

Abstract
The robustness of the circadian clock deteriorates with aging. Two new studies show that aging reprograms the circadian transcriptome in a cell-type-dependent manner and that such rewiring can be reversed by caloric restriction.
The subject of calorie restriction (CR) is of wide interest because it is the most robust intervention to extend mammalian lifespan. More impressively, it reverses aging-associated physiological decline and ameliorates a wide spectrum of diseases1,2. It was thought that this dietary regimen elicits its health benefits through a passive effect of reduced metabolic rate. However, recent advances in aging research favor the view that the organismal effects of mammalian CR are actively regulated processes3,4. Two recent studies by Sato et al.5 and Solanas et al.6 reveal that aging reprograms the circadian transcriptome in a cell-type-dependent manner and that such rewiring can be reversed by the administration of the CR diet. These findings provide high-resolution molecular ‘fingerprints’ of the circadian response to physiological aging and CR and add new evidence for the emerging concept that aging-associated conditions can be reversed7–9.
A large number of physiological events follow circadian rhythms. It has been speculated that the robustness of circadian rhythms deteriorates with age, accounting for much of aging-associated physiological decline10–12. Taking a systems biology approach, Sato et al. and Solanas et al. profiled the circadian transcriptome of the liver, epidermal and skeletal muscle stem cells of young and aged mice fed either ad libitum or a CR diet5,6. Strikingly, a large number of oscillatory transcripts (1,400–2,600) in young mice lost their rhythmic expression in old mice, whereas numerous de novo oscillating genes (~1,600 in all three cell types) were observed exclusively in old mice. These findings provide compelling molecular evidence that aging is associated with a significant reprogramming of the circadian clock and that this phenomenon is conserved across tissues and cell types.
An interesting finding came from direct comparison of the age-specific oscillatory transcripts from the three cell types. Very few oscillatory transcripts were consistently altered with age in all three cell types, illustrating a remarkable tissue- or cell-type specificity in aging-associated reprogramming of the clock. Aged epidermal and skeletal muscle stem cells lost oscillation of homeostasis genes but gained oscillation in genes mediating a stress response to DNA damage and inflammation or autophagy6. In contrast, oscillatory genes involved in NAD+ metabolism and protein acetylation figured prominently in the liver5 (Fig. 1).
Figure 1.
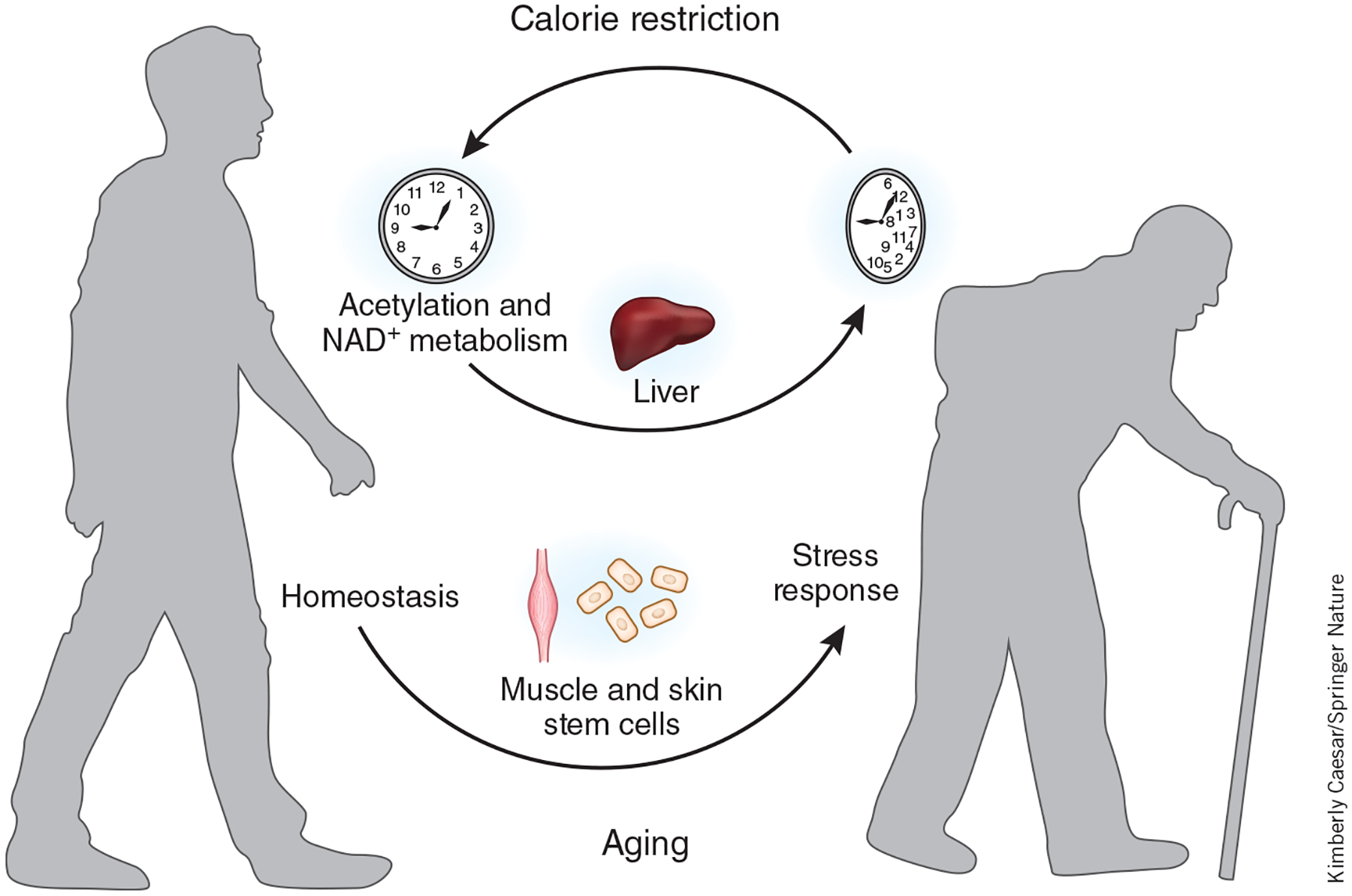
Aging reprograms the circadian transcriptome in a cell-type-dependent manner. Aged epidermal and skeletal muscle stem cells lose oscillating transcription of genes that regulate homeostasis but gain oscillating expression of genes mediating stress responses. Oscillatory genes involved in NAD+ metabolism and protein acetylation are enriched in the liver. Such rewiring can be reversed in part by the administration of the CR diet.
Surprisingly, the expression of core clock genes and clock-controlled genes remained unchanged with aging, despite the drastic circadian reprogramming5,6. Thus, the core clock machinery remains largely intact in old age, giving hope for the prospect of reversing aging-associated circadian reprogramming to potentially improve physiological functions. Indeed, CR-induced robust reprogramming of the circadian transcriptome partially overlaps with the circadian transcriptome in young mice5,6. Thus, the profound physiological impact of CR may be, in part, mediated by the reprogramming of the circadian clock.
How does rewiring of the circadian clock contribute to cellular aging? A closer look at the phases of the circadian genes and their regulated cellular processes in young and old mice might offer a clue. In young epidermal stem cells, DNA replication takes place at night, whereas maximal oxidative phosphorylation occurs during the day13,14. It was thought that temporal segregation of DNA replication and oxidative phosphorylation at different times of the 24-h cycle ensures that unwound DNA is exposed to minimal oxidative stress and thereby prevents DNA damage. Solanas et al. now found that in aged epidermal stem cells, DNA replication was delayed and extended into the daytime, whereas the phase of oxidative phosphorylation remained unchanged6. Thus, rewiring of the circadian clock with age can lead to misalignment of cellular activities and the resulting accumulation of cellular damage.
Taken together, these new findings also open up many questions. If the core clock machinery remains intact during aging, what are the driving forces of aging-associated circadian reprogramming, and what are the mediators of CR-induced circadian reprogramming? The observed enrichment of circadian genes related to NAD+ metabolism, protein acetylation, and stress resistance points to the sirtuin family of NAD+-dependent deacetylases as potential mediators of reprogramming. Sirtuins are known to extend mammalian lifespan and healthspan15,16, mediate aspects of the CR response3,4, and have increasingly been appreciated as stress-resistance regulators4,7,9,17 Fittingly, several sirtuins have been implicated in circadian control18 and there is some degree of overlap between SIRT1-dependent hepatic circadian genes and aging- or CR-associated hepatic circadian genes5. Further studies are needed to determine whether overexpression of SIRT1 or other mammalian sirtuins (SIRT2–SIRT7) prevents aging-associated circadian reprogramming and whether CR induces circadian reprogramming in the absence of sirtuins.
It is now widely accepted that the general cause of aging is the accumulation of cellular damage19,20. Aging-associated stress resistance is particularly relevant to adult stem cells, which persist throughout the organismal lifespan to repair and maintain tissues7,9. The aging-associated circadian reprogramming of both homeostasis and stress-resistance genes observed in adult stem cells suggests that increased cellular damage is a driver of aging-associated circadian reprogramming. Given that aging-associated accumulation of DNA damage in stem cells originates from exposure to mitochondrial stress6 and that the mitochondrial protective programs are repressed in aged adult stem cells7,9, it is tempting to speculate that reactivating the mitochondrial protective programs may provide a means to reduce the accumulation of cellular damage and reverse aging-associated circadian reprogramming.
ACKNOWLEDGMENTS
D.C. is supported by NIH grant R01DK101885, the National Institute of Food and Agriculture, the PackerWentz Endowment and the Chau Hoi Shuen Foundation. R.O. is supported by the ITO Scholarship and the Honjo International Scholarship.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Luo H, Chiang HH, Louw M, Susanto A & Chen D Trends Endocrinol. Metab 28, 449–460 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weindruch R & Walford RL The Retardation of Aging and Disease by Dietary Restriction (Charles C. Thomas Publisher, 1988). [Google Scholar]
- 3.Chen D, Steele AD, Lindquist S & Guarente L Science 310, 1641 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Qiu X, Brown K, Hirschey MD, Verdin E & Chen D Cell Metab. 12, 662–667 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Sato S et al. Cell 170, 664–677 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Solanas G et al. Cell 170, 678–692 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Brown K et al. Cell Rep. 3, 319–327 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goodell MA & Rando TA Science 350, 1199–1204 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Mohrin M et al. Science 347, 1374–1377 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV & Antoch MP Genes Dev. 20, 1868–1873 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakamura TJ et al. J. Neurosci 31, 10201–10205 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sellix MT et al. J. Neurosci 32, 16193–16202 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geyfman M et al. Proc. Natl. Acad. Sci. USA 109, 11758–11763 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stringari C et al. Cell Rep. 10, 1–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finkel T, Deng CX & Mostoslavsky R Nature 460, 587–591 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanfi Y et al. Nature 483, 218–221 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Shin J et al. Cell Rep. 5, 654–665 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masri S & Sassone-Corsi P Sci. Signal 7, re6 (2014). [DOI] [PubMed] [Google Scholar]
- 19.López-Otín C, Blasco MA, Partridge L, Serrano M & Kroemer G Cell 153, 1194–1217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vijg J & Campisi J Nature 454, 1065–1071 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]