

Abstract
Members of the caspase family of proteases play essential roles in the initiation and execution of apoptosis. These caspases are divided into two groups: the initiator caspases (caspase-2, −8, −9 and −10), which are the first to be activated in response to a signal, and the executioner caspases (caspase-3, −6, and −7) that carry out the demolition phase of apoptosis. Many conventional cancer therapies induce apoptosis to remove the cancer cell by engaging these caspases indirectly. Newer therapeutic applications have been designed, including those that specifically activate individual caspases using gene therapy approaches and small molecules that repress natural inhibitors of caspases already present in the cell. For such approaches to have maximal clinical efficacy, emerging insights into non-apoptotic roles of these caspases need to be considered. This review will discuss the roles of caspases as safeguards against cancer in the context of the advantages and potential limitations of targeting apoptotic caspases for the treatment of cancer.
Keywords: Caspase, cancer, apoptosis, IAPs, mitochondria
1. Introduction
Members of the caspase protease family play integral roles in the initiation and execution of apoptosis. Once activated, they cleave a number of structural and regulatory proteins to dismantle the cell from within [1]. These proteolytic events are responsible for the classical hallmarks of apoptosis including nuclear condensation, DNA fragmentation, and plasma membrane blebbing [2]. Apoptosis plays crucial roles in the maintenance of cellular homeostasis and the removal of damaged cells [3]. Not surprisingly, incorrect regulation of caspases and apoptosis underlies the pathogenesis of many human diseases [4, 5]. The most notable of these is cancer. Cancer is the second leading cause of death in the United States accounting for one in every 4 deaths in the United States (www.cdc.gov/uscs.). One of the hallmarks of human cancer is the evasion of apoptosis [6]. Therefore, many existing therapies for cancer aim to overcome apoptosis evasion by targeting different caspase pathways. While most of these do so indirectly, emerging insights into caspase functions and new therapeutic strategies based on directly targeting caspases are beginning to show some promise.
2. The caspase protease family
The identification of the mammalian caspase family of proteases and their roles in apoptotic cell death was made possible by the discovery that the Caenorhabditis elegans gene Ced3 was required for apoptosis in the nematode [7]. When the protein product of the Ced3 gene was cloned and sequenced, it was determined to be homologous to the mammalian cysteine protease caspase-1, then known as interleukin-1β converting enzyme (ICE) [8]. Multiple additional mammalian proteins were found to share similar homology with cell death protein 3 (CED-3) and this family of proteases became known as the caspases [9]. Each caspase has in common an active site cysteine (‘C’) and preferentially cleaves its target substrates after an aspartate residue (‘asp-ase’) [9]. There are 14 known mammalian caspases and 12 known human caspases. Caspases are zymogens comprised of three defined domains: the prodomain, the large (~p20) catalytic domain, and small catalytic domain (~p10) (Figure 1). Cleavage between the three domains is generally required to form the stable active enzyme [10]. In order for a caspase to be activated, it must be in a dimer configuration that is either preformed or induced [10]. The active enzyme is thus a heterotetramer of the catalytic domains comprised of two large and two small subunits (p20/p10)2 (Figure 2) [11]. Each caspase has a specific substrate specificity, but they all share the preference for an aspartate residue immediately prior to the cleavage site [12]. In rarer cases, caspases can also cleave after glutamate and phosphoserine residues [13]. For the cysteine protease activity to occur, the caspase recognizes four residues (P4-P1, where P1 is the aspartate) on the N-terminal side of a scissile bond (P1-P1’), resulting in cleavage of a peptide bond C-terminal to P1 [14].
Figure 1. The human apoptotic caspases.
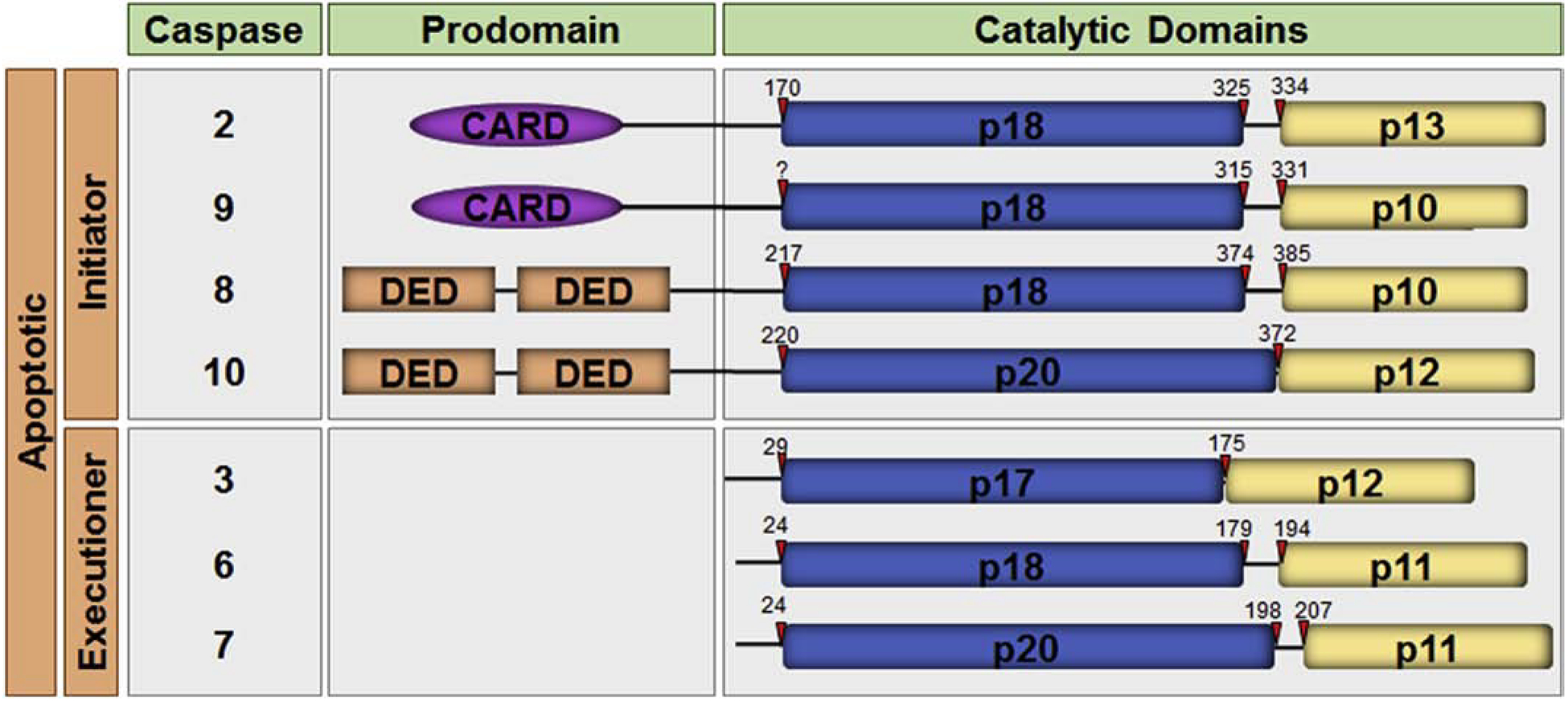
A schematic representation of the domain organization of the human apoptotic caspases is shown. The caspases are divided into the initiator caspases and the executioner caspases. Each caspase comprises a prodomain and a large and small catalytic subunit. Initiator caspases have long prodomains containing the protein:protein interaction motifs, caspase recruitment domain (CARD) or death effector domain (DED), as indicated. The cleavage sites for each caspase are depicted with red arrows.
Figure 2. Structure of the caspase-3 heterotetramer.
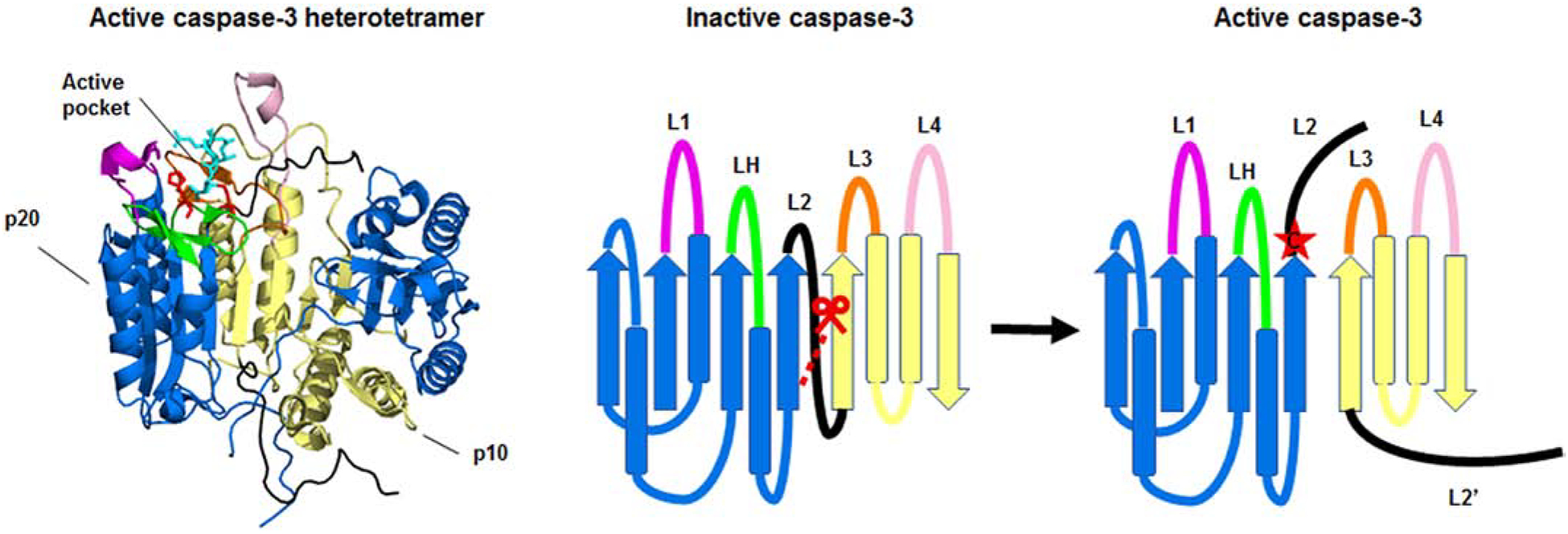
The crystal structure of the active caspase-2 heterodimer (left) and a ribbon diagram of the structure (right) are shown. The large (p20) and small (p10) subunits of the heterodimer are depicted in blue and yellow respectively. Caspase-3 is shown bound to the inhibitor z-DEVD-cmk (cyan). The catalytic histidine and cysteine are shown in red. The loops, L1 (magenta), L2 (black), LH (green), L3 (orange), and L4 (pale pink) make up the active pocket. Caspase-3 is activated by cleavage of the intersubunit linker (L2). Cleavage of the intersubunit linker opens up the active pocket, exposing the active cysteine (red star). For clarity, the loops, catalytic residues, and inhibitor are only highlighted in one half of the heterotetramer. The molecular models of the protein structures were created using PyMOL v1.3.Edu with the full length caspase-3 crystal structure (PDB:2DKO) [38].
The mammalian caspases can be divided into three broad categories termed executioner caspases, initiator caspases, and inflammatory caspases. The executioner caspases include caspase-3 and caspase-7. Caspase-6 is also in this group, but it has a less clearly defined role in apoptosis. Instead, it plays a unique role in neurodegenerative disease [15]. The executioner caspases all have in common a short prodomain of 20–30 residues that lacks any defined motif or protein interaction site. Executioner caspases are so-called because they bring about the execution phase of apoptosis by cleaving many different structural and regulatory proteins that shut down the functions of the cell and contribute to the morphological hallmarks of apoptosis [1]. The executioner caspases are activated when cleaved by an upstream initiator caspase or additional protease. Caspase-2, −8, −9, and −10 comprise the initiator caspases. They each have a long prodomain that encodes a protein:protein interaction motif used to facilitate dimerization of the catalytic domains. These motifs include the caspase recruitment domain (CARD) found in caspase-2 and caspase-9, and the death effector domain (DED), found in the prodomains of caspase-8 and caspase-10 [16] (Figure 1). These interaction domains allow the recruitment of the initiator caspases to large multi-protein complexes that serve as activation platforms for each caspase. Activation platforms are specific to each caspase and include the death inducing signaling complex (DISC) for caspase-8 and caspase-10 [17], the apoptotic peptidase activating factor 1 (APAF1) apoptosome for caspase-9 [18], and the p53-induced death domain protein (PIDD)osome for caspase-2 [19]. Each complex generally consists of a receptor protein that oligomerizes and recruits either the caspase or an adaptor protein. For example, the CD95/Fas DISC is formed when oligomers of CD95/Fas recruit molecules of the adaptor protein Fas-associated protein with death domain (FADD). FADD in turn binds to caspase-8, a binding that is mediated by the DED in FADD binding to the DED in caspase-8 [20]. Similarly, oligomerization of APAF1 forms the apoptosome, providing the activation platform for the recruitment of caspase-9 via the CARD present in caspase-9 and in APAF1 [21]. Recruitment of caspase monomers to their respective activation platform increases the local concentration of caspase molecules permitting dimerization. Dimerization is essential to initiates the activation of the caspase [22, 23]. This mechanism is referred to as the induced proximity model [24]. A third group of caspases is known as the inflammatory caspase subgroup. These caspases consist of human caspases-1, −4, −5, and 12, murine caspase-11, and bovine caspase-13. Each of these caspases has a long prodomain encoding a CARD and, thus, they can be considered initiator caspases. However, the inflammatory caspases are not generally involved in the initiation of apoptotic cell death. Rather, they are defined by their contribution to inflammation by regulating the proteolytic cleavage of the immature cytokines, proIL-1β and proIL-18, or the pore forming protein gasdermin D to induce an inflammatory form of cell death called pyroptosis [25].
In addition to their modes of activation, the separation into the initiator and executioner caspase groups is also based on their substrate preferences. Caspase-3 and caspase-7 have similar preferences for substrate sequences (DEXD), as determined using positional scanning peptide libraries. Caspase-6, in contrast, appears to prefer branched aliphatic residues in the P4 position. Caspase-8, caspase-9, caspase-10, and the inflammatory caspases all prefer substrates with an aliphatic residue at P4 such as leucine or valine. The substrate specificity of caspase-2 is similar to those of common with caspase-3 and −7 [12, 26]. For that reason, caspase-2 has sometimes been considered an executioner caspase. However, it contains a long prodomain in its structure, which places it within the initiator caspase subgroup. It must be noted that these substrate specificities are not always matched by the repertoire of natural substrates for each caspase [27]. For example, while caspase-3 and caspase-7 have similar peptide preferences they have few substrates in common. These common substrates include poly-ADP ribose polymerase (PARP), Rho GDP-disassociation inhibitor (RhoGDI) and Rho-associated coiled coil containing protein kinase 1 (ROCK1). Caspase-6 has a more limited substrate repertoire that includes lamin A and ceramide phosphoethanolamine synthase [28, 29]. Caspase-3 is responsible for the cleavage of the majority of apoptosis-associated substrates known [30, 31].
3. Mechanisms of caspase activation
The crystal structures of the catalytic domains of caspase-1, −2, −3, −6, −7, −8, and −9 have been determined [11, 32–38]. Many of the crystal structures of isolated caspases have demonstrated a stable structure as a homodimer of the catalytic domains representing the (p20/p10)2 heterotetramer. Each homodimer has an active site on either end of the dimer. The homodimer is stabilized by hydrophobic interactions between multiple beta-strands and one beta sheet of each monomer. The active sites are made of 5 loops (L1–4 and LH) with L1 and L3 being the more conserved of the loops and L2 and L4 more variant. L1 and L4 make up either side of the substrate-binding pocket and L3 comprises the center of the pocket. L2 contributes the catalytic cysteine [39] and LH contributes a histidine [40] that together make up the catalytic dyad (Figure 2). Executioner caspases are present in the cells as preformed dimers that are activated by cleavage [10]. They are activated when the linker between the large and small subunit is cleaved by an upstream caspase or another protease such as granzyme B [41]. When the caspase is in its uncleaved form, the active site is occupied blocking the formation of a functional substrate-binding site. The site is occupied by the interdomain linker in caspase-3 and caspase-7 [35, 42, 43]. Cleavage of the linker forms L2 and L2’. L2’ stabilizes the active site of the neighboring dimer in the heterotetramer [42]. The cleavage and the accompanying conformational change allows access to substrate binding pocket and stabilization of the active site (Figure 2).
Unlike the executioner caspases, the initiator caspases are activated by dimerization rather than cleavage. The active site pocket is thus created and exposed upon formation of the caspase dimer. Caspase-9 can be activated in the absence of cleavage [44], and dimeric caspase-2 retains 20% activity when cleavage is blocked [22]. It has been shown in vitro that enforced dimerization of caspase-8 and caspase-9 resulted enhanced activity. Blocking cleavage of caspase-8 impaired the stability of the dimeric species [10]. Thus dimerization of caspase-8 is required for activation and cleavage serves to stabilize and further enhance activity of the enzyme. Similarly, cleavage of the interchain linker of capase-9 yielded a 5-fold increase in activity and removal of the CARD resulted in a further 10-fold enhancement. However, autocleavage of caspase-9 at the level of the apoptosome has a different outcome. When caspase-9 is recruited to the APAF-1 apoptosome, it undergoes dimerization, which is sufficient for activation [45].
The two main pathways of caspase activation that result in apoptosis are termed the intrinsic pathway and the extrinsic pathway, so-called because they are generally engaged by stresses that come from outside the cells or within the cell respectively. The intrinsic pathway is primarily activated by cellular stresses including DNA damage, metabolic stress, and endoplasmic reticulum stress. Many chemotherapeutic drugs that are used to treat various cancers engage this pathway. These stimuli converge on the mitochondria leading to mitochondrial outer membrane permeabilization (MOMP) and the release of cytochrome c from the mitochondrial intermembrane space to the cytosol (Figure 2). Once in the cytosol, cytochrome c binds to the WD repeats of APAF1, inducing a conformational change that opens up the molecule. In the presence of dATP or ATP, APAF1 then oligomerizes to assemble the APAF1 apoptosome [46, 47]. In turn, the apoptosome recruits and activates caspase-9, which continues the caspase cascade by cleaving the executioner caspases-3 and −7. To be able to cleave caspase-3, caspase-9 needs to remain tethered to the apoptosome and caspase-9 autocleavage to the p35/p17 subunits induces dissociation from the apoptosome [48]. This has been proposed to act as a molecular timer for caspase activation [49] rather than increasing the enzyme’s activity as is seen with caspase-8.
The extrinsic pathway generally refers to apoptosis induced by the engagement of death receptors by their cognate ligands at the cell membrane. The death receptors are all members of the tumor necrosis factor receptor (TNFR) superfamily, but the death receptors have in common a death domain (DD) at their C-terminal. The DD is a protein interaction domain that shares structural features with the DED and CARD. Death receptors include CD95, TNFR1, and the TNF-related apoptosis-inducing ligand (TRAIL) receptors (TRAIL-R1/DR4 and TRAIL-R2/DR5). Each of these receptors is activated following multimerization upon binding of their cognate ligand: CD95L/Fas ligand (FasL), TNF, and TRAIL respectively, which ultimately leads to activation platform assembly and caspase-8 activation (Figure 3). CD95 and the TRAIL each assemble to form the DISC at the plasma membrane. In contrast, the caspase-8 activation platform induced by the TNFR1 does not initially assemble at the membrane. Upon engagement of TNFR1 by TNF, the receptor rapidly recruits TRADD, a FADD-like homolog, receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and TNF receptor associated factor 2 (TRAF2) at the plasma membrane [50]. Termed complex I, this complex induces nuclear factor kappa B (NFκB) activation. A second complex, complex IIa, is formed when TRADD/RIPK1/TRAF2 dissociates away from TNFR1 and recruits FADD and caspase-8 in the cytoplasm to induce apoptosis [50] (Figure 3). If caspase-8 is absent or inactive, complex IIb is formed between RIPK1 and RIPK3. RIPK3 induces phosphorylation of the pore forming protein, mixed lineage kinase domain like pseudokinase (MLKL), which forms pores in the plasma membrane to induce an alternative form of cell death called necroptosis [51] (Figure 3).
Figure 3. The mitochondrial pathway of apoptosis.
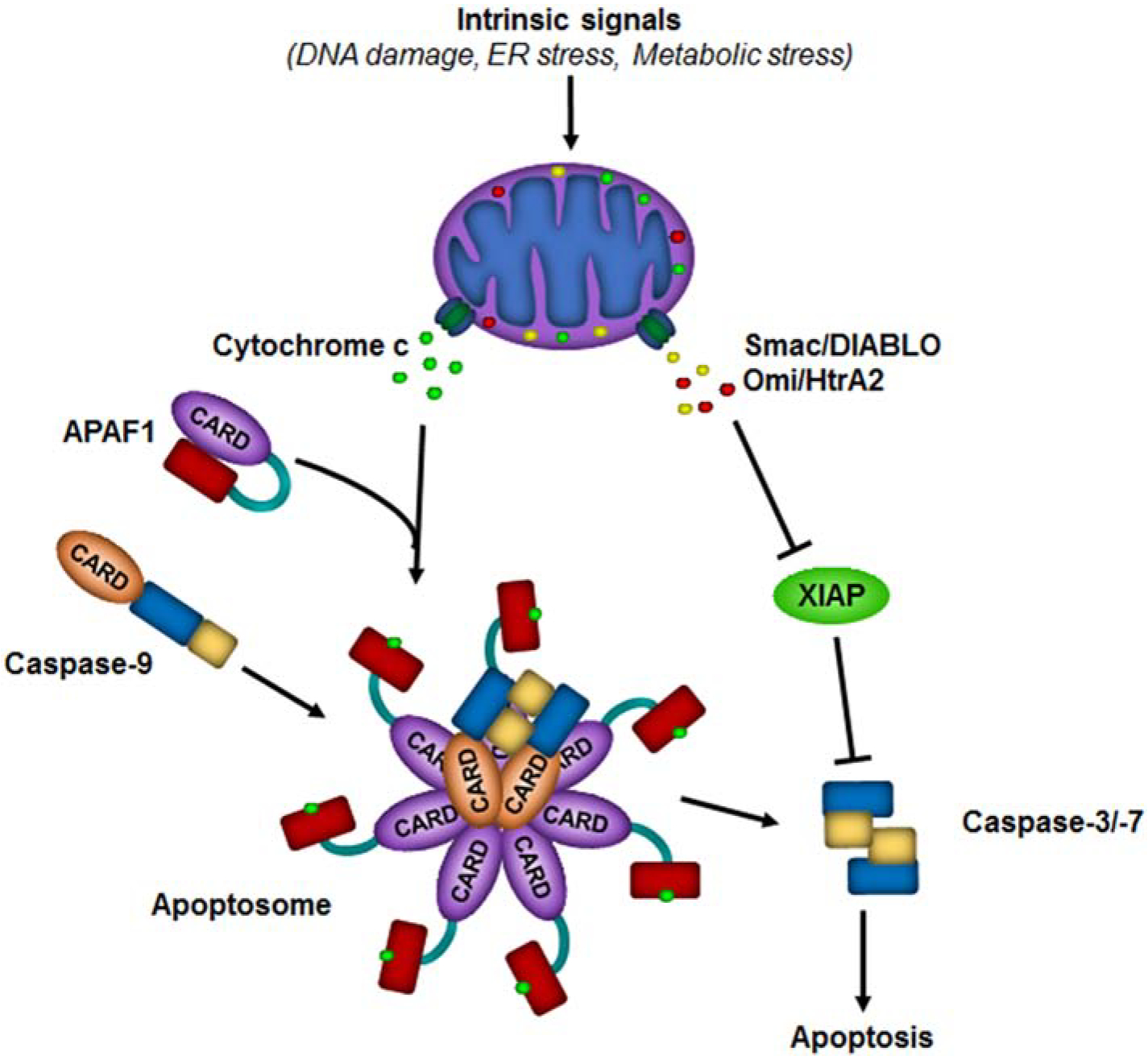
Diverse cellular stresses such as DNA damage, ER stress, and metabolic stress activate the intrinsic pathway leading to mitochondrial outer membrane permeabilization (MOMP). MOMP allows for the release of cytochrome c, second mitochondria-derived activator of caspases/ direct IAP binding protein with low pI (Smac/DIABLO), and Omi/ high temperature requirement protein A2 (HtrA2) from the mitochondrial intermembrane space. Cytochrome c binds to the WD repeats (red rectangle) of apoptotic peptidase activating factor 1 (APAF1), inducing a conformational change that opens up the molecule. This allows multiple molecules of APAF1 to oligomerize and assemble into the APAF1 apoptosome. The apoptosome recruits and activates caspase-9, which then activates caspase-3 and caspase-7 leading to apoptosis. Smac/DIABLO and Omi/HtrA2 bind to XIAP to prevent it from inhibiting active caspases.
The adaptor molecule FADD recruits caspase-8 to the DISC or TNFR complex II. Caspase-8 has two DEDs in its prodomain and the current model for its activation shows that, when recruited to the DISC, these DEDs form filaments. Cryo-electron microscopy has revealed that the caspase-8 DED filaments form ~20 nm wide structures [52]. The first DED of caspase-8 binds to FADD and the second DED in the same caspase-8 molecule binds to the first DED of an additional caspase-8 molecule through tandem linkage [53, 54]. Two additional interaction interfaces of the two caspase-8 DED domains further amplify the DED chains in two additional directions forming a helical filament assembly [52]. This provides a mechanism of amplifying the caspase-8 signal.
The pathway of caspase-2-induced apoptosis is somewhat distinct from the intrinsic and extrinsic pathway. Unlike caspase-8 and caspase-9, caspase-2 does not directly cleave the executioner caspases, but leads to their activation indirectly through the cleavage of the proapoptotic B-cell lymphoma 2 (BCL2) family protein, BH3-interacting domain death agonist (BID), leading to MOMP and subsequent caspase-9 and caspase-3 activation [55]. Caspase-8 can also cleave BID to induce MOMP, bridging the extrinsic and intrinsic apoptotic pathway [56]. Hence, in certain cell types, caspase-8-induced apoptosis can be inhibited by BCL2 expression. Cells in which caspase-8 induced apoptosis is not inhibited by anti-apoptotic BCL2-family proteins are termed type I cells and cells require MOMP for caspase-8-induced apoptosis are called type II cells [57].
4. Mechanisms of caspase inhibition
There are a number of inhibitors of caspases that are expressed by cells. While the function of these inhibitors is to limit uncontrolled caspase activation and unnecessary cell death, high expression of caspase inhibitors in cancer cells is often associated with chemoresistance. The first group of inhibitors comprises the inhibitor of apoptosis proteins (IAPs), which includes X-linked IAP (XIAP), cIAP1, and cIAP2. The IAPs are characterized by baculovirus IAP repeat (BIR) motifs containing 1–3 Zn fingers and a RING finger domain on the carboxy-terminal. Members of this group of proteins can inhibit caspases both directly and indirectly. Human XIAP, cIAP1 and cIAP2 have all been shown to bind to and inhibit the active forms of caspase-3, caspase-7, and caspase-9 in vitro [58, 59]. However, cIAP1 and cIAP2 bind caspases with much less efficiency because they lack important caspase interacting residues that allow for tight binding and direct inhibition [60]. Therefore, XIAP is the primary IAP to directly inhibit caspases in vivo. The crystal structure of caspase-3 or caspase-7 in complex with XIAP revealed that BIR2 domain in the N-terminal portion of the XIAP protein binds to and blocks the substrate binding pocket of the caspase, therefore obstructing recruitment of its substrates [61, 62]. Caspase-9 interacts with the BIR3 domain of XIAP [37]. XIAP is an E3 ligase. This provides a secondary means to inhibit caspase-induced apoptosis by targeting active caspase-3 to the proteasome for degradation [63].
XIAP mediated inhibition of caspases is regulated by second mitochondria-derived activator of caspases (Smac)/direct IAP binding protein with low pI (DIABLO) and the related protein Omi/High temperature requirement protein A2 (HtrA2) [64–67]. Both of these proteins are released from the mitochondria along with cytochrome c at the time of MOMP. Smac and Omi bind to the BIR2 or BIR3 domain of XIAP thereby preventing its binding to the active caspases (Figure 3) [67–69]. The ability of Smac to modulate XIAP activity plays an important role in the caspase-8 pathway. The difference between type I and type II cells is determined by the level of XIAP expression. In type I cells, XIAP expression is low or absent and therefore caspase-8-mediated activation of caspase-3 is unimpeded and cell death is rapid. In contrast, in type II cells, XIAP expression is high. In these cells Smac release from mitochondria is required to prevent XIAP from blocking caspase-8-mediated caspase-3 activation and apoptosis proceeds more slowly [70].
cIAP1 and cIAP2 inhibit caspases in an indirect manner. cIAP1/2 block the formation of complex II, the caspase-8 activation complex induced by TNF. Following complex I formation, TRAF2 serves a linker to recruit cIAP1 and cIAP2 to TNFR complex I [71] and, through ubiquitination of components of the complex, cIAP1/2 activates the canonical NFκB pathway. Additionally, cIAP1/2 blocks complex II formation by recruiting linear ubiquitin assembly complex (LUBAC) to complex I. LUBAC is a complex that consists of the proteins heme-oxidized IRP2 ubiquitin ligase 1 (HOIL1), HOIL1-interacting protein (HOIP) and SHANK Associated RH Domain Interactor (SHARPIN). LUBAC adds linear ubiquitin chains to inhibitor of nuclear factor kappa-B kinase subunit gamma/NF-kappa-B essential modulator (IKKγ/NEMO), the regulatory component of the IKK complex [72]. This brings the IKK complex into the TNFR1 complex, facilitating canonical NFκB activation resulting in the translocation of the NFκB p50/p65 complexes to the nucleus to induce transcription of multiple survival genes including cIAP2. LUBAC also adds linear ubiquitin chains to other components of the complex, stabilizing complex I and effectively inhibiting complex II formation (Figure 4) [73]. Therefore, cIAP1/2 are key factors in the inhibition of caspase-8 activation through the TNF pathway. cIAP1/2 also prevents non-canonical NFκB signaling by facilitating degradation of NFκB inducing kinase (NIK) [74]. In the absence of cIAP1/2, NIK phosphorylates IKKα inducing RelB/p52 complexes.
Figure 4. The death receptor pathway of apoptosis.
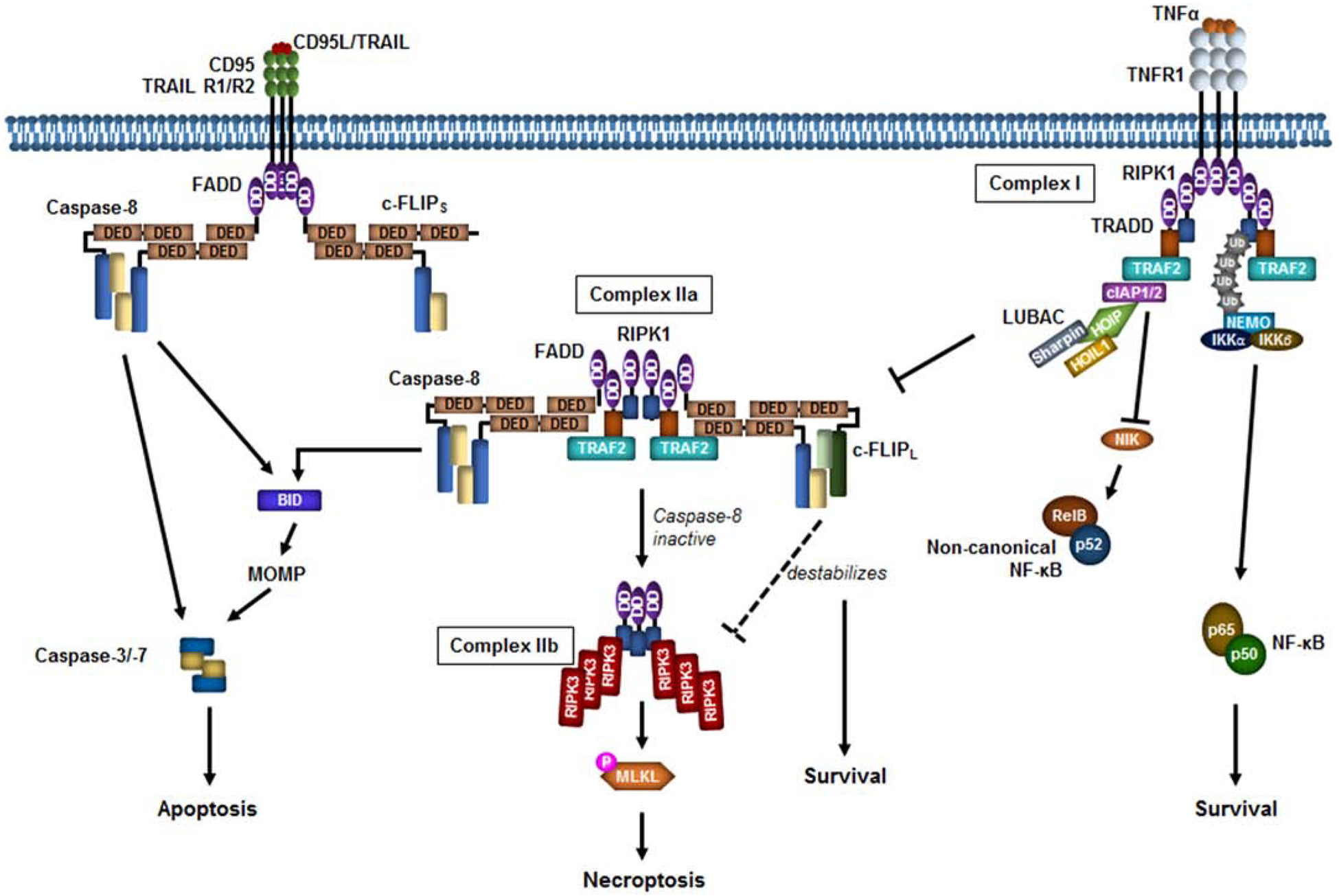
At the plasma membrane, the ligands CD95L, TNF-related apoptosis-inducing ligand (TRAIL), and tumor necrosis factor α (TNFα) induce multimerization and activation of the death receptors CD95, TRAIL R1/R2, and TNFR1 respectively leading to death-inducing signaling complex (DISC) assembly. The CD95 or TRAILR1/R2 DISC leads to caspase-8 induced proximity via fas-associated protein with death domain (FADD)binding the intracellular death domain (DD) of the receptor and the death effector domain (DED) of caspase-8. Caspase-8 activates the downstream caspases directly or indirectly through BID cleavage and mitochondrial outer membrane permeabilization (MOMP). TNFR1 recruits receptor-interacting serine/threonine-protein kinase 1 (RIPK1), TNFR1-associated death domain protein (TRADD), TNF receptor associated factor 2 (TRAF2), cellular inhibitor of apoptosis protein 1/2 (cIAP1/2), and linear ubiquitin assembly complex (LUBAC), comprised of HOIL1-interacting protein (HOIP), heme-oxidized IRP2 ubiquitin ligase 1 (HOIL1), and SHANK Associated RH Domain Interactor (Sharpin). LUBAC adds linear ubiquitin chains (Ub) to RIPK1 and NF-kappa-B essential modulator (NEMO)to recruit the inhibitor of nuclear factor kappa-B kinase (IKK)complex to activate nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) (p50/p65). cIAP1/2 induce NIK degradation to inhibit non-canonical NFκB (RelB/p50). In the absence of cIAP1/2, TRADD/RIPK1/TRAF2 disassociates from the plasma membrane and recruits FADD and caspase-8 (Complex IIa) to induce apoptosis. In the absence of caspase-8, RIPK1 forms a complex with RIPK3 (Complex IIb). RIPK3 induces phosphorylation of mixed lineage kinase domain like pseudokinase (MLKL), resulting in necroptosis. Cellular-FLICE inhibitory protein long or short (c-FLIPL/s) can form heterodimers with caspase-8. At high concentrations c-FLIPL and c-FLIPs inhibit caspase-8 activation. c-FLIPL interaction with caspase-8 within complex IIa destabilizes the complex IIb, promoting survival. For simplicity, c-FLIPs is shown in the DISC and c-FLIPL is shown in complex IIa but the isoforms can interact with both complexes.
A second mechanism that cells have evolved to negatively regulate caspases is by blocking dimerization of initiator caspases. FADD-like IL-1Beta-converting enzyme-inhibitory protein (FLIP) is a caspase inhibitor that has multiple isoforms. Cellular FLIP long (c-FLIPL) is a catalytically inactive homolog of caspase-8. Cellular FLIP short (c-FLIPS) [75], and cellular FLIP raji (c-FLIPR [76]) only comprise the DED containing prodomain and lack the catalytic domains. c-FLIP can form heterodimers with caspase-8 [23]. Heterodimers formed between caspase-8 and c-FLIPS are completely inactive, blocking caspase-8 activation [77] In contrast, heterodimers between c-FLIPL and caspase-8 have catalytic activity. Heterodimerization between c-FLIPL and caspase-8 enables the stabilization of the active site without caspase-8 cleavage and is energetically favoured when compared to the homodimerization of caspase-8 [78]. The resulting c-FLIPL/caspase-8 complex has a more limited substrate repertoire [79]. In the context of the TNFR complex II, c-FLIPL/caspase-8 heterodimers result in the destabilization of the RIPK1/RIPK3 complex (complex IIb), blocking RIPK3 signalling and necroptosis [80]. This is because the resulting c-FLIPL/caspase-8 heterodimers, while unable to initiate apoptosis, are able to cleave RIPK1 leading to disassembly of the complex. Conversely, c-FLIPs/caspase-8 heterodimers prevent the cleavage of RIPK1 by caspase-8 and, therefore, promote the formation of the RIPK1/RIPK3 complex [81].
Although c-FLIPL/S can directly bind to FADD, in vitro DISC reconstitution studies have shown that c- FLIPL/S is recruited to the DISC primarily by binding to the DED of caspase-8 [77]. Certain studies have shown that high levels of c-FLIPS and c-FLIPL result in shorter DED filaments [77, 82]. However, confocal imaging as well as mathematical and structural modelling propose that c-FLIP inhibits caspase-8 by increasing the proportion of caspase-8/FLIP heterodimers compared to caspase-8 homodimers in the chain without affecting filament length [52, 54, 83]. The reasons for these discrepancies are unclear but are likely due, in part, to the limitations of working with in vitro reconstituted complexes. When present at low concentrations or with high receptor stimulation, the formation of a c-FLIPL/caspase-8 heterodimer at the DISC promotes caspase-8 oligomers. Structural modeling suggests that c-FLIP isoforms may even stabilize the DISC by bridging neighboring DISCs together [82]. Thus, in these conditions, c-FLIPL augments apoptosis [84, 85].
Caspase-10 is a close homolog of caspase-8 that also appears to function as a negative regulator of the extrinsic pathway. Unlike, c-FLIPL, caspase-10 is catalytically active [86]. Caspase-10 has been shown to inhibit CD95L mediated death independently of c-FLIP by binding to caspase-8 and preventing caspase-8 activation within the DISC. Instead, recruitment of caspase-10 appears to have a pro-survival effect by promoting CD95L-mediated NFκB activation [87].
Increased expression levels of each of these inhibitors have been noted in a number of cancers. They also provide viable drug targets for removing existing brakes on caspase activation and lowering the threshold for apoptosis. Increased expression of c-FLIP or the specific c-FLIP isoforms has been demonstrated in multiple cancers including lymphoma and non-small-cell lung cancer (NSCLC) [88–91] and its overexpression has been linked to poor prognosis and lower survival rates [92–94]. Similarly, multiple lines of evidence show that IAP expression levels are an important determinant of chemoresistance in cancer. IAP overexpression has been noted in a number of human cancers and is associated with increased drug resistance and poorer outcomes [95, 96].
5. Can caspases act as tumor suppressors?
While many upstream regulators of caspases have been defined as oncogenes or tumor suppressors [97], there have only been a few cases where caspases themselves have been shown to function as tumor suppressors (see below). In addition, there are not an overwhelming number of published examples of deregulated caspase expression or mutation profoundly impacting cancer incidence or progression. This may be because of the essential nature of these proteins for development. Genetic deletion of many caspases leads to some form of abnormality or lethality. Deletion of caspase-9 or its upstream regulator, APAF1 results in perinatal lethality due to forebrain outgrowth and resistance to apoptotic inducers that trigger the intrinsic pathway [98, 99]. Caspase-3 knockouts generated on the murine 129 background showed a similar phenotype with exencephaly, perinatal lethality, and delayed growth [100]. Surprisingly, thymocytes from the Casp3−/− animals were not resistant to apoptosis in this model. When backcrossed to B6 mice, the gross craniofacial abnormalities of Casp3−/− were ablated and the mice were born at expected Mendelian ratios [101]. This indicates the existence of genetic modifiers in 129 mice that causes loss of caspase-3 to induce exencephaly. Casp6−/− mice are normal with only a slight resistance to anti-CD95-induced death [102]. Knockout of caspase-7 on the B6 background was viable but combined deletion of Casp3 and Casp7 resulted in lethality shortly after birth [103]. Exencephaly was largely absent in these double knockout animals but they had major defects in cardiac development and their cells were resistant to numerous inducers of apoptosis. Therefore, caspase-3 or caspase-7 can each compensate for the loss of the other during development. These studies also suggest that caspase-3 and −7 rather than caspase-6 are the main executioners of apoptosis. Caspase-8 deficient mice are also embryonic lethal [104]. However, the phenotype somewhat mirrors the Casp3/Casp7 double knockout, with a failure of the cardiovascular system to develop. Deletion of Ripk3 in Casp8 deficient mice rescued the embryonic lethality [105]. Therefore, the lethal developmental defect of Casp8−/− mice is not a result of disrupting caspase-8’s role in regulating apoptosis but due to its failure to suppress necroptosis.
Of the apoptotic caspases, caspase-2 is the only one not to result in a developmental abnormality when deleted in mice. Mice are born normal and at the expected Mendelian ratios [106, 107]. Although these mice do not show any evidence of spontaneous tumorigenesis, caspase-2 has been shown to function as a tumor suppressor when the Casp2−/− mice have been crossed to a number of established murine models of cancer. Compared to wild type mice, caspase-2 deficient mice showed significantly accelerated tumorigenesis in the Eμ-Myc lymphoma model and in ataxia-telangiectasia mutated (Atm)−/− lymphomas [108, 109]. Caspase-2 has also been shown to have a tumor suppressor role in non-hematologic tumors. Deficiency of caspase-2 significantly accelerated tumorigenesis in the MMTV/c-neu murine mammary carcinogenesis model, and in (LSL)-KrasG12D/+ lung adenocarcinoma [110, 111]. Finally, chemically-induced liver tumors have been reported to grow more aggressively in the absence of caspase-2 [112]. Despite this, caspase-2 does not appear to act universally as a tumor suppressor. For example, deletion of caspase-2 has no effect on the growth or incidence of chemically-induced fibrosarcoma or irradiation-induced lymphoma [113]. In a murine model of neuroblastoma, deletion of caspase-2 has actually been reported to delay TH-MYCN-induced neuroblastoma [114]. This may suggest that caspase-2 has both tumor promoting and tumor suppressive effects depending on the type of cancer.
At present, it is unclear if caspase-2 is unique in its tumor suppressive abilities or if similar functions of the other caspases are masked by their essential functions in development. Caspase-1 is another caspase that does not show a lethal phenotype when knocked out [115] and Casp1 deficiency has been shown to enhance tumor formation in an inflammation-induced colorectal cancer model [116]. Conditional knockout of caspase-8 in B cells resulted in B-cell lymphomagenesis, the incidence of which was further enhanced by co-deletion of Tp53 [117]. Similarly, neural crest specific deletion of Casp8 in TH-MYCN mice led to more aggressive neuroblastoma [118]. However, in the latter case, there was no difference in tumor incidence, latency or size. Rather, loss of caspase-8 was associated with a significant increased incidence of bone metastasis and rate of secondary tumors. Based on these observations, it is possible that the use of additional lineage specific deletions could uncover tumor suppressive roles for the additional caspases.
6. What are the consequences of deregulated caspase expression in cancer?
Evidence of significant associations between caspase expression levels and cancer severity has often been either slight or contradictory. For example, in pediatric neuroblastoma, caspase-8 expression was shown to be absent or deficient in 75% of primary neuroblastoma cases and this downregulation appeared to be due to gene silencing by methylation of the CpG rich 5’ flanking region of CASP8 [119, 120]. Although there was no correlation between caspase-8 protein expression and survival [119], neuroblastoma cell lines with methylated CASP8 were resistant to doxorubicin and CD95L-mediated apoptosis [120]. However, CASP8 methylation has not been reported in any other tumor type, and in glioblastoma, increased expression of caspase-8 protein was reported to be associated with a worse prognosis [121].
Although caspase-2 has been shown to function as a tumor suppressor in mouse models, corroborating evidence from human studies has been somewhat lacking. CASP2 is located on chromosome 7q34–35 that is frequently deleted in leukemia. Lower expression of caspase-2 was associated with drug resistance in childhood leukemia subtypes such as acute lymphoblastic leukemia (T-ALL) and acute myeloid leukemia (AML) [122], and reduced expression of caspase-2 in AML has been correlated with poor prognosis [123]. High levels of inactive caspase-2 protein have been linked to a worse prognosis in cancer patients with ALL and AML, suggesting that defects in caspase-2 activation may also contribute to patient outcomes [124, 125]. CASP2 is subject to epigenetic silencing in a high proportion of mesotheliomas and cases of adult ALL [126]. In contrast, low caspase-2 expression correlates with survival in MYCN non-amplified neuroblastoma [114]. This is somewhat consistent with the data from the mouse model, although, in mice, the delay in tumorigenesis associated with caspase-2 loss was observed in a MYCN amplified context. It must be noted that many of these studies were hampered by small sample sizes or the significance of the association was not overwhelmingly large. Therefore, the clinical significance of caspase-2 expression levels in human cancer has not been fully elucidated.
There are similar conflicting results concerning the validity of caspase-3 as a biomarker for human cancer. In breast tissue samples collected from 46 cancer patients, 31 of whom had adenocarcinoma, 75% of the tumor samples analyzed were found devoid of both caspase-3 protein and mRNA, [127]. However, some studies have shown a link between increased caspase-3 expression and poor cancer prognosis. For example, immunohistochemistry for caspase-3 on tissue microarrays from a large cohort of early stage breast cancer patients revealed a small but statistically significant association between high caspase-3 expression and lower rates of cancer-specific survival [128]. Similarly, increased levels of cleaved caspase-3, as measured by immunohistochemistry analysis, were significantly associated with lower survival rates in gastric cancer, ovarian cancer, cervical cancer, and colorectal cancer [129]. The association of poor cancer prognosis with increased levels of cleaved caspase-3 was also detected in studies on oral tongue squamous cell carcinoma [130]. However, stratification analysis of the results revealed that, in patients with advanced disease progression (i.e. larger tumor size and metastases to lymph nodes), a higher level of cleaved caspase-3 was associated with a better prognosis [130].
The reasons for these conflicting results are not very clear. As discussed, it may be that complete inactivation of an apoptotic caspase is rare because of its essential roles in development and other processes. Instead of direct inactivation of the caspase itself, it is more likely that caspases are compromised in cancer by mutation or deregulation of upstream regulatory factors impacting caspase activation. Another factor that could explain the contradictory associations of caspase expression with cancer incidence or severity in both human populations and murine models is that many caspases have been shown to have roles in addition to inducing apoptosis. The availability of distinct caspase substrate repertoires in different cell types or even disease stage could impact the downstream consequences of caspase activation. This may explain some of the differences seen in the correlations of caspase expression with prognosis or disease severity. It is thus possible that the specific conditions of the cell in which a caspase is activated determines whether that caspase induces apoptosis or an additional non-apoptotic process. This could potentially play a fundamental role in determining how a caspase can impact cancer. Therefore, it is important to understand both the mechanisms of action of caspases and the downstream functions of each caspase to be able to fully appreciate their role in cancer and how to effectively target these proteins for therapeutic success.
7. Targeting caspase pathways
One of the goals of cancer treatment is to design therapies that can remove the cancer cells without harming the healthy tissue. Compounds, such as TRAIL, that have been shown to selectively kill cancer cells [131] could achieve this. Another important challenge is designing strategies to overcome inherent resistance of tumor cells to conventional therapies. Leveraging the fact that caspase inhibitors including XIAP and c-FLIP are often overexpressed in cancer [95, 132] can provide a means to specifically target tumors.
TRAIL is a death receptor ligand with potential as a cancer therapy because it has been shown in mouse models and in cell lines to selectively kill cancer cells but not primary cells or tissue [131]. In contrast, treatment with CD95L induces a lethal level of hepatocyte cell death in mice [133]. The receptors for TRAIL, TRAIL-R1 and TRAIL-R2, are expressed widely in normal tissue [134, 135]. TRAIL also has two decoy receptors, TRAIL-R3/DcR1 and TRAIL-R4/DcR2, which do not express an intracellular DD and, thus, cannot transduce the apoptotic signal to caspase-8 [136]. An association between TRAIL-R4 expression and TRAIL resistance has been reported in breast cancer cell lines [137], suggesting that the decoy receptors provide a mechanism for the cancer-specific killing of TRAIL. However, in other cancers, such as multiple myeloma, no correlation between the level of decoy receptor expression and the efficiency of TRAIL-mediated killing has been shown [138]. A number of TRAIL agonists have been developed to clinically activate the TRAIL pathway for cancer therapies. These include agonistic antibodies against TRAIL-R1 and soluble recombinant forms of TRAIL. Unfortunately, most cancers have demonstrated resistance to these therapies [139]. The resistance factors appear to vary dependent on cell type and can be mediated through c-FLIP [140], IAPs [141] or anti-apoptotic BCL2 family proteins [142]. Combination therapies that include TRAIL plus Smac mimetics (see below) or BH3 mimetics (inhibitors of anti-apoptotic BCL2 family proteins) have shown promising preclinical efficacy (reviewed in [139]).
To overcome IAP-associated resistance in cancer, a range of drugs called Smac mimetics have been developed to target the IAPs. Smac mimetics were designed to disrupt the interaction between XIAP and active caspases as a means to induce apoptosis of cancer cells [143, 144] They are chemical derivatives of the peptide sequence AVPI, which is the region of Smac that binds to the BIR2 and BIR3 domains of XIAP [145, 146]. Although Smac mimetics were originally designed to target XIAP, they actually have improved efficiency in targeting cIAP1 and cIAP2. When bound to cIAP1/2, Smac mimetics enhance the proteosomal degradation of cIAP1/2 by inducing a conformational change that results in dimerization of the RING domain and facilitating autoubiquitination and degradation of the IAPs [147]. This facilitates complex II formation, caspase-8 activation and apoptosis.
Smac mimetics can be divided into two categories, monovalent or bivalent compounds. Bivalents are made up of two Smac mimetic motifs (monovalents) that are bridged by a linker. Monovalents have the advantage of being orally bioavailable. Bivalent compounds have higher potency and cytotoxicity [143, 144, 148]. Current Smac mimetics used in precilinical studies or in clinical trials include monovalent compounds, such as LCL161 and Debio1143 [149], and the bivalent compounds birinapant and AZD5582 [150]. In phase I clinical trials, these Smac mimetics have been shown to be tolerable with minimal severe side-effects or toxicities [151–153]. However, limited anti-tumor activity was observed in these investigations. Indeed, a phase II clinical trial using birinapant as a treatment for platinum-resistant or refractory epithelial ovarian cancer was terminated for lack of clinical benefit despite evidence of consistent cIAP1 protein depletion in tumors [154]. Although several groups have reported that Smac mimetics can be used effectively to kill cancer cells [143, 144, 148, 155], they currently show more clinical promise when used in combination with existing therapies. Smac mimetics used combination with therapeutic agents that target death receptors appear to be the most effective methods for cancer therapy and their use has enhanced the cytotoxicity of TRAIL [156]; CD95L [157] and TNFα [143].
The reason for the limited effectiveness of Smac mimetics as a monotherapy can be partially explained by a feedback loop leading to upregulation of cIAP2. Degradation of cIAP1/2 by Smac-mimetics facilitates non-canonical NFκB activation, triggering the upregulation of TNFα and, consequently, TNFα-mediated cell death [143]. cIAP1 and cIAP2 are functionally redundant, so only one needs to be present for their action [158]. Smac mimetic-induced depletion of cIAP1 can lead to activation non-canonical NFκB signaling, resulting in increased cIAP2 expression [159]. The de novo expressed cIAP2 is not further depleted by the Smac mimetic because cIAP2 degradation requires cIAP1 [160]. Thus, up-regulation of cIAP2 can lead to resistance to Smac mimetic-induced cell death. Another factor that confers resistance to Smac mimetics occurs in cancers that results from a chromosomal translocation resulting in the cIAP2-MALT1 oncogene. cIAP2-MALT1 is the most common translocation in Mucosa-associated lymphoid tissue (MALT) lymphoma and creates a fusion protein of the N-terminal portion of cIAP2 lacking the RING domain and the C-terminal caspase-like domain of the paracaspase MALT1. This fusion results in stabilized cIAP2 expression and constitutive canonical NFκB activation contributing to overall cell survival [161]. Thus, blocking NFκB signaling is an effective way to overcome resistance to Smac mimetics. For example, inhibition of IKKβ with the drug BMS-345541 blocked the upregulation of cIAP2 and sensitized H1299 NSCLC cells to apoptosis induced by the Smac mimetic plus TNFα [159].
Due to the relationship between c-FLIP overexpression with both poor patient outcomes and TRAIL resistance, inhibiting or downregulating c-FLIP has long been considered a potential means of overcoming resistance in cancer. While small molecule inhibitors of c-FLIP have been in development, their use as clinical agents has yet to be published. However, other methods have been used to either decrease c-FLIP levels or inhibit its expression. c-FLIP is a short-lived protein that is rapidly turned over by the proteasome [162]. A number of agents have been shown to enhance proteasome-mediated degradation of c-FLIP to overcome chemoresistance. These include cisplatin, which has been shown to induce ubiquitination and degradation of c-FLIP in a p53-dependent manner. Cisplatin-induces p53 expression and promotes p53 binding to c-FLIP and ITCH, an E3 ligase and key regulator of FLIP ubiquitination [163–165]. HDAC inhibitors have similarly been shown to induce FLIP depletion to overcome TRAIL resistance [166]. It has been proposed that HDAC inhibition disrupts c-FLIP binding to Ku70, which in turn promotes its degradation [167]. c-FLIP transcription is regulated by a number of factors including NFκB and AKT [168, 169]. Inhibition of NFκB in combination with TRAIL treatment induces apoptosis in part by downregulating FLIP expression [170]. Similarly, inhibition of AKT has been shown to overcome resistance to cisplatin-induced apoptosis that has been associated with c-FLIP overexpression [171]. Finally, c-FLIP function can be deregulated by interfering with its ability to bind to caspase-8. It has been demonstrated that mild heat stress induced aggregation of c-FLIP, leading to the depletion of the available cytosolic pool of c-FLIP that could be recruited to the DISC [172]. Generating chemical inhibitors that would disrupt c-FLIP binding to the DISC or complex II would be ideal. However, a number of challenges to this approach exist. The first is to identify compounds that do not simultaneously inhibit caspase-8. In addition, the mechanisms and outcomes of c-FLIP-associated inhibition of cell death are different for the different isoforms and, as discussed, can vary with c-FLIP concentration. Thus c-FLIP inhibitors may have different cellular effects depending on the isoform expressed and the relative levels of receptor to c-FLIP expression. Further clarification of the mechanism of FLIP-mediated caspase-8 inhibition will aid the rational drug design of FLIP specific inhibitors.
8. Using caspases in gene therapy approaches
The discovery that initiator caspase activation is induced by dimerization has directly led to the development of novel therapeutic approaches using an inducible version of caspase-9. This approach was first used as a safety switch in biological therapies used for the treatment of cancer. When haploidentical stem cell transplant is used for the treatment of leukemia, a major clinical side effect is graft-versus-host disease (GvHD), an often life-threatening allergic reaction to the transplanted T cells. To test if removing the cells following transplant could counteract GvHD, donor T cells were engineered to express an inducible version of caspase-9 (iCasp9). The catalytic domains of caspase-9 were fused to the FK-binding domain FKBP, dimerization of which can be induced by a small, otherwise bioinert, molecule AP1903 (referred to as chemical inducer of dimerization [CID]). Induced dimerization of FKBP serves to enforce dimerization and activation of caspase-9 in the absence of any other upstream signal. To test the effectiveness of this, T cells from a partially matched donor were engineered to express iCasp9 and were adoptively transferred into patients with high-risk malignancies who had received a T-cell-depleted hemopoietic stem cell transplant. Although such T cell depleted transplants produce a low incidence of graft versus host disease, the recipients have a higher risk of infection and relapse of their malignancy since they lack the benefit of T cell control of infectious agents and of residual host malignant cells. Accordingly, investigators gave iCasp9-expressing T cells after the transplant with the intent of restoring the beneficial effects of T cells, but this time adding the protection of iCasp9-mediated T cell killing, should GvHD emerge. Following any evidence of GvHD or transplant rejection, the dimerization agent was administered. This resulted in rapid apoptosis and elimination of more than 90% of the T cells. Remarkably, GvHD subsided in 24–28 hours in the 4/10 patients who had this complication [173]. The T cell reconstitution in patients given the dimerization agent after GvHD appeared unaffected when compared to patients who did not have GvHD and the T cells with iCasp9 in the non-GvHD patients remained for more than 2 years after treatment, showing lack of spontaneous iCasp9 dimerization and the elimination of cells that would result [174]. Further murine studies have shown the potential for using this approach to remove transplanted induced pluripotent stem cells or mesenchymal stromal cells (MSC) [175, 176].
The same inducible caspase-9 approach has been tested as a mechanism for directly targeting solid tumors. In this system, MSC were used to deliver an adenoviral-based iCasp9 gene to lung tumors [177, 178]. Upon infusion into mice or humans, MSC naturally home to lung vasculature and to lung tumors. This allows the iCasp9 gene to be directly and specifically delivered to NSCLC. Subsequent treatment with CID would induce caspase-9 activation and would be expected to kill the tumor cells. While this approach showed promise in NSCLC cell lines, resistance to CID in some of the lines revealed potential limitations. Two methods have been proposed to overcome this. First, inhibition of the proteasome by treatment with bortezomib was shown to enhance caspase-9-induced apoptosis in resistant lines. This appeared to be due to the inhibition of XIAP-mediated degradation of active caspases [177]. Second, using conditionally replication competent adenovirus to encode iCasp9 provided both an amplification step for inducible caspase-9 expression and an additional mechanism for tumor destruction, improving the overall effectiveness of the approach [178]. More recent studies have similarly used an adeno-associated virus-based delivery of iCasp9 to AML in a zebrafish xenograft model and to hepatocellular carcinoma in a murine xenograft model. When CID was administered systemically a significant reduction in survival and tumor size was observed respectively [179, 180].
Caspase-3 has similarly been proposed for use in gene therapy approaches. It has been proposed that caspase-3 can be targeted to gastric tumors by selectively delivering the caspase to tumors overexpressing human epidermal growth factor receptor (HER), an oncogene that is overexpressed in 10–40% human gastric carcinomas. For this, NIH3T3 mouse fibroblasts were engineered to express and secrete a fusion protein containing a secretion signal, an anti-HER2 monoclonal antibody fragment that would target HER2 expressing cells, a pseudomonas exotoxin A-derived transportation domain fragment, and a constitutively active caspase-3 that would induce apoptosis in the targeted cells (HER-PE-CP3). When cultured in the presence of medium from the engineered cells that contained the secreted active HER-PE-CP3, human gastric cancer cells underwent apoptosis at the level of approximately 20%. Injection of the recombinant protein producing cells into mice engrafted with SGC7901 gastric cancer cells resulted in a significant reduction in tumor size and a significant increase in survival. Importantly, there was no evidence of injury or cell death in the major organs, demonstrating the successful and specific delivery of the fusion protein to the tumor and not to the non-cancerous tissues [181].
A major advantage of directly engaging caspase-9 or caspase-3 in tumor cells is that they initiate apoptosis downstream of MOMP. Thus, these engineered cells would theoretically not be affected by overexpression of anti-apoptotic BCL2 family proteins that block MOMP. These anti-apoptotic proteins include BCL2, BCL-XL, and induced myeloid leukemia cell differentiation protein (MCL1) and are upregulated in many human cancers, providing a key mechanism for resistance to chemotherapy (reviewed in [182]). However, it has been shown that caspase-3 activation can induce a positive feedback loop to the mitochondria by cleaving BID [183]. Thus, it is conceivable that these caspase-9 and caspase-3-based therapies could induce MOMP to some degree, and this may enhance their cytotoxicity in tumors that do not overexpress the anti-apoptotic proteins. Consequently, it is important to measure the efficiency of such approaches in the presence and absence of high levels of BCL2, BCL-XL and MCL1 to determine their effectiveness in a wide array of cancers. The iCasp9 suicide could be further modified based on our knowledge of the caspase-9 activation pathway. For example, engineering a caspase-9 that is unable to bind to XIAP could be an effective means to overcome resistance. The use of additional initiator caspases in the inducible suicide gene system for cancer treatment has yet to be investigated. However, a similar approach was used for caspase-8 to selectively remove adipocytes and cardiac myocytes to develop mouse models of lipoatrophy and heart failure respectively [184, 185]. These studies support the concept that using an inducible caspase-8 for the induction of apoptosis could potentially be developed for cancer therapeutics. Given the differences in how the different initiator caspases are activated and their outcomes, it is possible that exploring the use of caspase-2 or caspase-8 in this system could have dramatically different effects on the efficiency of cancer cell death in a therapeutic setting.
10. Non-apoptotic functions of caspases
An important consideration when targeting caspase pathways for therapeutic benefit in cancer is that many caspases have non-apoptotic roles and these roles may help or hinder the efficacy of tumor killing depending on the context. For example, caspase-2 has been proposed to have a role in cellular proliferation. Caspase-2 deficient cells proliferate at higher rates [108, 186] and have evidence of aneuploidy and genomic instability [109, 110]. In MMTV/c-neu mice, mammary tumors from Casp2−/− mice showed little evidence of altered apoptosis, but displayed a number of indicators of impaired cell division and genomic instability including an increased mitotic index, abnormal mitoses, karyomegaly, and multinucleation [110]. Similar evidence of increased genomic instability was shown in Casp2−/−/Atm−/− lymphomas [109]. In the Kras lung tumor model, Casp2−/− lung tumors initially responded well to chemotherapy but the tumors promptly rebounded with increased proliferation compared to the Casp2+/+ mice [111]. In colon cancer, it has been shown that aneuploidy tolerance associated with somatic mutations in Bcl9L is largely a result of the impaired ability of BCL9L to induce caspase-2 expression [187]. Caspase-2 has been shown to cleave murine double minute 2 (MDM2) [188], which has been proposed to be a key effector of the role of caspase-2 in protecting from polyploidy and aneuploidy [187, 189]. MDM2 is an E3 ligase that targets p53 for proteasomal degradation. MDM2 is cleaved by caspase-2 at Asp-367, liberating the c-terminal RING domain, thereby preventing ubiquitination and degradation of p53 and promoting p53 stability and function [188]. Thus, in the absence of caspase-2, DNA-damage-induced expression of the p53 target genes Pidd, p21, and Mdm2 was decreased [110]. This promotion of the p53 pathway may underlie the proposed role of caspase-2 in regulating cell division. However, caspase-2 has been shown to function in the absence of p53 [190] so any role of caspase-2 on cell cycle in a p53-deficient context needs to be investigated.
While the roles of caspase-2 in inducing apoptosis and limiting cell proliferation may both be expected to have an overall positive impact on impairing cancer growth, non-apoptotic roles of other caspases have been shown to have opposing effects on cancer growth and treatment outcomes. In addition to inducing apoptosis, caspase-3 can impair tumor growth and progression by cleaving substrates that do not have a direct role in apoptosis. Ras GTPase (RasGAP) is a caspase-3 substrate that, when cleaved at positions 157 and 455, sensitizes tumor cells to apoptosis [191]. However, under conditions of low cellular stress that lead to a low level of caspase-3 activation, caspase-3 only cleaves RasGAP at position 455, producing an N-terminal fragment that activates the Ras-PI3K-AKT pathway, promoting survival [191]. The calcium-independent phospholipase A2 (iPLA2) is a caspase-3 substrate that, when cleaved, promotes cellular proliferation [192]. This appears to provide a mechanism whereby caspase-3 can trigger tumor cell repopulation. Immune compromised mice were engrafted with tumor cells in the presence of dying wild type or Casp3−/− mouse embryonic fibroblasts (MEF). The resulting tumors showed a strikingly lower rate of growth, caspase-3 activation, and cell proliferation in the presence co-injected cells deficient in caspase-3 [192]. This suggested the caspase-3 replete dying cells released a growth signal to boost proliferation of surrounding tumor cells. This signal was identified as cleaved iPLA2. Inactivation of caspase-3 has been shown to have additional anti-tumor effects. In the colon cancer cell line, HCT116, CRISPR/Cas9 knockout of caspase-3 resulted in reduced colony forming ability in soft agar and increased sensitivity to radiation and mitomycin C-induced cell death. Upon engraftment into mice, these cells showed similar rates of tumor growth but the caspase-3 deficient tumors responded significantly better to radiotherapy with lower rates of lung metastases. The caspase-3 deficient cells had reduced expression of N-cadherin, Snail, Slug, and zinc finger E-box-binding homeobox 1 (ZEB1), all proteins known to promote metastasis [193]. Thus, in certain conditions, caspase-3 appears to promote metastasis.
As discussed, caspase-8 can exert a pro-survival function by inhibiting RIPK3-dependent necrosis. While this mechanism is usually associated with immune defects, necroptosis of endothelial cells induced by tumor cells has been shown to induce metastasis. This process was enhanced when caspase-8 was inactivated in the endothelial cells [194]. It has been shown that under non-apoptotic conditions, caspase-8 in transformed cells can potentially contribute to metastasis by increasing cell motility through the promotion of calpain activation, an important driver of cytoskeletal remodeling through Rac activation, lamellipodia formation, and cell adhesion turnover [195]. Thus, different non-apoptotic functions of caspase-8 may both positively and negatively affect outcomes in cancer.
When the mitochondrial pathway is engaged, MOMP had long been considered to be a complete process, with all mitochondria being permeabilized at once [196]. More recent studies have challenged this view, showing that it is possible for only a small subset of the mitochondria in a cell to undergo MOMP. This process has been termed minority MOMP [197]. Minority MOMP has been shown to induce small amounts of caspase activation that are not sufficient to induce apoptosis. Instead, through cleavage of the caspase-3 substrate, ICAD, which is an inhibitor of the DNase CAD, CAD-mediated DNA damage ensues, which can lead to accumulation of genomic instability. The fact that low levels of caspase activity can result in outcomes that could either increase resistance to therapies or enhance tumor growth is an important consideration when targeting caspase pathways. As discussed, in trials using the caspase-9 suicide gene, up to 10% of the cells do not undergo apoptosis despite expressing the iCasp9. While it has not been investigated whether this small population of cells induced lower levels of caspase activation or if these cells show any evidence of genomic instability or proinflammatory markers, these events should be considered when translating these studies to more direct tumor killing mechanisms. To avoid relapse or developing resistance to these therapies, these approaches need to develop to the point that they induce maximal apoptosis in the tumors and the biological characteristics of the cells left behind need to be fully evaluated.
10. Caspase independent cell death
Under many circumstances, even if caspases are inhibited, cell death will still occur by some mechanism. This is especially true of the intrinsic pathway. MOMP is often considered the point of no return in cell death. For example, in a study of Apaf1-knockout embryonic stem cells, deficiency in APAF1 was not sufficient to block cell death but inhibition of MOMP by overexpression of BCL2 allowed cell survival [198]. Similarly, cells lacking both effectors of MOMP, BCL2 associated X apoptosis regulator (BAX) and BCL2-antagonist killer 1 (BAK), are completely resistant to stimuli that engage the mitochondrial pathway [199]. Thus, if caspases are blocked downstream of MOMP, caspase-independent cell death (CICD) will occur, whereas if MOMP is blocked, the cell will survive. CICD is thought to occur primarily as a result of eventual loss of ATP from the mitochondria due to MOMP. However, the release of apoptosis inducing factor (AIF) and endonuclease G (EndoG) from mitochondria has also been proposed to play a role. EndoG is an apoptotic DNase that, once released from the mitochondria, translocates to the nucleus to induce DNA fragmentation independent of caspases [200]. AIF similarly translocates to the nucleus to induce DNA fragmentation and peripheral chromatin condensation in the absence of caspases [201]. However, most documented forms of CICD are not characterized by fragmented nuclei and other mechanisms are likely involved.
There are a number of consequences of CICD. Studies have shown that, in the absence of caspases, both the NFκB and the cGAS/STING pathways are engaged. These independent pathways result in a proinflammatory response that have anti-tumor effects. For example, CICD-associated NFκB activation led to a profound reduction in the growth of BCL2-dependent tumors [202]. Release of mtDNA from the mitochondria as a result of mitochondrial inner membrane permeabilization following an apoptotic stimulus has been shown to activate cGAS/STING when caspases are blocked or under conditions of minority MOMP [203, 204]. cGAS/STING induces the type I interferon response. By inducing an immune response this pathway may also have an anti-tumor effect but this has not been directly investigated. Together, these studies show that inducing CICD rather than apoptosis may have a therapeutic benefit when treating certain cancers. If CICD was more effective at inducing an anti-tumorigenic response, then blocking caspase activation downstream of MOMP through APAF-1 removal would be expected to result in improved outcomes. Arguing against this is the fact that epigenetic silencing of APAF-1 in melanoma cell lines is associated with increased chemoresistance [205], and loss of heterozygosity of APAF-1 did not correlate with changes in overall survival [206]. Nonetheless, it is not clear if this would be the case in all cancer types. While many caspase inhibitors have been patented for use in diseases ranging from stroke to neurodegeneration [207], their efficacy, if any, in combination with chemotherapy for cancer treatment has not been explored.
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) has been shown to protect from CICD [208]. GAPDH is overexpressed in many human tumors [209] and its overexpression results in accelerated Eμ-Myc lymphomagenesis [210]. Overexpression of GAPDH increased glycolysis and enhanced autophagy by increasing expression of autophagy related 12 (Atg12). These two processes appear to cooperate to inhibit CICD [208]. In the absence of caspase activation, GAPDH has also been shown to stabilize the AKT pathway leading to a mildly increased BCL-XL expression. The increased expression of BCL-XL was sufficient to protect a subset of mitochondria from MOMP leading to increased clonogenic outgrowth [211]. This form of incomplete MOMP (iMOMP) can occur regardless of the presence or absence of caspase activity, but, when caspases are blocked, there is a strong correlation between iMOMP and recovery from CICD and transformation potential [212].
11. Conclusions
In summary, approaches that directly induce caspase activation or lift the brakes on existing inhibitors of caspase activation represent promising strategies for the treatment of different types of cancer. However, certain challenges remain. Since the discovery of the caspase family of proteases, our knowledge of their functions has expanded from being solely drivers of apoptosis to having multifaceted roles in cellular biology ranging from regulating cellular proliferation to inducing genomic instability. Adding to this complexity, different substrate repertoires of individual caspases that change with cell types and disease progression can significantly impact the outcome of activation of a specific caspase. Because of these diverse roles, insufficient or incomplete activation of caspases may even contribute to chemoresistance or resurgence of tumor growth. Thus, in order to completely leverage the apoptotic caspase pathways for therapeutic or preventative purposes both the mechanisms and full consequences of their activation need to be considered and their multiple critical roles in determining cell fate need to be fully understood. Great strides have been made in elucidating these biochemical pathways and will ensure that these challenges can be overcome to provide continued improvements in the development and application of new therapeutic strategies to prevent and cure cancer.
Highlights.
The caspase family of proteases are essential to initiate and execute apoptotic cell death.
Deregulation of caspase expression and activation contributes to cancer and resistance to cancer therapies.
Targeting apoptotic caspase pathways by gene therapy approaches or by targeting endogenous caspase inhibitors represents a promising therapeutic strategy for cancer treatment.
Many caspases have non-apoptotic functions that need to be considered when targeting caspase pathways in cancer.
Acknowledgements
We thank Alexandra Brown, Beatriz Bolivar, and Malcolm Brenner for careful reading of the manuscript and their helpful suggestions. Research in the Bouchier-Hayes laboratory is funded by a grant from the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM121389 and support from Texas Children’s Cancer and Hematology Centers.
Abbreviations
- AIF
apoptosis inducing factor
- ALL
acute lymphoblastic leukemia
- AML
acute myeloid leukemia
- APAF1
apoptotic peptidase activating factor 1
- ATM
ataxia-telangiectasia mutated
- BAK
BCL2-antagonist killer 1
- BAX
BCL2 associated X apoptosis regulator
- BCL
proapoptotic B-cell lymphoma
- BID
BH3-interacting domain death agonist
- BIR
baculovirus IAP repeat
- CAD
Caspase-activated DNase
- CARD
caspase recruitment domain
- c-FLIPL
cellular FLIP long
- c-FLIPS
cellular FLIP short
- cGAS
cyclic GMP-AMP synthase
- cIAP
cellular inhibitor of apoptosis protein
- CICD
caspase-independent cell death
- CID
chemical inducer of dimerization
- DD
death domain
- DED
death effector domain
- DIABLO
direct IAP binding protein with low pI
- DISC
death inducing signaling complex
- EndoG
endonuclease G
- FADD
fas-associated protein with death domain
- FLIP
FLICE (FADD-like IL-1β-converting enzyme)- inhibitory protein
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- GvHD
graft-versus-host disease
- HDAC
Histone deacetylase
- HER
human epidermal growth factor receptor
- HOIL1
heme-oxidized IRP2 ubiquitin ligase 1
- HOIP
HOIL1-interacting protein
- HtrA2
High temperature requirement protein A2
- IAP
inhibitor of apoptosis proteins
- iCasp9
inducible version of caspase-9
- IKKγ
inhibitor of nuclear factor kappa-B kinase subunit gamma
- iMOMP
incomplete MOMP
- iPL A2
calcium-independent phospholipase A2
- LUBAC
linear ubiquitin assembly complex
- MALT
Mucosa-associated lymphoid tissue
- MCL-1
myeloid leukemia cell differentiation protein
- MDM2
murine double minute 2
- MEF
mouse embryonic fibroblasts
- MLKL
mixed lineage kinase domain like pseudokinase
- MOMP
mitochondrial outer membrane permeabilization
- MSC
mesenchymal stromal cells
- NEMO
NF-kappa-B essential modulator
- NFκB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NIK
NFκB inducing kinase
- NSCLC
non-small-cell lung cancer
- PARP
poly-ADP ribose polymerase
- PIDD
p53-induced death domain protein
- RasGAP
Ras GTPase
- RhoGDI
Rho GDP-disassociation inhibitor
- RIPK1
receptor-interacting serine/threonine-protein kinase 1
- ROCK1
Rho-associated coiled coil containing protein kinase 1
- Sharpin
SHANK Associated RH Domain Interactor
- Smac
second mitochondria-derived activator of caspases
- STING
stimulator of interferon genes
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Martin SJ, Green DR, Protease activation during apoptosis: death by a thousand cuts?, Cell, 82 (1995) 349–352. [DOI] [PubMed] [Google Scholar]
- [2].Kerr JF, Wyllie AH, Currie AR, Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics, Br J Cancer, 26 (1972) 239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Arandjelovic S, Ravichandran KS, Phagocytosis of apoptotic cells in homeostasis, Nature immunology, 16 (2015) 907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].McIlwain DR, Berger T, Mak TW, Caspase functions in cell death and disease, Cold Spring Harb Perspect Biol, 5 (2013) a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Thompson CB, Apoptosis in the pathogenesis and treatment of disease, Science, 267 (1995) 1456–1462. [DOI] [PubMed] [Google Scholar]
- [6].Hanahan D, Weinberg RA, The hallmarks of cancer, Cell, 100 (2000) 57–70. [DOI] [PubMed] [Google Scholar]
- [7].Ellis HM, Horvitz HR, Genetic control of programmed cell death in the nematode C. elegans, Cell, 44 (1986) 817–829. [DOI] [PubMed] [Google Scholar]
- [8].Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR, The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme, Cell, 75 (1993) 641–652. [DOI] [PubMed] [Google Scholar]
- [9].Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J, Human ICE/CED-3 protease nomenclature, Cell, 87 (1996) 171. [DOI] [PubMed] [Google Scholar]
- [10].Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR, Salvesen GS, A unified model for apical caspase activation, Mol Cell, 11 (2003) 529–541. [DOI] [PubMed] [Google Scholar]
- [11].Walker NP, Talanian RV, Brady KD, Dang LC, Bump NJ, Ferenz CR, Franklin S, Ghayur T, Hackett MC, Hammill LD, et al. , Crystal structure of the cysteine protease interleukin-1 beta-converting enzyme: a (p20/p10)2 homodimer, Cell, 78 (1994) 343–352. [DOI] [PubMed] [Google Scholar]
- [12].Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, Chapman KT, Nicholson DW, A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis, J Biol Chem, 272 (1997) 17907–17911. [DOI] [PubMed] [Google Scholar]
- [13].Seaman JE, Julien O, Lee PS, Rettenmaier TJ, Thomsen ND, Wells JA, Cacidases: caspases can cleave after aspartate, glutamate and phosphoserine residues, Cell Death Differ, 23 (2016) 1717–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nicholson DW, Caspase structure, proteolytic substrates, and function during apoptotic cell death, Cell Death Differ, 6 (1999) 1028–1042. [DOI] [PubMed] [Google Scholar]
- [15].Graham RK, Ehrnhoefer DE, Hayden MR, Caspase-6 and neurodegeneration, Trends in neurosciences, 34 (2011) 646–656. [DOI] [PubMed] [Google Scholar]
- [16].Hofmann K, Bucher P, Tschopp J, The CARD domain: a new apoptotic signalling motif, Trends Biochem Sci, 22 (1997) 155–156. [DOI] [PubMed] [Google Scholar]
- [17].Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME, Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor, EMBO J, 14 (1995) 5579–5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pan G, O’Rourke K, Dixit VM, Caspase-9, Bcl-XL, and Apaf-1 form a ternary complex, J Biol Chem, 273 (1998) 5841–5845. [DOI] [PubMed] [Google Scholar]
- [19].Tinel A, Tschopp J, The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress, Science, 304 (2004) 843–846. [DOI] [PubMed] [Google Scholar]
- [20].Muzio M, Chinnaiyan AM, Kischkel FC, O’Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, Mann M, Krammer PH, Peter ME, Dixit VM, FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death--inducing signaling complex, Cell, 85 (1996) 817–827. [DOI] [PubMed] [Google Scholar]
- [21].Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X, Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade, Cell, 91 (1997) 479–489. [DOI] [PubMed] [Google Scholar]
- [22].Baliga BC, Read SH, Kumar S, The biochemical mechanism of caspase-2 activation, Cell Death Differ, 11 (2004) 1234–1241. [DOI] [PubMed] [Google Scholar]
- [23].Boatright KM, Deis C, Denault JB, Sutherlin DP, Salvesen GS, Activation of caspases-8 and −10 by FLIP(L), Biochem J, 382 (2004) 651–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Salvesen GS, Dixit VM, Caspase activation: the induced-proximity model, Proc Natl Acad Sci U S A, 96 (1999) 10964–10967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bolivar BE, Vogel TP, Bouchier-Hayes L, Inflammatory caspase regulation: maintaining balance between inflammation and cell death in health and disease, FEBS J, 286 (2019) 2628–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Stennicke HR, Renatus M, Meldal M, Salvesen GS, Internally quenched fluorescent peptide substrates disclose the subsite preferences of human caspases 1, 3, 6, 7 and 8, Biochem J, 350 Pt 2 (2000) 563–568. [PMC free article] [PubMed] [Google Scholar]
- [27].McStay GP, Salvesen GS, Green DR, Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways, Cell Death Differ, 15 (2008) 322–331. [DOI] [PubMed] [Google Scholar]
- [28].Cabukusta B, Nettebrock NT, Kol M, Hilderink A, Tafesse FG, Holthuis JCM, Ceramide phosphoethanolamine synthase SMSr is a target of caspase-6 during apoptotic cell death, Biosci Rep, 37 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Shahzidi S, Brech A, Sioud M, Li X, Suo Z, Nesland JM, Peng Q, Lamin A/C cleavage by caspase-6 activation is crucial for apoptotic induction by photodynamic therapy with hexaminolevulinate in human B-cell lymphoma cells, Cancer Lett, 339 (2013) 25–32. [DOI] [PubMed] [Google Scholar]
- [30].Walsh JG, Cullen SP, Sheridan C, Luthi AU, Gerner C, Martin SJ, Executioner caspase-3 and caspase-7 are functionally distinct proteases, Proc Natl Acad Sci U S A, 105 (2008) 12815–12819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Slee EA, Adrain C, Martin SJ, Executioner caspase-3, −6, and −7 perform distinct, non-redundant roles during the demolition phase of apoptosis, J Biol Chem, 276 (2001) 7320–7326. [DOI] [PubMed] [Google Scholar]
- [32].Schweizer A, Briand C, Grutter MG, Crystal structure of caspase-2, apical initiator of the intrinsic apoptotic pathway, J Biol Chem, 278 (2003) 42441–42447. [DOI] [PubMed] [Google Scholar]
- [33].Rotonda J, Nicholson DW, Fazil KM, Gallant M, Gareau Y, Labelle M, Peterson EP, Rasper DM, Ruel R, Vaillancourt JP, Thornberry NA, Becker JW, The three-dimensional structure of apopain/CPP32, a key mediator of apoptosis, Nature structural biology, 3 (1996) 619–625. [DOI] [PubMed] [Google Scholar]
- [34].Baumgartner R, Meder G, Briand C, Decock A, D’Arcy A, Hassiepen U, Morse R, Renatus M, The crystal structure of caspase-6, a selective effector of axonal degeneration, Biochem J, 423 (2009) 429–439. [DOI] [PubMed] [Google Scholar]
- [35].Riedl SJ, Fuentes-Prior P, Renatus M, Kairies N, Krapp S, Huber R, Salvesen GS, Bode W, Structural basis for the activation of human procaspase-7, Proc Natl Acad Sci U S A, 98 (2001) 14790–14795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Blanchard H, Kodandapani L, Mittl PR, Marco SD, Krebs JF, Wu JC, Tomaselli KJ, Grutter MG, The three-dimensional structure of caspase-8: an initiator enzyme in apoptosis, Structure, 7 (1999) 1125–1133. [DOI] [PubMed] [Google Scholar]
- [37].Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM, Alnemri ES, Fairman R, Shi Y, Mechanism of XIAP-mediated inhibition of caspase-9, Mol Cell, 11 (2003) 519–527. [DOI] [PubMed] [Google Scholar]
- [38].Ganesan R, Mittl PR, Jelakovic S, Grutter MG, Extended substrate recognition in caspase-3 revealed by high resolution X-ray structure analysis, J Mol Biol, 359 (2006) 1378–1388. [DOI] [PubMed] [Google Scholar]
- [39].Shi Y, Mechanisms of caspase activation and inhibition during apoptosis, Mol Cell, 9 (2002) 459–470. [DOI] [PubMed] [Google Scholar]
- [40].Thomsen ND, Koerber JT, Wells JA, Structural snapshots reveal distinct mechanisms of procaspase-3 and −7 activation, Proc Natl Acad Sci U S A, 110 (2013) 8477–8482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Darmon AJ, Nicholson DW, Bleackley RC, Activation of the apoptotic protease CPP32 by cytotoxic T-cell-derived granzyme B, Nature, 377 (1995) 446–448. [DOI] [PubMed] [Google Scholar]
- [42].Bose K, Pop C, Feeney B, Clark AC, An uncleavable procaspase-3 mutant has a lower catalytic efficiency but an active site similar to that of mature caspase-3, Biochemistry, 42 (2003) 12298–12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chai J, Wu Q, Shiozaki E, Srinivasula SM, Alnemri ES, Shi Y, Crystal structure of a procaspase-7 zymogen: mechanisms of activation and substrate binding, Cell, 107 (2001) 399–407. [DOI] [PubMed] [Google Scholar]
- [44].Stennicke HR, Deveraux QL, Humke EW, Reed JC, Dixit VM, Salvesen GS, Caspase-9 can be activated without proteolytic processing, J Biol Chem, 274 (1999) 8359–8362. [DOI] [PubMed] [Google Scholar]
- [45].Pop C, Timmer J, Sperandio S, Salvesen GS, The apoptosome activates caspase-9 by dimerization, Mol Cell, 22 (2006) 269–275. [DOI] [PubMed] [Google Scholar]
- [46].Adrain C, Slee EA, Harte MT, Martin SJ, Regulation of apoptotic protease activating factor-1 oligomerization and apoptosis by the WD-40 repeat region, J Biol Chem, 274 (1999) 20855–20860. [DOI] [PubMed] [Google Scholar]
- [47].Hu Y, Ding L, Spencer DM, Nunez G, WD-40 repeat region regulates Apaf-1 self-association and procaspase-9 activation, J Biol Chem, 273 (1998) 33489–33494. [DOI] [PubMed] [Google Scholar]
- [48].Bratton SB, Walker G, Srinivasula SM, Sun XM, Butterworth M, Alnemri ES, Cohen GM, Recruitment, activation and retention of caspases-9 and −3 by Apaf-1 apoptosome and associated XIAP complexes, EMBO J, 20 (2001) 998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Malladi S, Challa-Malladi M, Fearnhead HO, Bratton SB, The Apaf-1*procaspase-9 apoptosome complex functions as a proteolytic-based molecular timer, EMBO J, 28 (2009) 1916–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Micheau O, Tschopp J, Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes, Cell, 114 (2003) 181–190. [DOI] [PubMed] [Google Scholar]
- [51].Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG, Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis, Nat Cell Biol, 16 (2014) 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Fu TM, Li Y, Lu A, Li Z, Vajjhala PR, Cruz AC, Srivastava DB, DiMaio F, Penczek PA, Siegel RM, Stacey KJ, Egelman EH, Wu H, Cryo-EM Structure of Caspase-8 Tandem DED Filament Reveals Assembly and Regulation Mechanisms of the Death-Inducing Signaling Complex, Mol Cell, 64 (2016) 236–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Schleich K, Warnken U, Fricker N, Ozturk S, Richter P, Kammerer K, Schnolzer M, Krammer PH, Lavrik IN, Stoichiometry of the CD95 death-inducing signaling complex: experimental and modeling evidence for a death effector domain chain model, Mol Cell, 47 (2012) 306–319. [DOI] [PubMed] [Google Scholar]
- [54].Dickens LS, Boyd RS, Jukes-Jones R, Hughes MA, Robinson GL, Fairall L, Schwabe JW, Cain K, Macfarlane M, A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death, Mol Cell, 47 (2012) 291–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Guo Y, Srinivasula SM, Druilhe A, Fernandes-Alnemri T, Alnemri ES, Caspase-2 induces apoptosis by releasing proapoptotic proteins from mitochondria, J Biol Chem, 277 (2002) 13430–13437. [DOI] [PubMed] [Google Scholar]
- [56].Li H, Zhu H, Xu CJ, Yuan J, Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis, Cell, 94 (1998) 491–501. [DOI] [PubMed] [Google Scholar]
- [57].Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME, Two CD95 (APO-1/Fas) signaling pathways, EMBO J, 17 (1998) 1675–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Deveraux QL, Takahashi R, Salvesen GS, Reed JC, X-linked IAP is a direct inhibitor of cell-death proteases, Nature, 388 (1997) 300–304. [DOI] [PubMed] [Google Scholar]
- [59].Roy N, Deveraux QL, Takahashi R, Salvesen GS, Reed JC, The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases, EMBO J, 16 (1997) 6914–6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Eckelman BP, Salvesen GS, The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases, J Biol Chem, 281 (2006) 3254–3260. [DOI] [PubMed] [Google Scholar]
- [61].Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, Liddington RC, Salvesen GS, Structural basis for the inhibition of caspase-3 by XIAP, Cell, 104 (2001) 791–800. [DOI] [PubMed] [Google Scholar]
- [62].Chai J, Shiozaki E, Srinivasula SM, Wu Q, Datta P, Alnemri ES, Shi Y, Structural basis of caspase-7 inhibition by XIAP, Cell, 104 (2001) 769–780. [DOI] [PubMed] [Google Scholar]
- [63].Suzuki Y, Nakabayashi Y, Takahashi R, Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death, Proc Natl Acad Sci U S A, 98 (2001) 8662–8667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Du C, Fang M, Li Y, Li L, Wang X, Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition, Cell, 102 (2000) 33–42. [DOI] [PubMed] [Google Scholar]
- [65].Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL, Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins, Cell, 102 (2000) 43–53. [DOI] [PubMed] [Google Scholar]
- [66].Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi R, A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death, Mol Cell, 8 (2001) 613–621. [DOI] [PubMed] [Google Scholar]
- [67].Martins LM, Iaccarino I, Tenev T, Gschmeissner S, Totty NF, Lemoine NR, Savopoulos J, Gray CW, Creasy CL, Dingwall C, Downward J, The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif, J Biol Chem, 277 (2002) 439–444. [DOI] [PubMed] [Google Scholar]
- [68].Chai J, Du C, Wu JW, Kyin S, Wang X, Shi Y, Structural and biochemical basis of apoptotic activation by Smac/DIABLO, Nature, 406 (2000) 855–862. [DOI] [PubMed] [Google Scholar]
- [69].Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, Lee RA, Robbins PD, Fernandes-Alnemri T, Shi Y, Alnemri ES, A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis, Nature, 410 (2001) 112–116. [DOI] [PubMed] [Google Scholar]
- [70].Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, Huang DC, Bouillet P, Thomas HE, Borner C, Silke J, Strasser A, Kaufmann T, XIAP discriminates between type I and type II FAS-induced apoptosis, Nature, 460 (2009) 1035–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Rothe M, Pan MG, Henzel WJ, Ayres TM, Goeddel DV, The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins, Cell, 83 (1995) 1243–1252. [DOI] [PubMed] [Google Scholar]
- [72].Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, Kensche T, Uejima T, Bloor S, Komander D, Randow F, Wakatsuki S, Dikic I, Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation, Cell, 136 (2009) 1098–1109. [DOI] [PubMed] [Google Scholar]
- [73].Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, Feltham R, Vince J, Warnken U, Wenger T, Koschny R, Komander D, Silke J, Walczak H, Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction, Mol Cell, 36 (2009) 831–844. [DOI] [PubMed] [Google Scholar]
- [74].Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, Shiba T, Yang X, Yeh WC, Mak TW, Korneluk RG, Cheng G, Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK, Nature immunology, 9 (2008) 1371–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Krueger A, Schmitz I, Baumann S, Krammer PH, Kirchhoff S, Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex, J Biol Chem, 276 (2001) 20633–20640. [DOI] [PubMed] [Google Scholar]
- [76].Golks A, Brenner D, Fritsch C, Krammer PH, Lavrik IN, c-FLIPR, a new regulator of death receptor-induced apoptosis, J Biol Chem, 280 (2005) 14507–14513. [DOI] [PubMed] [Google Scholar]
- [77].Hughes MA, Powley IR, Jukes-Jones R, Horn S, Feoktistova M, Fairall L, Schwabe JW, Leverkus M, Cain K, MacFarlane M, Co-operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate, Mol Cell, 61 (2016) 834–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Yu JW, Jeffrey PD, Shi Y, Mechanism of procaspase-8 activation by c-FLIPL, Proc Natl Acad Sci U S A, 106 (2009) 8169–8174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Pop C, Oberst A, Drag M, Van Raam BJ, Riedl SJ, Green DR, Salvesen GS, FLIP(L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity, Biochem J, 433 (2011) 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR, Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis, Nature, 471 (2011) 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Hacker G, Leverkus M, cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms, Mol Cell, 43 (2011) 449–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Hillert LK, Ivanisenko NV, Espe J, Konig C, Ivanisenko VA, Kahne T, Lavrik IN, Long and short isoforms of c-FLIP act as control checkpoints of DED filament assembly, Oncogene, (2019). [DOI] [PubMed] [Google Scholar]
- [83].Schleich K, Buchbinder JH, Pietkiewicz S, Kahne T, Warnken U, Ozturk S, Schnolzer M, Naumann M, Krammer PH, Lavrik IN, Molecular architecture of the DED chains at the DISC: regulation of procaspase-8 activation by short DED proteins c-FLIP and procaspase-8 prodomain, Cell Death Differ, 23 (2016) 681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Chang DW, Xing Z, Pan Y, Algeciras-Schimnich A, Barnhart BC, Yaish-Ohad S, Peter ME, Yang X, c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis, EMBO J, 21 (2002) 3704–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Fricker N, Beaudouin J, Richter P, Eils R, Krammer PH, Lavrik IN, Model-based dissection of CD95 signaling dynamics reveals both a pro- and antiapoptotic role of c-FLIPL, J Cell Biol, 190 (2010) 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Fernandes-Alnemri T, Armstrong RC, Krebs J, Srinivasula SM, Wang L, Bullrich F, Fritz LC, Trapani JA, Tomaselli KJ, Litwack G, Alnemri ES, In vitro activation of CPP32 and Mch3 by Mch4, a novel human apoptotic cysteine protease containing two FADD-like domains, Proc Natl Acad Sci U S A, 93 (1996) 7464–7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Horn S, Hughes MA, Schilling R, Sticht C, Tenev T, Ploesser M, Meier P, Sprick MR, MacFarlane M, Leverkus M, Caspase-10 Negatively Regulates Caspase-8-Mediated Cell Death, Switching the Response to CD95L in Favor of NF-kappaB Activation and Cell Survival, Cell reports, 19 (2017) 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Li X, Pan X, Zhang H, Lei D, Liu D, Xu F, Luan X, Overexpression of cFLIP in head and neck squamous cell carcinoma and its clinicopathologic correlations, Journal of cancer research and clinical oncology, 134 (2008) 609–615. [DOI] [PubMed] [Google Scholar]
- [89].Riley JS, Hutchinson R, McArt DG, Crawford N, Holohan C, Paul I, Van Schaeybroeck S, Salto-Tellez M, Johnston PG, Fennell DA, Gately K, O’Byrne K, Cummins R, Kay E, Hamilton P, Stasik I, Longley DB, Prognostic and therapeutic relevance of FLIP and procaspase-8 overexpression in non-small cell lung cancer, Cell Death Dis, 4 (2013) e951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Valente G, Manfroi F, Peracchio C, Nicotra G, Castino R, Nicosia G, Kerim S, Isidoro C, cFLIP expression correlates with tumour progression and patient outcome in non-Hodgkin lymphomas of low grade of malignancy, British journal of haematology, 132 (2006) 560–570. [DOI] [PubMed] [Google Scholar]
- [91].Valnet-Rabier MB, Challier B, Thiebault S, Angonin R, Margueritte G, Mougin C, Kantelip B, Deconinck E, Cahn JY, Fest T, c-Flip protein expression in Burkitt’s lymphomas is associated with a poor clinical outcome, British journal of haematology, 128 (2005) 767–773. [DOI] [PubMed] [Google Scholar]
- [92].McLornan D, Hay J, McLaughlin K, Holohan C, Burnett AK, Hills RK, Johnston PG, Mills KI, McMullin MF, Longley DB, Gilkes A, Prognostic and therapeutic relevance of c-FLIP in acute myeloid leukaemia, British journal of haematology, 160 (2013) 188–198. [DOI] [PubMed] [Google Scholar]
- [93].Ullenhag GJ, Mukherjee A, Watson NF, Al-Attar AH, Scholefield JH, Durrant LG, Overexpression of FLIPL is an independent marker of poor prognosis in colorectal cancer patients, Clin Cancer Res, 13 (2007) 5070–5075. [DOI] [PubMed] [Google Scholar]
- [94].Zhou XD, Yu JP, Liu J, Luo HS, Chen HX, Yu HG, Overexpression of cellular FLICE-inhibitory protein (FLIP) in gastric adenocarcinoma, Clinical science (London, England : 1979), 106 (2004) 397–405. [DOI] [PubMed] [Google Scholar]
- [95].Obexer P, Ausserlechner MJ, X-linked inhibitor of apoptosis protein - a critical death resistance regulator and therapeutic target for personalized cancer therapy, Frontiers in oncology, 4 (2014) 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Tamm I, Kornblau SM, Segall H, Krajewski S, Welsh K, Kitada S, Scudiero DA, Tudor G, Qui YH, Monks A, Andreeff M, Reed JC, Expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias, Clin Cancer Res, 6 (2000) 1796–1803. [PubMed] [Google Scholar]
- [97].Canman CE, Kastan MB, Induction of apoptosis by tumor suppressor genes and oncogenes, Seminars in cancer biology, 6 (1995) 17–25. [DOI] [PubMed] [Google Scholar]
- [98].Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW, Apaf1 is required for mitochondrial pathways of apoptosis and brain development, Cell, 94 (1998) 739–750. [DOI] [PubMed] [Google Scholar]
- [99].Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA, Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9, Cell, 94 (1998) 325–337. [DOI] [PubMed] [Google Scholar]
- [100].Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA, Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice, Nature, 384 (1996) 368–372. [DOI] [PubMed] [Google Scholar]
- [101].Leonard JR, Klocke BJ, D’Sa C, Flavell RA, Roth KA, Strain-dependent neurodevelopmental abnormalities in caspase-3-deficient mice, Journal of neuropathology and experimental neurology, 61 (2002) 673–677. [DOI] [PubMed] [Google Scholar]
- [102].Zheng TS, Hunot S, Kuida K, Momoi T, Srinivasan A, Nicholson DW, Lazebnik Y, Flavell RA, Deficiency in caspase-9 or caspase-3 induces compensatory caspase activation, Nat Med, 6 (2000) 1241–1247. [DOI] [PubMed] [Google Scholar]
- [103].Lakhani SA, Masud A, Kuida K, Porter GA Jr., Booth CJ, Mehal WZ, Inayat I, Flavell RA, Caspases 3 and 7: key mediators of mitochondrial events of apoptosis, Science, 311 (2006) 847–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, Lapidot T, Soffer D, Sobe T, Avraham KB, Goncharov T, Holtmann H, Lonai P, Wallach D, Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally, Immunity, 9 (1998) 267–276. [DOI] [PubMed] [Google Scholar]
- [105].Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES, RIP3 mediates the embryonic lethality of caspase-8-deficient mice, Nature, 471 (2011) 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A, Varmuza S, Latham KE, Flaws JA, Salter JC, Hara H, Moskowitz MA, Li E, Greenberg A, Tilly JL, Yuan J, Defects in regulation of apoptosis in caspase-2-deficient mice, Genes Dev, 12 (1998) 1304–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].O’Reilly LA, Ekert P, Harvey N, Marsden V, Cullen L, Vaux DL, Hacker G, Magnusson C, Pakusch M, Cecconi F, Kuida K, Strasser A, Huang DC, Kumar S, Caspase-2 is not required for thymocyte or neuronal apoptosis even though cleavage of caspase-2 is dependent on both Apaf-1 and caspase-9, Cell Death Differ, 9 (2002) 832–841. [DOI] [PubMed] [Google Scholar]
- [108].Ho LH, Taylor R, Dorstyn L, Cakouros D, Bouillet P, Kumar S, A tumor suppressor function for caspase-2, Proc Natl Acad Sci U S A, 106 (2009) 5336–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Puccini J, Shalini S, Voss AK, Gatei M, Wilson CH, Hiwase DK, Lavin MF, Dorstyn L, Kumar S, Loss of caspase-2 augments lymphomagenesis and enhances genomic instability in Atm-deficient mice, Proc Natl Acad Sci U S A, 110 (2013) 19920–19925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Parsons MJ, McCormick L, Janke L, Howard A, Bouchier-Hayes L, Green DR, Genetic deletion of caspase-2 accelerates MMTV/c-neu-driven mammary carcinogenesis in mice, Cell Death Differ, 20 (2013) 1174–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Terry MR, Arya R, Mukhopadhyay A, Berrett KC, Clair PM, Witt B, Salama ME, Bhutkar A, Oliver TG, Caspase-2 impacts lung tumorigenesis and chemotherapy response in vivo, Cell Death Differ, 22 (2015) 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Shalini S, Nikolic A, Wilson CH, Puccini J, Sladojevic N, Finnie J, Dorstyn L, Kumar S, Caspase-2 deficiency accelerates chemically induced liver cancer in mice, Cell Death Differ, 23 (2016) 1727–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Manzl C, Peintner L, Krumschnabel G, Bock F, Labi V, Drach M, Newbold A, Johnstone R, Villunger A, PIDDosome-independent tumor suppression by Caspase-2, Cell Death Differ, 19 (2012) 1722–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Dorstyn L, Puccini J, Nikolic A, Shalini S, Wilson CH, Norris MD, Haber M, Kumar S, An unexpected role for caspase-2 in neuroblastoma, Cell Death Dis, 5 (2014) e1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA, Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme, Science, 267 (1995) 2000–2003. [DOI] [PubMed] [Google Scholar]
- [116].Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, Eisenbarth SC, Flavell RA, Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4, Proc Natl Acad Sci U S A, 107 (2010) 21635–21640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Hakem A, El Ghamrasni S, Maire G, Lemmers B, Karaskova J, Jurisicova A, Sanchez O, Squire J, Hakem R, Caspase-8 is essential for maintaining chromosomal stability and suppressing B-cell lymphomagenesis, Blood, 119 (2012) 3495–3502. [DOI] [PubMed] [Google Scholar]
- [118].Teitz T, Inoue M, Valentine MB, Zhu K, Rehg JE, Zhao W, Finkelstein D, Wang YD, Johnson MD, Calabrese C, Rubinstein M, Hakem R, Weiss WA, Lahti JM, Th-MYCN mice with caspase-8 deficiency develop advanced neuroblastoma with bone marrow metastasis, Cancer Res, 73 (2013) 4086–4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Fulda S, Poremba C, Berwanger B, Hacker S, Eilers M, Christiansen H, Hero B, Debatin KM, Loss of caspase-8 expression does not correlate with MYCN amplification, aggressive disease, or prognosis in neuroblastoma, Cancer Res, 66 (2006) 10016–10023. [DOI] [PubMed] [Google Scholar]
- [120].Teitz T, Wei T, Valentine MB, Vanin EF, Grenet J, Valentine VA, Behm FG, Look AT, Lahti JM, Kidd VJ, Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN, Nat Med, 6 (2000) 529–535. [DOI] [PubMed] [Google Scholar]
- [121].Fianco G, Mongiardi MP, Levi A, De Luca T, Desideri M, Trisciuoglio D, Del Bufalo D, Cina I, Di Benedetto A, Mottolese M, Gentile A, Centonze D, Ferre F, Barila D, Caspase-8 contributes to angiogenesis and chemotherapy resistance in glioblastoma, eLife, 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Holleman A, den Boer ML, Kazemier KM, Beverloo HB, von Bergh AR, Janka-Schaub GE, Pieters R, Decreased PARP and procaspase-2 protein levels are associated with cellular drug resistance in childhood acute lymphoblastic leukemia, Blood, 106 (2005) 1817–1823. [DOI] [PubMed] [Google Scholar]
- [123].Dawar S, Shahrin NH, Sladojevic N, D’Andrea RJ, Dorstyn L, Hiwase DK, Kumar S, Impaired haematopoietic stem cell differentiation and enhanced skewing towards myeloid progenitors in aged caspase-2-deficient mice, Cell Death Dis, 7 (2016) e2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Estrov Z, Thall PF, Talpaz M, Estey EH, Kantarjian HM, Andreeff M, Harris D, Van Q, Walterscheid M, Kornblau SM, Caspase 2 and caspase 3 protein levels as predictors of survival in acute myelogenous leukemia, Blood, 92 (1998) 3090–3097. [PubMed] [Google Scholar]
- [125].Faderl S, Thall PF, Kantarjian HM, Talpaz M, Harris D, Van Q, Beran M, Kornblau SM, Pierce S, Estrov Z, Caspase 2 and caspase 3 as predictors of complete remission and survival in adults with acute lymphoblastic leukemia, Clin Cancer Res, 5 (1999) 4041–4047. [PubMed] [Google Scholar]
- [126].Christensen BC, Marsit CJ, Houseman EA, Godleski JJ, Longacker JL, Zheng S, Yeh RF, Wrensch MR, Wiemels JL, Karagas MR, Bueno R, Sugarbaker DJ, Nelson HH, Wiencke JK, Kelsey KT, Differentiation of lung adenocarcinoma, pleural mesothelioma, and nonmalignant pulmonary tissues using DNA methylation profiles, Cancer Res, 69 (2009) 6315–6321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Devarajan E, Sahin AA, Chen JS, Krishnamurthy RR, Aggarwal N, Brun AM, Sapino A, Zhang F, Sharma D, Yang XH, Tora AD, Mehta K, Down-regulation of caspase 3 in breast cancer: a possible mechanism for chemoresistance, Oncogene, 21 (2002) 8843–8851. [DOI] [PubMed] [Google Scholar]
- [128].Pu X, Storr SJ, Zhang Y, Rakha EA, Green AR, Ellis IO, Martin SG, Caspase-3 and caspase-8 expression in breast cancer: caspase-3 is associated with survival, Apoptosis, 22 (2017) 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Hu Q, Peng J, Liu W, He X, Cui L, Chen X, Yang M, Liu H, Liu S, Wang H, Elevated cleaved caspase-3 is associated with shortened overall survival in several cancer types, Int J Clin Exp Pathol, 7 (2014) 5057–5070. [PMC free article] [PubMed] [Google Scholar]
- [130].Liu PF, Hu YC, Kang BH, Tseng YK, Wu PC, Liang CC, Hou YY, Fu TY, Liou HH, Hsieh IC, Ger LP, Shu CW, Expression levels of cleaved caspase-3 and caspase-3 in tumorigenesis and prognosis of oral tongue squamous cell carcinoma, PLoS One, 12 (2017) e0180620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, Chin W, Jones J, Woodward A, Le T, Smith C, Smolak P, Goodwin RG, Rauch CT, Schuh JC, Lynch DH, Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo, Nat Med, 5 (1999) 157–163. [DOI] [PubMed] [Google Scholar]
- [132].Humphreys L, Espona-Fiedler M, Longley DB, FLIP as a therapeutic target in cancer, FEBS J, 285 (2018) 4104–4123. [DOI] [PubMed] [Google Scholar]
- [133].Ogasawara J, Watanabe-Fukunaga R, Adachi M, Matsuzawa A, Kasugai T, Kitamura Y, Itoh N, Suda T, Nagata S, Lethal effect of the anti-Fas antibody in mice, Nature, 364 (1993) 806–809. [DOI] [PubMed] [Google Scholar]
- [134].Pan G, O’Rourke K, Chinnaiyan AM, Gentz R, Ebner R, Ni J, Dixit VM, The receptor for the cytotoxic ligand TRAIL, Science, 276 (1997) 111–113. [DOI] [PubMed] [Google Scholar]
- [135].Walczak H, Degli-Esposti MA, Johnson RS, Smolak PJ, Waugh JY, Boiani N, Timour MS, Gerhart MJ, Schooley KA, Smith CA, Goodwin RG, Rauch CT, TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL, EMBO J, 16 (1997) 5386–5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D, Ramakrishnan L, Gray CL, Baker K, Wood WI, Goddard AD, Godowski P, Ashkenazi A, Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors, Science, 277 (1997) 818–821. [DOI] [PubMed] [Google Scholar]
- [137].Sanlioglu AD, Dirice E, Aydin C, Erin N, Koksoy S, Sanlioglu S, Surface TRAIL decoy receptor-4 expression is correlated with TRAIL resistance in MCF7 breast cancer cells, BMC Cancer, 5 (2005) 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Lincz LF, Yeh TX, Spencer A, TRAIL-induced eradication of primary tumour cells from multiple myeloma patient bone marrows is not related to TRAIL receptor expression or prior chemotherapy, Leukemia, 15 (2001) 1650–1657. [DOI] [PubMed] [Google Scholar]
- [139].Lemke J, von Karstedt S, Zinngrebe J, Walczak H, Getting TRAIL back on track for cancer therapy, Cell Death Differ, 21 (2014) 1350–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].MacFarlane M, Harper N, Snowden RT, Dyer MJ, Barnett GA, Pringle JH, Cohen GM, Mechanisms of resistance to TRAIL-induced apoptosis in primary B cell chronic lymphocytic leukaemia, Oncogene, 21 (2002) 6809–6818. [DOI] [PubMed] [Google Scholar]
- [141].Finlay D, Vamos M, Gonzalez-Lopez M, Ardecky RJ, Ganji SR, Yuan H, Su Y, Cooley TR, Hauser CT, Welsh K, Reed JC, Cosford ND, Vuori K, Small-molecule IAP antagonists sensitize cancer cells to TRAIL-induced apoptosis: roles of XIAP and cIAPs, Mol Cancer Ther, 13 (2014) 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Hinz S, Trauzold A, Boenicke L, Sandberg C, Beckmann S, Bayer E, Walczak H, Kalthoff H, Ungefroren H, Bcl-XL protects pancreatic adenocarcinoma cells against CD95- and TRAIL-receptor-mediated apoptosis, Oncogene, 19 (2000) 5477–5486. [DOI] [PubMed] [Google Scholar]
- [143].Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJ, Flygare JA, Fairbrother WJ, Deshayes K, Dixit VM, Vucic D, IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis, Cell, 131 (2007) 669–681. [DOI] [PubMed] [Google Scholar]
- [144].Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, Benetatos CA, Chunduru SK, Condon SM, McKinlay M, Brink R, Leverkus M, Tergaonkar V, Schneider P, Callus BA, Koentgen F, Vaux DL, Silke J, IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis, Cell, 131 (2007) 682–693. [DOI] [PubMed] [Google Scholar]
- [145].Huang Y, Rich RL, Myszka DG, Wu H, Requirement of both the second and third BIR domains for the relief of X-linked inhibitor of apoptosis protein (XIAP)-mediated caspase inhibition by Smac, J Biol Chem, 278 (2003) 49517–49522. [DOI] [PubMed] [Google Scholar]
- [146].Liu Z, Sun C, Olejniczak ET, Meadows RP, Betz SF, Oost T, Herrmann J, Wu JC, Fesik SW, Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain, Nature, 408 (2000) 1004–1008. [DOI] [PubMed] [Google Scholar]
- [147].Dueber EC, Schoeffler AJ, Lingel A, Elliott JM, Fedorova AV, Giannetti AM, Zobel K, Maurer B, Varfolomeev E, Wu P, Wallweber HJ, Hymowitz SG, Deshayes K, Vucic D, Fairbrother WJ, Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination, Science, 334 (2011) 376–380. [DOI] [PubMed] [Google Scholar]
- [148].Probst BL, Liu L, Ramesh V, Li L, Sun H, Minna JD, Wang L, Smac mimetics increase cancer cell response to chemotherapeutics in a TNF-alpha-dependent manner, Cell Death Differ, 17 (2010) 1645–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Cai Q, Sun H, Peng Y, Lu J, Nikolovska-Coleska Z, McEachern D, Liu L, Qiu S, Yang CY, Miller R, Yi H, Zhang T, Sun D, Kang S, Guo M, Leopold L, Yang D, Wang S, A potent and orally active antagonist (SM-406/AT-406) of multiple inhibitor of apoptosis proteins (IAPs) in clinical development for cancer treatment, J Med Chem, 54 (2011) 2714–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Hennessy EJ, Adam A, Aquila BM, Castriotta LM, Cook D, Hattersley M, Hird AW, Huntington C, Kamhi VM, Laing NM, Li D, MacIntyre T, Omer CA, Oza V, Patterson T, Repik G, Rooney MT, Saeh JC, Sha L, Vasbinder MM, Wang H, Whitston D, Discovery of a novel class of dimeric Smac mimetics as potent IAP antagonists resulting in a clinical candidate for the treatment of cancer (AZD5582), J Med Chem, 56 (2013) 9897–9919. [DOI] [PubMed] [Google Scholar]
- [151].Amaravadi RK, Schilder RJ, Martin LP, Levin M, Graham MA, Weng DE, Adjei AA, A Phase I Study of the SMAC-Mimetic Birinapant in Adults with Refractory Solid Tumors or Lymphoma, Mol Cancer Ther, 14 (2015) 2569–2575. [DOI] [PubMed] [Google Scholar]
- [152].Infante JR, Dees EC, Olszanski AJ, Dhuria SV, Sen S, Cameron S, Cohen RB, Phase I dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors, Journal of clinical oncology : official journal of the American Society of Clinical Oncology, 32 (2014) 3103–3110. [DOI] [PubMed] [Google Scholar]
- [153].Hurwitz HI, Smith DC, Pitot HC, Brill JM, Chugh R, Rouits E, Rubin J, Strickler J, Vuagniaux G, Sorensen JM, Zanna C, Safety, pharmacokinetics, and pharmacodynamic properties of oral DEBIO1143 (AT-406) in patients with advanced cancer: results of a first-in-man study, Cancer chemotherapy and pharmacology, 75 (2015) 851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Noonan AM, Bunch KP, Chen JQ, Herrmann MA, Lee JM, Kohn EC, O’Sullivan CC, Jordan E, Houston N, Takebe N, Kinders RJ, Cao L, Peer CJ, Figg WD, Annunziata CM, Pharmacodynamic markers and clinical results from the phase 2 study of the SMAC mimetic birinapant in women with relapsed platinum-resistant or -refractory epithelial ovarian cancer, Cancer, 122 (2016) 588–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Petersen SL, Wang L, Yalcin-Chin A, Li L, Peyton M, Minna J, Harran P, Wang X, Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis, Cancer cell, 12 (2007) 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Fakler M, Loeder S, Vogler M, Schneider K, Jeremias I, Debatin KM, Fulda S, Small molecule XIAP inhibitors cooperate with TRAIL to induce apoptosis in childhood acute leukemia cells and overcome Bcl-2-mediated resistance, Blood, 113 (2009) 1710–1722. [DOI] [PubMed] [Google Scholar]
- [157].Geserick P, Hupe M, Moulin M, Wong WW, Feoktistova M, Kellert B, Gollnick H, Silke J, Leverkus M, Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment, J Cell Biol, 187 (2009) 1037–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, Arora V, Mak TW, Lacasse EC, Waring J, Korneluk RG, Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation, Proc Natl Acad Sci U S A, 105 (2008) 11778–11783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Petersen SL, Peyton M, Minna JD, Wang X, Overcoming cancer cell resistance to Smac mimetic induced apoptosis by modulating cIAP-2 expression, Proc Natl Acad Sci U S A, 107 (2010) 11936–11941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Darding M, Feltham R, Tenev T, Bianchi K, Benetatos C, Silke J, Meier P, Molecular determinants of Smac mimetic induced degradation of cIAP1 and cIAP2, Cell Death Differ, 18 (2011) 1376–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Varfolomeev E, Wayson SM, Dixit VM, Fairbrother WJ, Vucic D, The inhibitor of apoptosis protein fusion c-IAP2.MALT1 stimulates NF-kappaB activation independently of TRAF1 AND TRAF2, J Biol Chem, 281 (2006) 29022–29029. [DOI] [PubMed] [Google Scholar]
- [162].Poukkula M, Kaunisto A, Hietakangas V, Denessiouk K, Katajamaki T, Johnson MS, Sistonen L, Eriksson JE, Rapid turnover of c-FLIPshort is determined by its unique C-terminal tail, J Biol Chem, 280 (2005) 27345–27355. [DOI] [PubMed] [Google Scholar]
- [163].Abedini MR, Muller EJ, Brun J, Bergeron R, Gray DA, Tsang BK, Cisplatin induces p53-dependent FLICE-like inhibitory protein ubiquitination in ovarian cancer cells, Cancer Res, 68 (2008) 4511–4517. [DOI] [PubMed] [Google Scholar]
- [164].Song JH, Song DK, Herlyn M, Petruk KC, Hao C, Cisplatin down-regulation of cellular Fas-associated death domain-like interleukin-1beta-converting enzyme-like inhibitory proteins to restore tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human melanoma cells, Clin Cancer Res, 9 (2003) 4255–4266. [PubMed] [Google Scholar]
- [165].Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, Liu YC, Karin M, The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover, Cell, 124 (2006) 601–613. [DOI] [PubMed] [Google Scholar]
- [166].Venza I, Visalli M, Oteri R, Teti D, Venza M, Class I-specific histone deacetylase inhibitor MS-275 overrides TRAIL-resistance in melanoma cells by downregulating c-FLIP, International immunopharmacology, 21 (2014) 439–446. [DOI] [PubMed] [Google Scholar]
- [167].Kerr E, Holohan C, McLaughlin KM, Majkut J, Dolan S, Redmond K, Riley J, McLaughlin K, Stasik I, Crudden M, Van Schaeybroeck S, Fenning C, O’Connor R, Kiely P, Sgobba M, Haigh D, Johnston PG, Longley DB, Identification of an acetylation-dependant Ku70/FLIP complex that regulates FLIP expression and HDAC inhibitor-induced apoptosis, Cell Death Differ, 19 (2012) 1317–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Panka DJ, Mano T, Suhara T, Walsh K, Mier JW, Phosphatidylinositol 3-kinase/Akt activity regulates c-FLIP expression in tumor cells, J Biol Chem, 276 (2001) 6893–6896. [DOI] [PubMed] [Google Scholar]
- [169].Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J, NF-kappaB signals induce the expression of c-FLIP, Mol Cell Biol, 21 (2001) 5299–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Jani TS, DeVecchio J, Mazumdar T, Agyeman A, Houghton JA, Inhibition of NF-kappaB signaling by quinacrine is cytotoxic to human colon carcinoma cell lines and is synergistic in combination with tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) or oxaliplatin, J Biol Chem, 285 (2010) 19162–19172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Abedini MR, Muller EJ, Bergeron R, Gray DA, Tsang BK, Akt promotes chemoresistance in human ovarian cancer cells by modulating cisplatin-induced, p53-dependent ubiquitination of FLICE-like inhibitory protein, Oncogene, 29 (2010) 11–25. [DOI] [PubMed] [Google Scholar]
- [172].Morle A, Garrido C, Micheau O, Hyperthermia restores apoptosis induced by death receptors through aggregation-induced c-FLIP cytosolic depletion, Cell Death Dis, 6 (2015) e1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, Liu H, Cruz CR, Savoldo B, Gee AP, Schindler J, Krance RA, Heslop HE, Spencer DM, Rooney CM, Brenner MK, Inducible apoptosis as a safety switch for adoptive cell therapy, N Engl J Med, 365 (2011) 1673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Zhou X, Di Stasi A, Tey SK, Krance RA, Martinez C, Leung KS, Durett AG, Wu MF, Liu H, Leen AM, Savoldo B, Lin YF, Grilley BJ, Gee AP, Spencer DM, Rooney CM, Heslop HE, Brenner MK, Dotti G, Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene, Blood, 123 (2014) 3895–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Yagyu S, Hoyos V, Del Bufalo F, Brenner MK, An Inducible Caspase-9 Suicide Gene to Improve the Safety of Therapy Using Human Induced Pluripotent Stem Cells, Mol Ther, 23 (2015) 1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Ramos CA, Asgari Z, Liu E, Yvon E, Heslop HE, Rooney CM, Brenner MK, Dotti G, An inducible caspase 9 suicide gene to improve the safety of mesenchymal stromal cell therapies, Stem Cells, 28 (2010) 1107–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Ando M, Hoyos V, Yagyu S, Tao W, Ramos CA, Dotti G, Brenner MK, Bouchier-Hayes L, Bortezomib sensitizes non-small cell lung cancer to mesenchymal stromal cell-delivered inducible caspase-9-mediated cytotoxicity, Cancer Gene Ther, 21 (2014) 472–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Hoyos V, Del Bufalo F, Yagyu S, Ando M, Dotti G, Suzuki M, Bouchier-Hayes L, Alemany R, Brenner MK, Mesenchymal Stromal Cells for Linked Delivery of Oncolytic and Apoptotic Adenoviruses to Non-small-cell Lung Cancers, Mol Ther, 23 (2015) 1497–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Khan N, Bammidi S, Jayandharan GR, A CD33 Antigen-Targeted AAV6 Vector Expressing an Inducible Caspase-9 Suicide Gene Is Therapeutic in a Xenotransplantation Model of Acute Myeloid Leukemia, Bioconjug Chem, 30 (2019) 2404–2416. [DOI] [PubMed] [Google Scholar]
- [180].Khan N, Bammidi S, Chattopadhyay S, Jayandharan GR, Combination Suicide Gene Delivery with an Adeno-Associated Virus Vector Encoding Inducible Caspase-9 and a Chemical Inducer of Dimerization Is Effective in a Xenotransplantation Model of Hepatocellular Carcinoma, Bioconjug Chem, 30 (2019) 1754–1762. [DOI] [PubMed] [Google Scholar]
- [181].Zhang DX, Zhao PT, Xia L, Liu LL, Liang J, Zhai HH, Zhang HB, Guo XG, Wu KC, Xu YM, Jia LT, Yang AG, Chen SY, Fan DM, Potent inhibition of human gastric cancer by HER2-directed induction of apoptosis with anti-HER2 antibody and caspase-3 fusion protein, Gut, 59 (2010) 292–299. [DOI] [PubMed] [Google Scholar]
- [182].Delbridge AR, Grabow S, Strasser A, Vaux DL, Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies, Nat Rev Cancer, 16 (2016) 99–109. [DOI] [PubMed] [Google Scholar]
- [183].Slee EA, Keogh SA, Martin SJ, Cleavage of BID during cytotoxic drug and UV radiation-induced apoptosis occurs downstream of the point of Bcl-2 action and is catalysed by caspase-3: a potential feedback loop for amplification of apoptosis-associated mitochondrial cytochrome c release, Cell Death Differ, 7 (2000) 556–565. [DOI] [PubMed] [Google Scholar]
- [184].Pajvani UB, Trujillo ME, Combs TP, Iyengar P, Jelicks L, Roth KA, Kitsis RN, Scherer PE, Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy, Nat Med, 11 (2005) 797–803. [DOI] [PubMed] [Google Scholar]
- [185].Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, Shirani J, Armstrong RC, Kitsis RN, A mechanistic role for cardiac myocyte apoptosis in heart failure, The Journal of clinical investigation, 111 (2003) 1497–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Ren K, Lu J, Porollo A, Du C, Tumor-suppressing function of caspase-2 requires catalytic site Cys-320 and site Ser-139 in mice, J Biol Chem, 287 (2012) 14792–14802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Lopez-Garcia C, Sansregret L, Domingo E, McGranahan N, Hobor S, Birkbak NJ, Horswell S, Gronroos E, Favero F, Rowan AJ, Matthews N, Begum S, Phillimore B, Burrell R, Oukrif D, Spencer-Dene B, Kovac M, Stamp G, Stewart A, Danielsen H, Novelli M, Tomlinson I, Swanton C, BCL9L Dysfunction Impairs Caspase-2 Expression Permitting Aneuploidy Tolerance in Colorectal Cancer, Cancer cell, 31 (2017) 79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Oliver TG, Meylan E, Chang GP, Xue W, Burke JR, Humpton TJ, Hubbard D, Bhutkar A, Jacks T, Caspase-2-Mediated Cleavage of Mdm2 Creates a p53-Induced Positive Feedback Loop, Mol Cell, 43 (2011) 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Fava LL, Schuler F, Sladky V, Haschka MD, Soratroi C, Eiterer L, Demetz E, Weiss G, Geley S, Nigg EA, Villunger A, The PIDDosome activates p53 in response to supernumerary centrosomes, Genes Dev, 31 (2017) 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Sidi S, Sanda T, Kennedy RD, Hagen AT, Jette CA, Hoffmans R, Pascual J, Imamura S, Kishi S, Amatruda JF, Kanki JP, Green DR, D’Andrea AA, Look AT, Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3, Cell, 133 (2008) 864–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Yang JY, Walicki J, Michod D, Dubuis G, Widmann C, Impaired Akt activity down-modulation, caspase-3 activation, and apoptosis in cells expressing a caspase-resistant mutant of RasGAP at position 157, Mol Biol Cell, 16 (2005) 3511–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Huang Q, Li F, Liu X, Li W, Shi W, Liu FF, O’Sullivan B, He Z, Peng Y, Tan AC, Zhou L, Shen J, Han G, Wang XJ, Thorburn J, Thorburn A, Jimeno A, Raben D, Bedford JS, Li CY, Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy, Nat Med, 17 (2011) 860–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Zhou M, Liu X, Li Z, Huang Q, Li F, Li CY, Caspase-3 regulates the migration, invasion and metastasis of colon cancer cells, Int J Cancer, 143 (2018) 921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Strilic B, Yang L, Albarran-Juarez J, Wachsmuth L, Han K, Muller UC, Pasparakis M, Offermanns S, Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis, Nature, 536 (2016) 215–218. [DOI] [PubMed] [Google Scholar]
- [195].Helfer B, Boswell BC, Finlay D, Cipres A, Vuori K, Bong Kang T, Wallach D, Dorfleutner A, Lahti JM, Flynn DC, Frisch SM, Caspase-8 promotes cell motility and calpain activity under nonapoptotic conditions, Cancer Res, 66 (2006) 4273–4278. [DOI] [PubMed] [Google Scholar]
- [196].Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR, The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant, Nat Cell Biol, 2 (2000) 156–162. [DOI] [PubMed] [Google Scholar]
- [197].Ichim G, Lopez J, Ahmed SU, Muthalagu N, Giampazolias E, Delgado ME, Haller M, Riley JS, Mason SM, Athineos D, Parsons MJ, van de Kooij B, Bouchier-Hayes L, Chalmers AJ, Rooswinkel RW, Oberst A, Blyth K, Rehm M, Murphy DJ, Tait SWG, Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death, Mol Cell, 57 (2015) 860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Haraguchi M, Torii S, Matsuzawa S, Xie Z, Kitada S, Krajewski S, Yoshida H, Mak TW, Reed JC, Apoptotic protease activating factor 1 (Apaf-1)-independent cell death suppression by Bcl-2, J Exp Med, 191 (2000) 1709–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, Chen Y, Wei M, Eng VM, Adelman DM, Simon MC, Ma A, Golden JA, Evan G, Korsmeyer SJ, MacGregor GR, Thompson CB, The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues, Mol Cell, 6 (2000) 1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Li LY, Luo X, Wang X, Endonuclease G is an apoptotic DNase when released from mitochondria, Nature, 412 (2001) 95–99. [DOI] [PubMed] [Google Scholar]
- [201].Susin SA, Daugas E, Ravagnan L, Samejima K, Zamzami N, Loeffler M, Costantini P, Ferri KF, Irinopoulou T, Prevost MC, Brothers G, Mak TW, Penninger J, Earnshaw WC, Kroemer G, Two distinct pathways leading to nuclear apoptosis, J Exp Med, 192 (2000) 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Giampazolias E, Zunino B, Dhayade S, Bock F, Cloix C, Cao K, Roca A, Lopez J, Ichim G, Proics E, Rubio-Patino C, Fort L, Yatim N, Woodham E, Orozco S, Taraborrelli L, Peltzer N, Lecis D, Machesky L, Walczak H, Albert ML, Milling S, Oberst A, Ricci JE, Ryan KM, Blyth K, Tait SWG, Mitochondrial permeabilization engages NF-kappaB-dependent anti-tumour activity under caspase deficiency, Nat Cell Biol, 19 (2017) 1116–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Riley JS, Quarato G, Cloix C, Lopez J, O’Prey J, Pearson M, Chapman J, Sesaki H, Carlin LM, Passos JF, Wheeler AP, Oberst A, Ryan KM, Tait SW, Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis, EMBO J, 37 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Brokatzky D, Dorflinger B, Haimovici A, Weber A, Kirschnek S, Vier J, Metz A, Henschel J, Steinfeldt T, Gentle IE, Hacker G, A non-death function of the mitochondrial apoptosis apparatus in immunity, EMBO J, 38 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Soengas MS, Capodieci P, Polsky D, Mora J, Esteller M, Opitz-Araya X, McCombie R, Herman JG, Gerald WL, Lazebnik YA, Cordon-Cardo C, Lowe SW, Inactivation of the apoptosis effector Apaf-1 in malignant melanoma, Nature, 409 (2001) 207–211. [DOI] [PubMed] [Google Scholar]
- [206].Fujimoto A, Takeuchi H, Taback B, Hsueh EC, Elashoff D, Morton DL, Hoon DS, Allelic imbalance of 12q22–23 associated with APAF-1 locus correlates with poor disease outcome in cutaneous melanoma, Cancer Res, 64 (2004) 2245–2250. [DOI] [PubMed] [Google Scholar]
- [207].Lee H, Shin EA, Lee JH, Ahn D, Kim CG, Kim JH, Kim SH, Caspase inhibitors: a review of recently patented compounds (2013–2015), Expert opinion on therapeutic patents, 28 (2018) 47–59. [DOI] [PubMed] [Google Scholar]
- [208].Colell A, Ricci JE, Tait S, Milasta S, Maurer U, Bouchier-Hayes L, Fitzgerald P, Guio-Carrion A, Waterhouse NJ, Li CW, Mari B, Barbry P, Newmeyer DD, Beere HM, Green DR, GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation, Cell, 129 (2007) 983–997. [DOI] [PubMed] [Google Scholar]
- [209].Altenberg B, Greulich KO, Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes, Genomics, 84 (2004) 1014–1020. [DOI] [PubMed] [Google Scholar]
- [210].Chiche J, Pommier S, Beneteau M, Mondragon L, Meynet O, Zunino B, Mouchotte A, Verhoeyen E, Guyot M, Pages G, Mounier N, Imbert V, Colosetti P, Goncalves D, Marchetti S, Briere J, Carles M, Thieblemont C, Ricci JE, GAPDH enhances the aggressiveness and the vascularization of non-Hodgkin’s B lymphomas via NF-kappaB-dependent induction of HIF-1alpha, Leukemia, 29 (2015) 1163–1176. [DOI] [PubMed] [Google Scholar]
- [211].Jacquin MA, Chiche J, Zunino B, Beneteau M, Meynet O, Pradelli LA, Marchetti S, Cornille A, Carles M, Ricci JE, GAPDH binds to active Akt, leading to Bcl-xL increase and escape from caspase-independent cell death, Cell Death Differ, 20 (2013) 1043–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]