permission
(f) 2D 1H-1H NOESY spectrum of PIM1 glycan (4.6-fold excess) in the presence of wild-type ZG16p collected with a mixing time of 250 ms at 10 °C (upper panel). Key inter-residual correlation was provided between Man-H1 and Ino-H2 and intra-residual correlations (Man-H1 and Man-H2) as negative NOEs. The atomic distance of inter-residue Man-H1-Ino-H2 was determined as 2.2 Å from the relative intensity of the signal. No correlation was observed when using the inactive ZG16p mutant (D151N) (lower panel). This figure is reproduced from the paper (Hanashima et al. 2015) with permission
The second example is the interaction of a C-type lectin receptor mDCIR2 and bisected N-glycan (Nagae et al. 2013b). Bisected and non-bisected N-glycans (glycans a and b) were analyzed. Concentration-dependent spectral shifts in the 1H-NMR spectra were only observed in the presence of bisected glycan (Fig. 7.6b). The interaction involved slow exchange in terms of chemical shift, and the dissociation constant was estimated to be 3.0 × 10−5 M, a ligand concentration at which there is an equal amount of ligand-free and ligand-bound lectin. The specificity is well explained from the crystal structure of mDCIR2-bisected glycan complex, in which the bisected GlcNAc residue is directly interacting with the protein (Fig. 7.6b, right).
Transferred Nuclear Overhauser Effects (trNOE)
The nuclear Overhauser effect (NOE) describes the phenomenon of magnetization transfer from one spin to another. This process proceeds through cross relaxation originating from dipolar interaction between two spins that are spatially close (less than 5 Å apart). Roughly, the NOE is related to the distance between the two protons and shows a 1/r 6 dependence.
If the glycan ligand binds to lectin with slow exchange, both intermolecular lectin-glycan NOEs and intramolecular NOEs of the glycan are important in elucidating the bound conformation of the ligand. However, in most lectin-glycan interactions, the exchange is fast because the affinity is weak. In such cases, transferred- NOESY (tr-NOESY) is the suitable measurement for conformational analysis of bound glycan ligand. trNOE analysis is generally valid for ligands that bind in the μM-mM range (Fig. 7.6c).
TrNOE experiments were applied to the conformational analysis of bisected N-glycan bound to a lectin, Calsepa (Nagae et al. 2016c). The NOE build-up curve shows linearity up to 200 ms for the glycan-Calsepa mixture and to 500 ms for glycan alone (Fig. 7.6d). The mixing time was then chosen as 200 ms for the trNOE experiments. In the presence of Calsepa lectin, long-range inter-residual trNOEs were observed between core β-mannose (Man-3) and GlcNAc-5′ on the outer branch (Fig. 7.6e). The presence of this long-range trNOE indicates that the glycan assumes a compact back-fold conformation when bound to Calsepa lectin. In a second example, the interaction between ZG16p and phosphatidylinositol mannoside glycans (PIM1 and PIM2) was investigated by trNOE (Hanashima et al. 2015). In the presence of ZG16p, several inter- and intra-residual NOE correlations were observed in PIM1 (Fig. 7.6f). There were none with the inactive ZG16p mutant D151N. From these correlations, the conformation of PIM1 in ZG16p-bound state was elucidated.
The structural information obtained from trNOE is usually limited by low proton density and by the inherently flexible nature of carbohydrate chains. Thus, obtained distances are typically further evaluated by molecular dynamics simulations and/or X-ray crystallographic analysis.
Other NMR Techniques
Residual dipolar coupling (RDC) measured in liquid crystalline alignment medium offer a viable alternative to traditional NOE-based approaches for structural analysis. RDC provides a way to constrain the relative orientation of two molecules in complex with each other by aligning their independently determined order tensors (Jain 2009). Proton-proton and proton-carbon RDC of heparin tetrasaccharide has been measured (Jin et al. 2009), which, when combined with molecular dynamics simulations, clarified ring conformer and global shape of the glycan.
Paramagnetic probes attached to the reducing ends of oligosaccharides cause paramagnetic relaxation enhancement (PRE) and/or pseudocontact shift (PCS) which can resolve the peak overlap problem (Kato and Yamaguchi 2015). These spatial perturbations can be sources of long-range atomic distance information (~10 Å), which complements the local conformational information derived from J-couplings and NOEs (~5 Å).
Molecular Dynamics Simulation Studies of Lectin-Glycan Interactions
Theoretical approaches such as docking models and molecular dynamics (MD) simulation complement experimental information and have great possibilities for visualizing the highly mobile glycans. Docking simulations of lectin-glycan complexes provide a theoretical foundation and reduce ambiguities in experimental results. We validated the accuracy of docking simulation by applying it to a crystal structure of POMK-ligand complex, in which the electron density of ligand was partially observed (Fig. 7.7a). The position of the trisaccharide ligand in the docking model coincides well with the fragmented electron density in the crystal structure, underscoring the usefulness of docking and MD simulations in determining bound structures (Nagae et al. 2017b).
Fig. 7.7.

Molecular dynamics simulations of protein-glycan complexes
(a) Top five docking poses of the core M3 trisaccharide ( GalNAcβ1–3GlcNAcβ1-4Man, shown in yellow) in POMK binding site (i-v) by AutoDock Vina. The mannose residue assigned in the crystal structure (PDB code: 5GZ9) is overlaid in cyan. The table contains docking results for each model (i)-(v). Calculated binding affinity (in kcal/mol) together with root mean square deviation (r.m.s.d. in Å) from top scoring docking pose (i) is shown for all five models. This figure is adapted from the paper (Nagae et al. 2017b) with permission
(b) Three major conformations (Conformers 1, 2, and 3) of the α1–6 branch of glycan A bound to Orysata lectin in MD simulations. The simulation was performed using the crystal structure of Orysata in complex with glycan A (PDB code: 5XFH). This figure is adapted from the paper (Nagae et al. 2017a) with permission
(c) Bar graph and table of calculated average enthalpy (∆H), entropy (T∆S), and total binding free energy (∆G) for the biantennary glycans A–D. Experimental dissociation constant (K D = 1/K A) determined by FAC analysis is also indicated. N.D.: K D was not determined due to weak or no binding
MD simulation is also useful for validation of possible binding modes with the estimation of binding free energy. Orysata lectin, one of the mannose-binding Jacalin-related lectins, shows a preference for complex-type glycans with a α1-6 branch extension rather than a α1-3 branch. It was not easy to understand the structural basis for the preference for the α1-6 branch from the “snap-shot” crystal structure of Orysata in complex with biantennary glycan. MD simulations yield the dynamic behavior of the glycan attached to Orysata lectin (Fig. 7.7b). Several docking models of Orysata in complex with a series of N-glycans were constructed, and the calculated binding free energies suggest that multiple binding modes exist in the solution, and the binding preference seems somehow to originate from an average of multiple binding modes that favors α1-6 branch binding (Fig. 7.7c). MD simulations are especially useful for analyzing such multiple ligand-binding modes .
Future Perspective: Application of Cryo-electron Microscopy Analysis to Investigate Lectin-Glycan Interaction
Recent improvement in cryo- electron microscopy (cryo-EM) is a great breakthrough for structural biology (Cheng et al. 2015). EM analysis has an inherent advantage for investigating the global conformations of macromolecules, rather than atomic details of local interfaces. The structural changes of glycans on glycoproteins affect the physiological functions of carrier glycoproteins. This has been clearly exemplified by EM analysis. For example, EM has shown that mutation or elimination of N-glycans affects the conformational equilibrium of α5β1 integrin (Li et al. 2017). Likewise, the inter-domain angle of LDL-receptor-related protein 6 (LRP6) is regulated by the evolutionarily conserved N-glycan (Matoba et al. 2017).
In X-ray crystallography, the trimming of surface glycans on glycoproteins by glycosidase is usually necessary to produce diffraction quality crystals (Chang et al. 2007). Mutations at N-glycosylation sites can also produce highly uniform proteins (Nagae et al. 2017b). However, such treatments prevent visualization of the atomic structure of glycans, although some native glycoproteins provide good glycan electron density due to extensive intramolecular interactions between glycan and carrier glycoproteins (Nagae and Yamaguchi 2012). In contrast, there is no need to eliminate the surface glycans of glycoproteins in single particle analysis of cryo-EM. Thus, cryo-EM has great potential to provide the atomic structure of native glycans. Actually, cryo-EM structures of glycoproteins, such as γ-secretase and human coronavirus, often include the coordinates of attached glycans (Bai et al. 2015; Walls et al. 2016) (Fig. 7.8a and b). Even tetra-antennary N-glycan structures are visible in the EM map of the HIV-1 envelope glycoprotein trimer (Env) (Lee et al. 2016) (Fig. 7.8c).
Fig. 7.8.
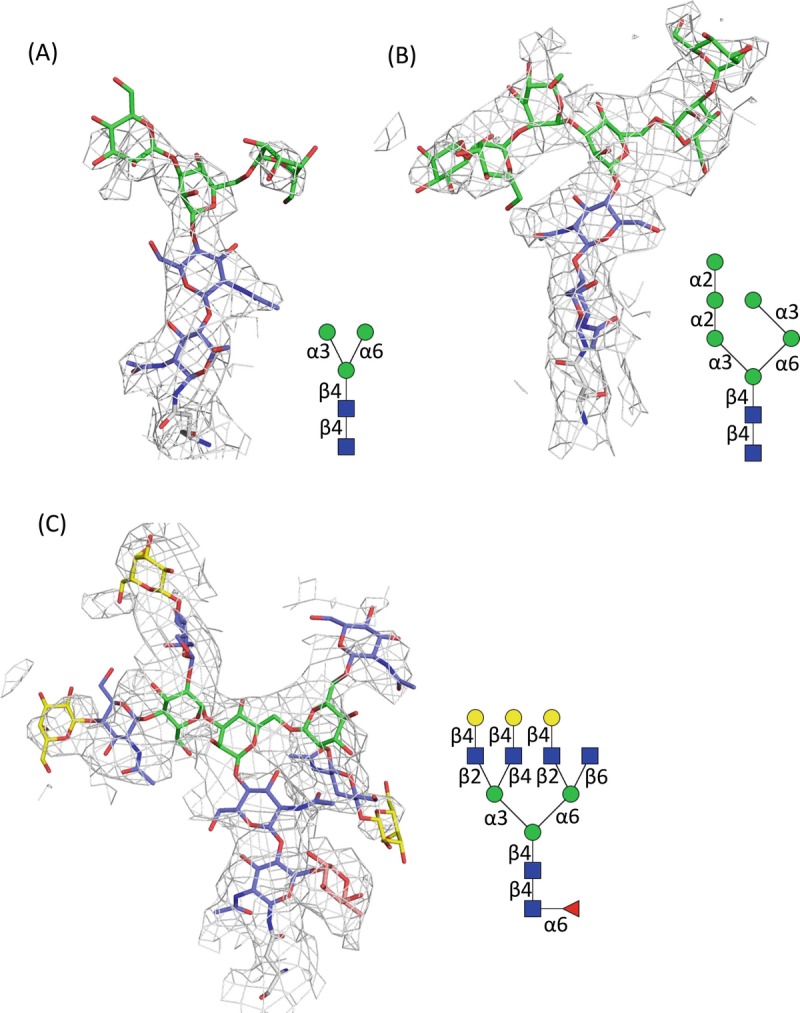
Electron microscopic images of carbohydrates covalently attached onto proteins
(a) N-glycan core attached onto N55 of human nicastrin, a component of γ-secretase complex ((Bai et al. 2015), PDB code: 5A63, EMDB: 3061). The density map is depicted at 5.0 σ level cutoff. Schematic representation of the observed glycan is shown in right panel
(b) High-mannose-type N-glycan attached onto N426 of human coronavirus NL63 (HCoV-NL63) ((Walls et al. 2016), PDB code, 5SZS; EMDB, 8331). The density map is contoured at 7.0 σ level
(c) Tetra-antennary glycan attached onto N637 of HIV-1 Env trimer ((Lee et al. 2016), PDB code, 5FUU; EMDB, 3308). The density map is depicted in cyan mesh contoured at 5.5 σ level
Due to limitation of molecular weight, it is normally difficult to directly visualize lectin-glycan interactions using single particle analysis by cryo-EM. However, population analysis of many particles is the great advantage of EM analysis. Although the flexibility of the glycan part remains a serious problem for visualization (averaging problem), we can expect that cryo-EM will become the method of choice for analyzing the structure of native glycan on glycoproteins. The quaternary structural analyses of elaborate complex systems have recently been reported, such as human MHC-I peptide-loading complex (PLC), which includes TAP1, TAP2, tapasin, MHC-I heavy chain, β2m, calreticulin, and ERp57 in the complex (Blees et al. 2017). It is likely that multicomponent glycoprotein complexes and lectin receptor-glycoprotein complexes will be analyzed by cryo-EM at atomic resolution, coupled with other biophysical methods in the very near future.
Acknowledgment
Most of the work described here was done with many collaborators as well as our lab members, all of whom we wish to acknowledge greatly. Optimization of recombinant protein expression was conducted by Drs. Ganesh P. Subedi and Hari P. Dulal, glycan microarray analysis was conducted by Dr. Yan Liu and Prof. Ten Feizi (Imperial College London), NMR study by Dr. Shinya Hanashima, FAC analysis by Dr. Hiroaki Tateno (AIST, Japan), ITC analysis by Drs. Masato Kikkawa and Seiji Yamamoto (GE Healthcare, Japan), and MD simulations by Dr. Sushil K. Mishra. SPR analysis was performed at One-Stop Facility Center, the University of Tokyo, by courtesy of Prof. Nobuaki Higashi. We also thank Noriko Tanaka for secretarial assistance.
Abbreviations
- DIS
Deuterium-induced isotope shift
- EM
Electron microscopy
- FAC
Frontal affinity chromatography
- GlcNAc
N-Acetylglucosamine
- ITC
Isothermal titration calorimetry
- MD
Molecular dynamics
- NMR
Nuclear magnetic resonance
- SPR
Surface plasmon resonance
- STD-NMR
Saturation transfer difference NMR
- trNOE
Transferred nuclear Overhauser effect
Contributor Information
Yoshiki Yamaguchi, Email: yyoshiki@riken.jp.
Koichi Kato, Email: kkato@phar.nagoya-cu.ac.jp.
Yoshiki Yamaguchi, Email: yyoshiki@riken.jp.
References
- Angulo J, Nieto PM. STD-NMR: application to transient interactions between biomolecules-a quantitative approach. Eur Biophys J. 2011;40(12):1357–1369. doi: 10.1007/s00249-011-0749-5. [DOI] [PubMed] [Google Scholar]
- Angulo J, Díaz I, Reina JJ, Tabarani G, Fieschi F, Rojo J, Nieto PM. Saturation transfer difference (STD) NMR spectroscopy characterization of dual binding mode of a mannose disaccharide to DC-SIGN. Chembiochem. 2008;9(14):2225–2227. doi: 10.1002/cbic.200800361. [DOI] [PubMed] [Google Scholar]
- Angulo J, Enríquez-Navas PM, Nieto PM. Ligand-receptor binding affinities from saturation transfer difference (STD) NMR spectroscopy: the binding isotherm of STD initial growth rates. Chemistry (Weinheim an der Bergstrasse, Germany) 2010;16(26):7803–7812. doi: 10.1002/chem.200903528. [DOI] [PubMed] [Google Scholar]
- Bai XC, Yan C, Yang G, Lu P, Ma D, Sun L, Zhou R, Scheres SHW, Shi Y. An atomic structure of human γ-secretase. Nature. 2015;525(7568):212–217. doi: 10.1038/nature14892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battistel MD, Azurmendi HF, Freedberg DI. Glycan OH exchange rate determination in aqueous solution: Seeking evidence for transient hydrogen bonds. J Phys Chem B. 2017;121(4):683–695. doi: 10.1021/acs.jpcb.6b10594. [DOI] [PubMed] [Google Scholar]
- Blees A, Januliene D, Hofmann T, Koller N, Schmidt C, Trowitzsch S, Moeller A, Tampe R. Structure of the human MHC-I peptide-loading complex. Nature. 2017;551(7681):525–528. doi: 10.1038/nature24627. [DOI] [PubMed] [Google Scholar]
- Burnouf D, Ennifar E, Guedich S, Puffer B, Hoffmann G, Bec G, Disdier F, Baltzinger M, Dumas P. kinITC: a new method for obtaining joint thermodynamic and kinetic data by isothermal titration calorimetry. J Am Chem Soc. 2012;134(1):559–565. doi: 10.1021/ja209057d. [DOI] [PubMed] [Google Scholar]
- Chang VT, Crispin M, Aricescu AR, Harvey DJ, Nettleship JE, Fennelly JA, Yu C, Boles KS, Evans EJ, Stuart DI, Dwek RA, Jones EY, Owens RJ, Davis SJ. Glycoprotein structural genomics: solving the glycosylation problem. Structure. 2007;15(3):267–273. doi: 10.1016/j.str.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Grigorieff N, Penczek PA, Walz T. A primer to single-particle cryo-electron microscopy. Cell. 2015;161(3):438–449. doi: 10.1016/j.cell.2015.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings RD, Pierce JM. The challenge and promise of glycomics. Chem Biol. 2014;21(1):1–15. doi: 10.1016/j.chembiol.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dam TK, Talaga ML, Fan N, Brewer CF. Measuring Multivalent Binding Interactions by Isothermal Titration Calorimetry. Methods Enzymol. 2016;567:71–95. doi: 10.1016/bs.mie.2015.08.013. [DOI] [PubMed] [Google Scholar]
- Dulal HP, Nagae M, Ikeda A, Morita-Matsumoto K, Adachi Y, Ohno N, Yamaguchi Y. Enhancement of solubility and yield of a β-glucan receptor Dectin-1 C-type lectin-like domain in Escherichia coli with a solubility-enhancement tag. Protein Expr Purif. 2016;123:97–104. doi: 10.1016/j.pep.2016.04.002. [DOI] [PubMed] [Google Scholar]
- Eggens I, Fenderson B, Toyokuni T, Dean B, Stroud M, Hakomori S. Specific interaction between Lex and Lex determinants. A possible basis for cell recognition in preimplantation embryos and in embryonal carcinoma cells. J Biol Chem. 1989;264(16):9476–9484. [PubMed] [Google Scholar]
- Enríquez-Navas PM, Chiodo F, Marradi M, Angulo J, Penadés S. STD NMR study of the interactions between antibody 2G12 and synthetic oligomannosides that mimic selected branches of gp120 glycans. Chembiochem. 2012;13(9):1357–1365. doi: 10.1002/cbic.201200119. [DOI] [PubMed] [Google Scholar]
- Feizi T, Chai W. Oligosaccharide microarrays to decipher the glyco code. Nat Rev Mol Cell Biol. 2004;5(7):582–588. doi: 10.1038/nrm1428. [DOI] [PubMed] [Google Scholar]
- Geissner A, Seeberger PH. Glycan arrays: From basic biochemical research to bioanalytical and biomedical applications. Annu Rev Anal Chem (Palo Alto, Calif) 2016;9(1):223–247. doi: 10.1146/annurev-anchem-071015-041641. [DOI] [PubMed] [Google Scholar]
- Hanashima S, Sato K, Naito Y, Takematsu H, Kozutsumi Y, Ito Y, Yamaguchi Y. Synthesis and binding analysis of unique AG2 pentasaccharide to human Siglec-2 using NMR techniques. Bioorg Med Chem. 2010;18(11):3720–3725. doi: 10.1016/j.bmc.2010.03.062. [DOI] [PubMed] [Google Scholar]
- Hanashima S, Kato K, Yamaguchi Y. 13C-NMR quantification of proton exchange at LewisX hydroxyl groups in water. Chem Commun (Camb) 2011;47(38):10800–10802. doi: 10.1039/c1cc13310a. [DOI] [PubMed] [Google Scholar]
- Hanashima S, Sato C, Tanaka H, Takahashi T, Kitajima K, Yamaguchi Y. NMR study into the mechanism of recognition of the degree of polymerization by oligo/polysialic acid antibodies. Bioorg Med Chem. 2013;21(19):6069–6076. doi: 10.1016/j.bmc.2013.07.023. [DOI] [PubMed] [Google Scholar]
- Hanashima S, Ikeda A, Tanaka H, Adachi Y, Ohno N, Takahashi T, Yamaguchi Y. NMR study of short β(1-3)-glucans provides insights into the structure and interaction with Dectin-1. Glycoconj J. 2014;31(3):199–207. doi: 10.1007/s10719-013-9510-x. [DOI] [PubMed] [Google Scholar]
- Hanashima S, Korekane H, Taniguchi N, Yamaguchi Y. Synthesis of N-glycan units for assessment of substrate structural requirements of N-acetylglucosaminyltransferase III. Bioorg Med Chem Lett. 2014;24(18):4533–4537. doi: 10.1016/j.bmcl.2014.07.074. [DOI] [PubMed] [Google Scholar]
- Hanashima S, Götze S, Liu Y, Ikeda A, Kojima-Aikawa K, Taniguchi N, Varon Silva D, Feizi T, Seeberger PH, Yamaguchi Y. Defining the interaction of human soluble lectin ZG16p and mycobacterial phosphatidylinositol mannosides. Chembiochem. 2015;16(10):1502–1511. doi: 10.1002/cbic.201500103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haselhorst T, Lamerz AC, Itzstein M. Saturation transfer difference NMR spectroscopy as a technique to investigate protein-carbohydrate interactions in solution. Methods Mol Biol. 2009;534:375–386. doi: 10.1007/978-1-59745-022-5_26. [DOI] [PubMed] [Google Scholar]
- Horan N, Yan L, Isobe H, Whitesides GM, Kahne D. Nonstatistical binding of a protein to clustered carbohydrates. Proc Natl Acad Sci U S A. 1999;96(21):11782–11786. doi: 10.1073/pnas.96.21.11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TY, Irene D, Zulueta MM, Tai TJ, Lain SH, Cheng CP, Tsai PX, Lin SY, Chen ZG, Ku CC, Hsiao CD, Chyan CL, Hung SC. Structure of the complex between a heparan sulfate octasaccharide and mycobacterial heparin-binding hemagglutinin. Angew Chem Int Ed Engl. 2017;56(15):4192–4196. doi: 10.1002/anie.201612518. [DOI] [PubMed] [Google Scholar]
- Hushegyi A, Tkac J. Are glycan biosensors an alternative to glycan microarrays? Anal Methods. 2014;6(17):6610–6620. doi: 10.1039/C4AY00692E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun JY, Pai J, Shin I. The glycan microarray story from construction to applications. Acc Chem Res. 2017;50(4):1069–1078. doi: 10.1021/acs.accounts.7b00043. [DOI] [PubMed] [Google Scholar]
- Jain NU. Use of residual dipolar couplings in structural analysis of protein-ligand complexes by solution NMR spectroscopy. Methods Mol Biol. 2009;544:231–252. doi: 10.1007/978-1-59745-483-4_15. [DOI] [PubMed] [Google Scholar]
- Jin L, Hricovíni M, Deakin JA, Lyon M, Uhrín D. Residual dipolar coupling investigation of a heparin tetrasaccharide confirms the limited effect of flexibility of the iduronic acid on the molecular shape of heparin. Glycobiology. 2009;19(11):1185–1196. doi: 10.1093/glycob/cwp105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanagawa M, Liu Y, Hanashima S, Ikeda A, Chai W, Nakano Y, Kojima-Aikawa K, Feizi T, Yamaguchi Y. Structural basis for multiple sugar recognition of Jacalin-related human ZG16p lectin. J Biol Chem. 2014;289(24):16954–16965. doi: 10.1074/jbc.M113.539114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K, Yamaguchi T. Paramagnetic NMR probes for characterization of the dynamic conformations and interactions of oligosaccharides. Glycoconj J. 2015;32(7):505–513. doi: 10.1007/s10719-015-9599-1. [DOI] [PubMed] [Google Scholar]
- Kato Y, Kaneko MK, Kunita A, Ito H, Kameyama A, Ogasawara S, Matsuura N, Hasegawa Y, Suzuki-Inoue K, Inoue O, Ozaki Y, Narimatsu H. Molecular analysis of the pathophysiological binding of the platelet aggregation-inducing factor podoplanin to the C-type lectin-like receptor CLEC-2. Cancer Sci. 2008;99(1):54–61. doi: 10.1111/j.1349-7006.2007.00634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Ozorowski G, Ward AB. Cryo-EM structure of a native, fully glycosylated, cleaved HIV-1 envelope trimer. Science. 2016;351(6277):1043–1048. doi: 10.1126/science.aad2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Su Y, Xia W, Qin Y, Humphries MJ, Vestweber D, Cabañas C, Lu C, Springer TA. Conformational equilibria and intrinsic affinities define integrin activation. EMBO J. 2017;36(5):629–645. doi: 10.15252/embj.201695803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang PH, Wang SK, Wong CH. Quantitative analysis of carbohydrate-protein interactions using glycan microarrays: determination of surface and solution dissociation constants. J Am Chem Soc. 2007;129(36):11177–11184. doi: 10.1021/ja072931h. [DOI] [PubMed] [Google Scholar]
- Liang CH, Wang SK, Lin CW, Wang CC, Wong CH, Wu CY. Effects of neighboring glycans on antibody-carbohydrate interaction. Angew Chem Int Ed Engl. 2011;50(7):1608–1612. doi: 10.1002/anie.201003482. [DOI] [PubMed] [Google Scholar]
- Machon O, Baldini SF, Ribeiro JP, Steenackers A, Varrot A, Lefebvre T, Imberty A. Recombinant fungal lectin as a new tool to investigate O-GlcNAcylation processes. Glycobiology. 2017;27(2):123–128. doi: 10.1093/glycob/cww105. [DOI] [PubMed] [Google Scholar]
- Matoba K, Mihara E, Tamura-Kawakami K, Miyazaki N, Maeda S, Hirai H, Thompson S, Iwasaki K, Takagi J. Conformational freedom of the LRP6 ectodomain is regulated by N-glycosylation and the binding of the Wnt antagonist Dkk1. Cell Rep. 2017;18(1):32–40. doi: 10.1016/j.celrep.2016.12.017. [DOI] [PubMed] [Google Scholar]
- Mayer M, Meyer B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J Am Chem Soc. 2001;123(25):6108–6117. doi: 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]
- Meyer B, Peters T. NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew Chem Int Ed Engl. 2003;42(8):864–890. doi: 10.1002/anie.200390233. [DOI] [PubMed] [Google Scholar]
- Nagae M, Yamaguchi Y. Function and 3D structure of the N-glycans on glycoproteins. Int J Mol Sci. 2012;13(7):8398–8429. doi: 10.3390/ijms13078398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagae M, Yamaguchi Y. Three-dimensional structural aspects of protein-polysaccharide interactions. Int J Mol Sci. 2014;15(3):3768–3783. doi: 10.3390/ijms15033768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagae M, Yamaguchi Y. Sugar recognition and protein-protein interaction of mammalian lectins conferring diverse functions. Curr Opin Struct Biol. 2015;34:108–115. doi: 10.1016/j.sbi.2015.08.005. [DOI] [PubMed] [Google Scholar]
- Nagae M, Ikeda A, Hane M, Hanashima S, Kitajima K, Sato C, Yamaguchi Y. Crystal structure of anti-polysialic acid antibody single chain Fv fragment complexed with octasialic acid: insight into the binding preference for polysialic acid. J Biol Chem. 2013;288(47):33784–33796. doi: 10.1074/jbc.M113.496224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagae M, Yamanaka K, Hanashima S, Ikeda A, Morita-Matsumoto K, Satoh T, Matsumoto N, Yamamoto K, Yamaguchi Y. Recognition of bisecting N-acetylglucosamine: structural basis for asymmetric interaction with the mouse lectin dendritic cell inhibitory receptor 2. J Biol Chem. 2013;288(47):33598–33610. doi: 10.1074/jbc.M113.513572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagae M, Ikeda A, Kitago Y, Matsumoto N, Yamamoto K, Yamaguchi Y. Crystal structures of carbohydrate recognition domain of blood dendritic cell antigen-2 (BDCA2) reveal a common domain-swapped dimer. Proteins. 2014;82(7):1512–1518. doi: 10.1002/prot.24504. [DOI] [PubMed] [Google Scholar]
- Nagae M, Morita-Matsumoto K, Kato M, Kaneko MK, Kato Y, Yamaguchi Y. A platform of C-type lectin-like receptor CLEC-2 for binding O-glycosylated podoplanin and nonglycosylated rhodocytin. Structure. 2014;22(12):1711–1721. doi: 10.1016/j.str.2014.09.009. [DOI] [PubMed] [Google Scholar]
- Nagae M, Soga K, Morita-Matsumoto K, Hanashima S, Ikeda A, Yamamoto K, Yamaguchi Y. Phytohemagglutinin from Phaseolus vulgaris (PHA-E) displays a novel glycan recognition mode using a common legume lectin fold. Glycobiology. 2014;24(4):368–378. doi: 10.1093/glycob/cwu004. [DOI] [PubMed] [Google Scholar]
- Nagae M, Hirata T, Morita-Matsumoto K, Theiler R, Fujita M, Kinoshita T, Yamaguchi Y. 3D structure and interaction of p24β and p24δ Golgi dynamics domains: Implication for p24 complex formation and cargo transport. J Mol Biol. 2016;428(20):4087–4099. doi: 10.1016/j.jmb.2016.08.023. [DOI] [PubMed] [Google Scholar]
- Nagae M, Ikeda A, Hanashima S, Kojima T, Matsumoto N, Yamamoto K, Yamaguchi Y. Crystal structure of human dendritic cell inhibitory receptor C-type lectin domain reveals the binding mode with N-glycan. FEBS Lett. 2016;590(8):1280–1288. doi: 10.1002/1873-3468.12162. [DOI] [PubMed] [Google Scholar]
- Nagae M, Kanagawa M, Morita-Matsumoto K, Hanashima S, Kizuka Y, Taniguchi N, Yamaguchi Y. Atomic visualization of a flipped-back conformation of bisected glycans bound to specific lectins. Sci Rep. 2016;6:22973. doi: 10.1038/srep22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagae M, Mishra SK, Hanashima S, Tateno H, Yamaguchi Y. Distinct roles for each N-glycan branch interacting with mannose-binding type Jacalin-related lectins Orysata and Calsepa. Glycobiology. 2017;27(12):1120–1133. doi: 10.1093/glycob/cwx081. [DOI] [PubMed] [Google Scholar]
- Nagae M, Mishra SK, Neyazaki M, Oi R, Ikeda A, Matsugaki N, Akashi S, Manya H, Mizuno M, Yagi H, Kato K, Senda T, Endo T, Nogi T, Yamaguchi Y. 3D structural analysis of protein O-mannosyl kinase, POMK, a causative gene product of dystroglycanopathy. Genes Cells. 2017;22(4):348–359. doi: 10.1111/gtc.12480. [DOI] [PubMed] [Google Scholar]
- Nimrichter L, Gargir A, Gortler M, Altstock RT, Shtevi A, Weisshaus O, Fire E, Dotan N, Schnaar RL. Intact cell adhesion to glycan microarrays. Glycobiology. 2004;14(2):197–203. doi: 10.1093/glycob/cwh022. [DOI] [PubMed] [Google Scholar]
- Pai J, Hyun JY, Jeong J, Loh S, Cho EH, Kang YS, Shin I. Carbohydrate microarrays for screening functional glycans. Chem Sci. 2016;7(3):2084–2093. doi: 10.1039/c5sc03789a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma AS, Feizi T, Childs RA, Chai W, Liu Y. The neoglycolipid (NGL)-based oligosaccharide microarray system poised to decipher the meta-glycome. Curr Opin Chem Biol. 2014;18:87–94. doi: 10.1016/j.cbpa.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer PE, Valentine KM, Parrish FW. Deuterium-induced differential isotope shift 13C NMR.1. Resonance reassignments of mono- and disaccharides. J Am Chem Soc. 1979;101(5):1265–1274. doi: 10.1021/ja00499a036. [DOI] [Google Scholar]
- Ribeiro JP, Pau W, Pifferi C, Renaudet O, Varrot A, Mahal LK, Imberty A. Characterization of a high-affinity sialic acid-specific CBM40 from Clostridium perfringens and engineering of a divalent form. Biochem J. 2016;473(14):2109–2118. doi: 10.1042/BCJ20160340. [DOI] [PubMed] [Google Scholar]
- Song X, Yu H, Chen X, Lasanajak Y, Tappert MM, Air GM, Tiwari VK, Cao H, Chokhawala HA, Zheng H, Cummings RD, Smith DF. A sialylated glycan microarray reveals novel interactions of modified sialic acids with proteins and viruses. J Biol Chem. 2011;286(36):31610–31622. doi: 10.1074/jbc.M111.274217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subedi GP, Satoh T, Hanashima S, Ikeda A, Nakada H, Sato R, Mizuno M, Yuasa N, Fujita-Yamaguchi Y, Yamaguchi Y. Overproduction of anti-Tn antibody MLS128 single-chain Fv fragment in Escherichia coli cytoplasm using a novel pCold-PDI vector. Protein Expr Purif. 2012;82(1):197–204. doi: 10.1016/j.pep.2011.12.010. [DOI] [PubMed] [Google Scholar]
- Tateno H, Nakamura-Tsuruta S, Hirabayashi J. Frontal affinity chromatography: sugar-protein interactions. Nat Protoc. 2007;2(10):2529–2537. doi: 10.1038/nprot.2007.357. [DOI] [PubMed] [Google Scholar]
- Vander Meulen KA, Horowitz S, Trievel RC, Butcher SE. Measuring the kinetics of molecular association by isothermal titration calorimetry. Methods Enzymol. 2016;567:181–213. doi: 10.1016/bs.mie.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A, Cummings RD, Aebi M, Packer NH, Seeberger PH, Esko JD, Stanley P, Hart G, Darvill A, Kinoshita T, Prestegard JJ, Schnaar RL, Freeze HH, Marth JD, Bertozzi CR, Etzler ME, Frank M, Vliegenthart JF, Lütteke T, Perez S, Bolton E, Rudd P, Paulson J, Kanehisa M, Toukach P, Aoki-Kinoshita KF, Dell A, Narimatsu H, York W, Taniguchi N, Kornfeld S. Symbol nomenclature for graphical representations of glycans. Glycobiology. 2015;25(12):1323–1324. doi: 10.1093/glycob/cwv091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedadi M, Niesen FH, Allali-Hassani A, Fedorov OY, Finerty PJ, Jr, Wasney GA, Yeung R, Arrowsmith C, Ball LJ, Berglund H, Hui R, Marsden BD, Nordlund P, Sundstrom M, Weigelt J, Edwards AM. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc Natl Acad Sci U S A. 2006;103(43):15835–15840. doi: 10.1073/pnas.0605224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls AC, Tortorici MA, Frenz B, Snijder J, Li W, Rey FA, DiMaio F, Bosch BJ, Veesler D. Glycan shield and epitope masking of a coronavirus spike protein observed by cryo-electron microscopy. Nat Struct Mol Biol. 2016;23(10):899–905. doi: 10.1038/nsmb.3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesener DA, Wangkanont K, McBride R, Song X, Kraft MB, Hodges HL, Zarling LC, Splain RA, Smith DF, Cummings RD, Paulson JC, Forest KT, Kiessling LL. Recognition of microbial glycans by human intelectin-1. Nat Struct Mol Biol. 2015;22(8):603–610. doi: 10.1038/nsmb.3053. [DOI] [PMC free article] [PubMed] [Google Scholar]