

This report highlights the importance of longitudinal, parallel characterization of oral fungi and bacteria in order to better elucidate the dynamic changes in microbial community structure and interkingdom functional interactions during the injury of chemotherapy and antibiotic exposure as well as the clinical consequences of these interrelated alterations.
KEYWORDS: mycobiome, interkingdom interactions, leukemia, induction chemotherapy, Malassezia
ABSTRACT
Although the term “microbiome” refers to all microorganisms, the majority of microbiome studies focus on the bacteriome. Here, we characterize the oral mycobiome, including mycobiome-bacteriome interactions, in the setting of remission-induction chemotherapy (RIC) for acute myeloid leukemia (AML). Oral samples (n = 299) were prospectively collected twice weekly from 39 AML patients during RIC until neutrophil recovery. Illumina MiSeq 16S rRNA gene (V4) and internal transcribed spacer 2 (ITS2) sequencing were used to determine bacterial and fungal diversity and community composition. Intrakingdom and interkingdom network connectivity at baseline (T1) and at midpoint (T3) and a later time point (T6) were assessed via SPIEC-EASI (sparse inverse covariance estimation for ecological association inference). In this exploratory study, mycobiome α-diversity was not significantly associated with antibiotic or antifungal receipt. However, postchemotherapy mycobiome α-diversity was lower in subjects receiving high-intensity chemotherapy. Additionally, greater decreases in Malassezia levels were seen over time among patients on high-intensity RIC compared to low-intensity RIC (P = 0.003). A significantly higher relative abundance of Candida was found among patients who had infection (P = 0.008), while a significantly higher relative abundance of Fusarium was found among patients who did not get an infection (P = 0.03). Analyses of intrakingdom and interkingdom relationships at T1, T3, and T6 indicated that interkingdom connectivity increased over the course of IC as bacterial α-diversity diminished. In (to our knowledge) the first longitudinal mycobiome study performed during AML RIC, we found that mycobiome-bacteriome interactions are highly dynamic. Our study data suggest that inclusion of mycobiome analysis in the design of microbiome studies may be necessary to optimally understand the ecological and functional role of microbial communities in clinical outcomes.
IMPORTANCE This report highlights the importance of longitudinal, parallel characterization of oral fungi and bacteria in order to better elucidate the dynamic changes in microbial community structure and interkingdom functional interactions during the injury of chemotherapy and antibiotic exposure as well as the clinical consequences of these interrelated alterations.
INTRODUCTION
There is increasing appreciation that microbiome composition affects treatment-related complications during cytotoxic chemotherapy and hematopoietic stem cell transplantation (HSCT) in patients with hematologic malignancy, including infections, gastrointestinal (GI) toxicities, and the development of graft-versus-host disease (GvHD) (1–3). For example, several studies have established an association between the gut microbiota and HSCT outcomes, including transplant-related mortality, relapse rates, and overall survival (4, 5). Although the term “microbiome” technically refers to all microorganisms, nearly all studies to date have focused on the role of bacteria rather than fungi (6).
Although still markedly understudied compared to the bacteriome, there are increasing numbers of reports linking the commensal fungi, or the “mycobiome,” to the health of patients with hematologic malignancy. For example, researchers have found that patients colonized with Candida sp. developed significantly more grade II to IV acute GvHD and GI GvHD than noncolonized patients (7). We previously found both the mouth and gut to be dominated by Candida albicans and Candida glabrata prior to mucormycosis during induction remission chemotherapy (8). Similarly, patients with a baseline microbiome community dominated by Candida and aciduric bacteria were more likely to develop oral candidiasis during antineoplastic treatment (9). Recently, variation in mycobiome composition was linked to treatment-related toxicities after autologous HSCT for multiple myeloma (10). Specifically, the composition of the baseline oral mycobiome was associated with vomiting as well as culture-negative neutropenic fever, while the fungal community on day 7 posttransplantion was associated with the incidence and severity of mucositis.
Patients with hematologic malignancy receive multiple types of drugs that may impact both fungal and bacterial communities. There is some evidence that the oral and intestinal bacterial microbiota in humans may be altered by cytotoxic chemotherapy (11), although the effects on the fungal microbiota are less clear. It has been previously shown that cytotoxic and cisplatin compounds have direct anti-Candida activity and also inhibit filamentation/conidation (12). More recently, investigators demonstrated that the oral microbiome is disrupted by 5-fluorouracil (5-FU) and doxorubicin chemotherapy, but they did not observe major changes in the composition of the mycobiome (13). Moreover, mice receiving both 5-fluorouracil and C. albicans had a loss of bacterial diversity and showed endogenous overgrowth of organisms such as Enterococcus and Stenotrophomonas in the oral mucosa (14). In addition to chemotherapy, antimicrobial therapy, a very common practice in this patient population, impacts mycobiome composition. For example, it was observed that antibiotics with antianaerobic activity affected the oral fungal community composition posttransplantion in multiple myeloma patients (10). Also, bacteremia with fluconazole-resistant Candida has been associated with exposure to several different antimicrobials, including carbapenems, clindamycin, and colistin (15). It has long been known that some antibacterial agents promote fungal growth via indirect effects (16), many of which promote Candida colonization (17, 18).
Given that the members of the mycobiome interact with the bacteriome in ways that can be beneficial, neutral, or detrimental to the host (19), we aimed to determine the changes in the oral mycobiome and in fungus-bacterium relationships in the setting of leukemia remission-induction chemotherapy (RIC) given that these patients typically receive combined antibiotics, antifungals, and cytotoxic chemotherapy (7). Given the key role of Candida in the health of the oropharynx and because oral microaspiration is the most common pathological mechanism of bacterial pneumonia, the most severe infection during RIC in this patient population, we focused on the oral mycobiome and its connectivity with the oral bacteriome (20, 21).
RESULTS AND DISCUSSION
Longitudinal oral mycobiome characteristics of acute myeloid leukemia (AML) patients undergoing RIC from baseline until neutrophil recovery.
Patient demographic and clinical data are shown in Table 1 and include approximately equal numbers of males (51%) and females (49%), with a mean age of 54 years. At the genus level, the most abundant genus observed across all time points was Malassezia, followed by Candida, Saccharomyces, Fusarium, and Cladosporium. This is very similar to the core set of oral mycobiome taxa previously identified among healthy individuals, which includes Candida, Cladosporium, Saccharomycetales, Aspergillus, Fusarium, Cryptococcus, and Malassezia (22, 23). Although Aspergillus was seen in at least one oral sample in over 70% of the patients, it had a detectable abundance in only 13% of all longitudinal samples. Additionally, among the samples which had a detectable abundance of Aspergillus, the median relative abundance was only 1.8% (interquartile range [IQR], 0.03% to 8.4%). This observation was interesting given that Aspergillus is the primary cause of invasive mold infection among patients with hematologic malignancy (24). The abundances of the top ten most abundant genera showed significant intrapatient and interpatient variability across time (Fig. 1). However, the β-diversity appeared to be more homogeneous within each patient across time, with the mean intrapatient Bray-Curtis dissimilarity between samples within the same patient being 0.649 among the cohort members versus an average interpatient β-diversity value of 0.745 for all samples across time points. This is consistent with another study that analyzed the oral mycobiome stability over 30 days, which revealed high interindividual diversity and yet indicated intraindividual stability (25). Using the results of principal-coordinate analysis (PCoA) shown in Fig. 2, we found no distinct grouping by patient or time point based on Bray-Curtis dissimilarity. We next sought to describe the oral mycobiome changes over the course of RIC. Through mixed-effects models, we found that mycobiome α-diversity did not significantly change over time (Chao1, P = 0.4639; Shannon, P = 0.8049; Simpson, P = 0.6637) (Fig. 3), unlike what has been previously been noted for the bacteriome, where diversity decreased from baseline until neutrophil recovery among leukemia patients receiving RIC (2, 26).
TABLE 1.
Patient demographics and clinical characteristics
| Characteristic | Valuesa |
|---|---|
| Demographics | |
| Mean age in yrs (range) | 54 (23–84) |
| Male | 20 (51) |
| Female | 19 (49) |
| Chemotherapy intensity | |
| High | 28 (72) |
| Low | 11 (28) |
| Chemotherapy type | |
| Fludarabine-containing regimenb | 17 (44) |
| Nonfludarabine high-intensity regimenc | 11 (28) |
| Hypomethylatord | 5 (13) |
| Othere | 6 (15) |
| Infection | 21 (54) |
| Microbiologically defined | 9 (23) |
| Bacteremia | 4 (10) |
| Urinary tract infection | 3 (8) |
| Other | 2 (5) |
| Clinically defined | 12 (31) |
| Cellulitis | 5 (13) |
| Pneumonia | 3 (8) |
| Other | 4 (10) |
| Antimicrobial administration | |
| Antifungals | 39 (100) |
| Triazoles | 31 (79) |
| Echinocandins | 30 (77) |
| Amphotericin B | 6 (15) |
| Broad-spectrum antibiotics | 34 (87) |
| Carbapenem | 26 (67) |
| Piperacillin-tazobactam | 11 (28) |
| Cephalosporin | 17 (44) |
Values represent number (percent) of patients except where otherwise indicated.
Includes FIA and FLAG-Ida-based regimens.
Includes CIA, IA, or CLIA-based regimens.
Includes azacitidine or decitabine-based regimens.
Includes LDAC-based regimens.
FIG 1.

Abundances of top 10 fungal genera and in each patient across time. Stacked bar plots show the relative abundances of the 10 most abundant taxa identified to the genus level for each patient from baseline (T1) until the sixth sample collected (T6). Other, taxa not included in the 10 most abundant genera. Fungi sp, unknown/unidentified fungal genus.
FIG 2.

Mycobiome β-diversity over time. Results of principal-coordinate analysis (PCoA) of ITS2 OTUs via Bray-Curtis dissimilarity are presented. All patients are represented by distinctly colored data according to the inlaid figure legend. Increasing longitudinal durations per patient from baseline until neutrophil recovery are represented using increasingly larger circles.
FIG 3.

Mycobiome α-diversity over time. Mixed-effects models were used to plot the α-diversity metrics of Chao1, Shannon index, and Simpson index data over time for each patient (colored dark blue to light blue). Time point data represent the sequential time points at which samples were collected from baseline until neutrophil recovery approximately every 96 h.
Mycobiome characteristics correlate with chemotherapy subtype and antifungal administration.
We next sought to determine if mycobiome profiles were associated with clinical variables among the patients in the cohort, to include chemotherapeutic or antimicrobial administration. In this sampling of patients, 72% were on high-intensity chemotherapy, while 28% received low-intensity chemotherapy (Table 1). The majority of patients (44%) received fludarabine-based regimens, with the majority of the regimens being FIA (idarubicin, cytarabine, and fludarabine) or FLAG-Ida (fludarabine, idarubicin, and granulocyte-colony-stimulating factor). Twenty-eight percent of patients received nonfludarabine high-intensity regimens, which typically included CIA (idarubicin, cytarabine, and clofarabine)-based, IA (idarubicin and cytarabine)-based, or CLIA (cladribine, idarubicin, and cytarabine)-based regimens. Patients also receive hypomethylator-based treatments (such as decitabine or azacitadine) (13%) or other low-dose therapies to include LDAC (low-dose antimetabolite cytarabine)-based regimens (14%). Given the uneven sampling time (time to neutrophil recovery) among patients, we evaluated the mycobiome structure and composition at the baseline (T1), a midpoint (T3), and a later time point (T6), common among most patients. At T6, patients who received low-intensity RIC had significantly higher mycobiome α-diversity than patients who received high-intensity RIC (observed operational taxonomic units [OTUs], P = 0.02; Shannon, P = 0.054) (see Fig. S1 in the supplemental material). Although Kruskal-Wallis tests indicated that the relative abundances of the five most abundant fungal genera did not significantly differ between the four different chemotherapy subtypes at any single time point (see Table S1 in the supplemental material), we did determine chemotherapy subtypes to be significantly associated with the relative abundance of specific taxa over time. Specifically, patients who received high-intensity chemotherapies experienced a decrease in Malassezia abundance over time (P = 0.003, Fig. 4). This effect was not seen in other taxa analyzed (data not shown). To our knowledge, the changes in the oral mycobiome occurring as a result of chemotherapy have been studied only in regard to their role in chemotherapy-induced mucositis in the setting of 5-fluorouracil (5-FU) or doxorubicin-based chemotherapy in mice and humans (13, 14). In patients, although the authors expected chemotherapy to affect the oral mycobiome, no major changes in the composition of fungal communities were observed (13). Thus, our study would be the first to show specific changes in the oral mycobiome associated with chemotherapeutic intensity and type.
FIG 4.

Changes in Malassezia over time stratified by chemotherapy intensity and chemotherapy type. Centered log ratio (clr) data represent transformed relative abundances of Malassezia with fitted lines from the mixed-effects model over time according to each variable of interest. P value data were derived from ANOVA F-test results. Panel A has fitted lines representing the intensities of the chemotherapy regimens, with high-intensity regimens indicated in coral and low-intensity regimens in aqua. Panel B further dissects these by chemotherapy subtype, with high-intensity fludarabine-containing regimens indicated in coral, hypomethylator-based therapies in green, non-fludarabine-containing high-intensity regimens in aqua, and other low-intensity/low-dose cytarabine-based therapies in purple.
Differences in mycobiome α-diversity (T6) based on chemotherapy intensity. A box plot compares observed OTUs and Shannon diversity at T6 between those who received high-intensity and low-intensity chemotherapy regimens. The P values are derived from a Mann-Whitney test. Download FIG S1, JPG file, 0.1 MB (129.6KB, jpg) .
{kind=link}
Copyright © 2020 Robinson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Taxon abundance differences by chemotherapy subtype. P values represent results of Kruskal-Wallis test performed with data from each time point. Listed are the top 5 taxa by abundance across all 6 time points. Download Table S1, DOCX file, 0.02 MB (17.5KB, docx) .
Copyright © 2020 Robinson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We next sought to determine if antimicrobial administration resulted in significant mycobiome changes during RIC for AML. We examined the three primary empirical broad-spectrum antibiotics received in this cohort, which were carbapenems, cephalosporins, and piperacillin-tazobactam, and found that 87% of the patients were administered at least one of these three broad-spectrum antimicrobials. Some patients received more than one broad-spectrum antibiotic prior to neutrophil recovery, with 67% receiving a carbapenem, 44% a cephalosporin, and 28% piperacillin-tazobactam. We also evaluated the three primary treatment antifungals, consisting of echinocandins, amphotericin B, and triazoles. All patients were administered at least one of these three antifungals, with 79% receiving an azole, 77% an echinocandin, and 15% amphotericin B. We analyzed the later time point during RIC (T6) given that all antimicrobials analyzed were administered by T6 and that it was the latest time point common among most patients. Surprisingly, there was no significant association between the relative abundance of the top 5 most abundant fungal taxa at time point 6 and receipt of specific antimicrobials (Tables S2 toS7). However, amphotericin B seemed to have a significant effect on the composition of the oral fungal community over time. Patients who received amphotericin B experienced an increase in the relative abundance of Fusarium over time (P = 0.034) (Fig. 5). There is biological plausibility to this observation inasmuch as Fusarium species are associated with high amphotericin B MICs (27). Amphotericin B had no significant effect on the relative abundance of other taxa over time.
FIG 5.
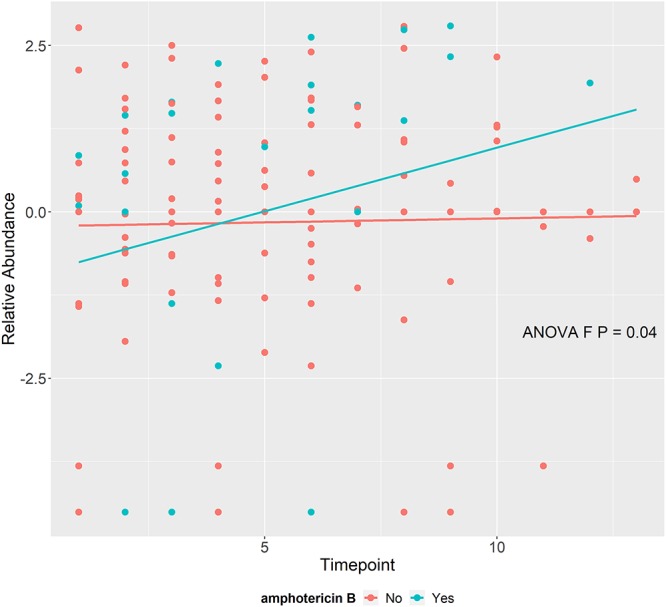
Changes in Fusarium over time stratified by amphotericin B administration. Centered log ratio (clr) data represent transformed relative abundances of Fusarium with fitted lines from the mixed-effects model over time according to whether amphotericin B was administered (aqua) or not (coral) during the study. P values were derived from the ANOVA F-test.
Taxon abundance differences at T6 by receipt of piperacillin-tazobactam. P values represent results of Mann-Whitney test comparing the relative abundances of designated taxa between those who did and did not receive piperacillin-tazobactam. Listed are the top 5 taxa by abundance at T6; however, all genera were tested. Download Table S2, DOCX file, 0.01 MB (12KB, docx) .
Copyright © 2020 Robinson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Taxon abundance differences at T6 by receipt of carbapenem. P values represent results of Mann-Whitney test comparing the relative abundances of designated taxa between those who did and did not receive a carbapenem. Listed are the top 5 taxa by abundance at T6; however, all genera were tested. Download Table S3, DOCX file, 0.01 MB (11.9KB, docx) .
Copyright © 2020 Robinson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Taxon abundance differences at T6 by receipt of cephalosporin. P values represent results of Mann-Whitney test comparing the relative abundances of designated taxa between those who did and did not receive a cephalosporin. Listed are the top 5 taxa by abundance at T6; however, all genera were tested. Download Table S4, DOCX file, 0.01 MB (11.9KB, docx) .
Copyright © 2020 Robinson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Taxon abundance differences at T6 by receipt of echinocandin. P values represent results of Mann-Whitney test comparing the relative abundances of designated taxa between those who did and did not receive an echinocandin. Listed are the top 5 taxa by abundance at T6; however, all genera were tested. Download Table S5, DOCX file, 0.01 MB (12KB, docx) .
Copyright © 2020 Robinson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Taxon abundance differences at T6 by receipt of amphotericin B. P values represent results of Mann-Whitney test comparing the relative abundances of designated taxa between those who did and did not receive amphotericin B. Listed are the top 5 taxa by abundance at T6; however, all genera were tested. Download Table S6, DOCX file, 0.01 MB (12KB, docx) .
Copyright © 2020 Robinson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Taxon abundance differences at T6 by receipt of triazole. P values represent results of Mann-Whitney test comparing the relative abundances of designated taxa between those who did and did not receive a triazole. Listed are the top 5 taxa by abundance at T6; however, all genera were tested. Download Table S7, DOCX file, 0.01 MB (12KB, docx) .
Copyright © 2020 Robinson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Mycobiome characteristics are correlated with bacterial infection outcomes.
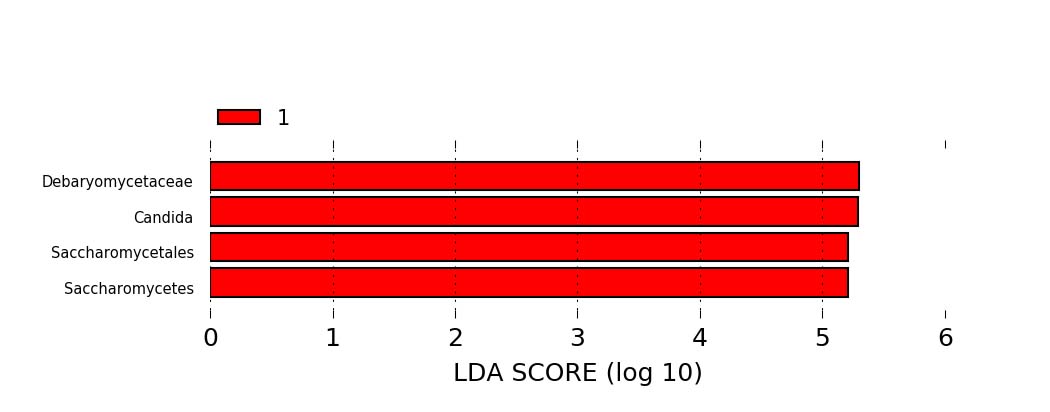
In light of the complex interplay between fungal populations and opportunistic bacterial pathogens (28), we sought to determine the relationships between the mycobiome and infections in AML patients during RIC. A total of 23% of patients had a microbiologically defined infection with a positive bacterial culture, which included mostly bacteremia or urinary tract infections (Table 1). We also considered the 31% of patients who had a clinically defined infection, such a pneumonia or cellulitis, in the absence of positive culture in our analysis. In analyzing the three major time points (T1, T3, and T6), we found that the relative abundance of Candida at T3 was significantly higher in patients that contracted microbiologically or clinically defined infections prior to neutrophil recovery (P = 0.008), while there was a significantly higher relative abundance of Fusarium (P = 0.03) at T6 in those that did not develop an infection prior to neutrophil recovery (Fig. 6). These results were corroborated using the LefSe (linear discriminant analysis of effect size) algorithm, where Candida was more differentially abundant at T3 among patients who developed an infection prior to neutrophil recovery (Fig. S2) and Fusarium more abundant at T6 among patients who did not have an infection (Fig. S3). No other statistical associations were observed for other genera at the three time points or at T1 (data not shown).
FIG 6.

Relative abundance differences of taxa at individual time points among AML patients who did and did not experience bacterial infection during RIC. Box plots depict the taxon abundance differences of Candida at T3 and Fusarium at T6 between those patients that did (aqua) and did not (coral) contract bacterial infections prior to neutrophil recovery following RIC. P values are derived from Mann-Whitney test.
Differential abundance of specific taxa at T3 for acute leukemia patients who had infectious complications during induction chemotherapy. The LefSe (linear discriminant analysis effect size) algorithm was used to determine which taxonomic units were most differentially abundant among acute leukemia patients who had infectious complications during induction chemotherapy (“1”) and those who did not experience infection (“0”) at T3. Download FIG S2, JPG file, 0.04 MB (46.9KB, jpg) .
{kind=link}
Copyright © 2020 Robinson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Differential abundance of specific taxa at T6 for acute leukemia patients who had infectious complications during induction chemotherapy. The LefSe (linear discriminant analysis effect size) algorithm was used to determine which taxonomic units were most differentially abundant among acute leukemia patients who had infectious complications during induction chemotherapy (“1”) and those who did not experience infection (“0”) at T6. Download FIG S3, JPG file, 0.04 MB (46.3KB, jpg) .
{kind=link}
Copyright © 2020 Robinson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Interkingdom interactions of the oral microbiome over time in AML patients receiving RIC.
Cross-domain association networks were constructed at T1, T3, and T6 using matching bacterial and fungal microbiome samples (n = 272). The three SPIEC-EASI (sparse inverse covariance estimation for ecological association inference) networks show vast differences in intra- and interkingdom connectivity between time points, highlighting the dynamic nature of bacterial and fungal interactions induced by changes in the community during RIC (Fig. 7). The average node degrees were 0.43 (standard deviation [SD], ±0.48), 0.84 (SD, ±0.92), and 0.5 (SD, ±0.56) for T1, T3, and T6, respectively.
FIG 7.

Cross-domain association networks across time. SPEIC-EASI networks were generated for time points 1, 3, and 6. Circles indicate bacterial taxa, while squares indicate fungal taxa. Cooccurrence (positive) relationships are indicated with black edges. Coexclusion (negative) relationships are indicated with red edges. Colors are indicated for each taxon in the inlaid legend and are constant across networks. Nodes that are black have no connection in any network.
At baseline (T1), we observed one cross-domain connection (Ramularia with Haemophilus), two bacterial connections (Fusobacterium with Alloprevotella and Enterococcus with Rothia), and two fungal connections (Malassezia with Candida and “Fungi sp.” [unknown/unidentified fungal genus] with Malasseziales sp.). The T3 network contained nine cross-domain connections (Cladosporium with Stenotrophomonas, Aspergillus with Fusobacterium, Aspergillus with Alloprevotella, Saccharomyces with Prevotellaceae [Unc00qrn], Saccharomyces with Neisseria, Meyerozyma with Acinetobacter, Meyerozyma with Stenotrophomonas, Pichia with Campylobacter, and Ramularia with Capnocytophaga), four bacterial connections (Acinetobacter with Stenotrophomonas, Prevotellaceae [Unc00qrn] with Prevotella, Alloprevotella with Prevotellaceae [Unid1919], and Prevotellaceae [Unid1919] with Prevotella), and three fungal connections (Cladosporium with Pichia, Cordyceps with Fungi sp., and Candida with Malassezia). At the latest time point analyzed (T6), we observed seven cross-domain connections (Alternaria with Streptococcus, Cladosporium with Rothia, Saccharomyces with Mycoplasma, Sporidiobolales sp. with Pseudomonas, Fungi sp. with Lactobacillus, Fungi sp. with Actinomyces, and Starmerella with Veillonella) and two bacterial connections (Enterococcus with Prevotella and Prevotellaceae [Unc00bms] with Leptotrichia).
Some of the intrakingdom and interkingdom interactions have been previously observed. Specifically, Malassezia and Candida are often coisolated from infectious samples from the skin infections (29) and onychomycosis (30), but to our knowledge cooccurrence has not been shown in the oral cavity. Interestingly, we found a negative correlation between Malassezia and Candida at T1 and T3 in the oral cavity. In a study of the oral microbiome of HIV patients versus uninfected individuals (31), it was found that Cladosporidium and Rothia had a positive correlation in uninfected individuals. We observed the same cooccurrence at T6 in treated leukemia patients. Also, in individuals uninfected with HIV, there was a negative correlation between Aspergillus and Prevotella (31). We observed the same cross-domain connection, but as a cooccurrence. In the same study, the authors observed cocolonization of Cladosporidium and Pichia in uninfected individuals (31), which is a fungal relationship that we detected at T3. Additionally, we saw a bacterial cooccurrence relationship between Prevotellaceae and Leptotrichia at T6, which has also been observed in subjects with apical periodontitis (32) and in subjects with halitosis, as well as in healthy individuals (33).
Only one connection overlapped any two networks. Interestingly however, time point 3 showed more-dependent connectivity (multiple nodes linked to each other), while time points 3 and 6 had increased connectivity overall. As we had previously reported loss of bacterial diversity over time among the patients included in these analyses (2), this suggests that either loss of bacterial diversity or changes in the oral environment under conditions of chemotherapeutic and antimicrobial pressure or both affect the bacterium-fungus interplay. This may potentially result in clinical consequences, as organisms that do not typically interact would inhabit the niche. This could potentially have acute consequences for functional metabolites, genetic exchange, and alterations in the virulence of the pathogen or in the immune status of the host (28, 34).
Limitations.
Some fungus-bacterium interactions in the oral cavity of diseased and healthy individuals (namely, Candida-bacterium interactions) have been intensely studied; however, interactions of bacteria with other known fungal inhabitants of the oral cavity have yet to be described. Thus, the impact of fungus-bacterium interactions in the oral cavity and on overall health is not completely understood. Although this exploratory study had a relatively small sample size and assessed only oral samples, the data shown here still highlight the dynamic nature of mycobiome and bacteriome communities and their interactions over the course of RIC. Unfortunately, the study design did not include information on diet, the metabolic milieu (e.g., other relevant comorbidities such as diabetes), oral health information (including oropharyngeal candidiasis), or occurrence of mucositis. These clinical factors/outcomes could potentially account for the heterogeneity seen within the data or might also be associated with specific mycobiome or bacteriome compositions/relationships. Despite not including a comprehensive record of clinical covariates, we were able to determine that specific changes in the mycobiome community structure appear to have clinical consequences in terms of bacterial infections. Moreover, interkingdom dynamics are likely driven by changes in the mycobiome due to chemotherapy intensity and subtype, as well as bacteriome changes driven by antibiotic exposures. Although this report highlights the underappreciated complex and dynamic nature of the cross-kingdom interactions that occur over time and result from the stress of chemotherapy and antibiotic therapy, these data are limited by our having used only marker gene analysis (16S rRNA gene and internal transcribed spacer 2 [ITS2] sequencing) to identify bacterium-fungus dynamics. Target gene analysis is limited in its ability to differentiate individual taxa, specifically to lower taxonomic levels such as the species level. Although the ability to assemble and differentiate fungal reads at the whole-genome level is currently restricted by the number of fungal genomes available in public databases, this information could be vastly improved in the future via shotgun metagenomics sequencing, particularly in regard to functional pathway analysis. An alternative approach would be to layer on metabolomics or metaproteomics to understand the functional consequences of bacterial and fungal changes as well as the interkingdom metabolic pathway interactions. Again, however, methods for data integration of shotgun metagenomic and metabolomics data, particularly in regard to cross-domain networks, are still in their infancy.
Conclusions.
Here, we provide the first longitudinal analysis of the oral mycobiome in patients with acute leukemia combined with parallel oral bacteriome data. Incorporation of fungal analyses into microbiome studies may help improve mechanistic understanding of how commensal flora impact clinical outcomes. These data suggest that simultaneous mycobiome and bacteriome longitudinal analyses may add additional insight in order to fully understand how the microbiome impacts infectious risk in immunocompromised patients. We anticipate these data will inspire future studies aimed to further elucidate these relationships in the cancer treatment setting.
MATERIALS AND METHODS
Patient sample collection, DNA extraction, sequencing, and bioinformatics.
In this pilot exploratory study, we collected oral (buccal swab) samples (n = 299) from 39 newly diagnosed adult AML patients undergoing IC at the MD Anderson Cancer Center (MDACC) in Houston, TX, from September 2013 to August 2015. These oral samples and accompanying 16S rRNA gene sequences were derived from a previously reported cohort (2). The study protocol was approved by the MDACC Institutional Review Board (PA13-0339), and the study was conducted in compliance with the Declaration of Helsinki. Written informed consent was obtained from all participants before enrollment. Buccal swabs were collected by swabbing the inside the cheek 3 times on each side using a Catch-All sample collection swab (Epicentre), and the swab tips were then placed in sterile 2-ml cryovials and maintained at −80°C until DNA extraction. Collection of samples from each patient was performed at baseline, continued approximately every 96 h, and stopped upon neutrophil recovery. Clinical data, including antibiotic and antifungal administration, chemotherapy subtypes, and infection status, were extracted from the electronic medical records. Infections prior to neutrophil recovery were classified as microbiologically defined infections (MDI) or clinically defined infections (CDI) using established guidelines (35). Patients were categorized as receiving (i) non-fludarabine-based high-intensity regimens, (ii) high-intensity fludarabine-containing regimens, (iii) low-intensity hypomethylating-agent-based therapy, or (iv) other low-intensity/low-dose cytarabine-based regimens.
Microbial DNA was extracted from oral swabs using the MO BIO PowerSoil DNA isolation kit protocol as described previously (36). We performed ITS sequencing on a subset of patients (the first sequential 39 patients enrolled on study with available residual sample) in which we had previously characterized the microbiome using 16S rRNA gene sequencing (2, 37–39). The internal transcribed spacer 2 (ITS2) region was amplified from oral swab DNA using primers ITS3 and ITS4 as described previously (36). Each primer included an Illumina adapter and linker sequence, and the reverse primer (ITS4) also contained a unique 12-bp Golay barcode (40). Amplicons were sequenced by the use of an Illumina MiSeq platform with the 2 × 300-bp paired-end protocol. The ITS2 read pairs were demultiplexed based on the unique barcodes. Reads were merged and filtered using default settings in USEARCH v7.0.1090 (41). ITS2 sequences were clustered into operational taxonomic units (OTUs) at a similarity cutoff of 97% using the UPARSE pipeline (42). Chimeras were removed using USEARCH v8.0.1517 and UCHIME (43). OTUs were aligned against sequences from the NCBI GenBank Plant and UNITE databases (44). Abundance data were recovered by mapping the demultiplexed reads to the UPARSE OTUs. A custom script developed at the Center for Metagenomics and Microbiome Research at Baylor College of Medicine was used to construct an OTU table. Unmapped (<80% identity or <95% coverage) OTUs were manually analyzed by BLASTN (37). After we performed filtering that included only the samples which had corresponding clinical and 16S data to match the ITS data, 290 samples were obtained for further analyses.
Statistical methods.
For β-diversity, principal-coordinate analysis (PCoA) plots were produced using Bray-Curtis distances. A mixed-effects model was applied to determine if time was a significant predictor of α-diversity. A random intercept for patients was included. A Kruskal-Wallis test was performed to compare α-diversity and taxon abundance data between chemotherapy subtype groups. In association testing, we focused on the top five most abundant fungal genera based on average abundance: Candida, Cladosporium, Fusarium, Malassezia, and Saccharomyces. Similarly, a Mann-Whitney test was performed to compare both alpha-diversity data and taxon abundance data corresponding to chemotherapy intensity (high versus low), use of amphotericin B (yes versus no), and infection status (yes versus no). The linear discriminant analysis effect size (LEfSe) algorithm was used to test whether taxon abundance predicted infection status. The LEfSe algorithm was run using the Galaxy module with default settings. Mixed-effects models were used to determine whether taxon abundance significantly changed over time. Analyses based on these models were performed on the centered log ratio (clr) transformed relative abundance data to address the compositional characteristics of the data. Each model contained the following variables: time point, the variable of interest, and an interaction between the two. To account for repeated measures, a random intercept for patient was included in the model. An analysis of variance (ANOVA) F-test was applied to determine whether time and the variable of interest were significantly associated. The statistical analyses performed as a part of this study were exploratory in nature. In general, P values of less than 0.05 were considered significant; for analyses across the mycobiome, we computed false-discovery-rate (FDR)-adjusted P values, which are provided in the supplemental tables. Data analysis and visualization were performed using R, mainly using functions from the R packages ggplot2 and phyloseq (26, 45).
Cross-domain association networks.
SPIEC-EASI was used to construct the cross-domain microbial networks at the genus level for both bacteria and fungi. For the tables listing bacterial and fungal genera, the data corresponding to time points 1 (T1), 3 (T3), and 6 (T6) were separated (46). As statistically sound methods for longitudinal analyses/trends are underdeveloped for microbiome network analyses, we aimed at analyzing samples at baseline prior to chemotherapy and antimicrobial administration (T1), at a time point after chemotherapy administration (T3), and at a time point after antimicrobial administration (T6). As we had significant sample dropout after T6, and given our already small sample size, T6 was the last time point which retained most samples. In addition, all patients who received broad-spectrum antibiotics had received at least one type of broad-spectrum antibiotic by T6. For each OTU table, taxa are included in the network analysis if the mean abundance was greater than 0.01. T1 had 35 samples with 16 bacterial taxa and 13 fungal taxa. T3 was represented by 36 samples, with 22 bacterial taxa and 16 fungal taxa. T6 contained 31 samples with 21 bacterial taxa and 15 fungal taxa. The sparsity parameter λ was set to 0.3, and the value corresponding to λ was set to 50. We verified the stability of our results for a single value of λ. We also examined sensitivity across multiple λ values and found that the networks were not sensitive to the choice of λ.
Data availability.
All 16S rRNA gene sequences were deposited in the NCBI Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) under BioProject identifiers (IDs) PRJNA352060 and PRJNA526551, whereas ITS2 sequences were submitted under the BioProject ID PRJNA599151 (the authors could not make this BioProject record available at the time of this paper’s publication due to circumstances related to the COVID-19 pandemic, but it will be made accessible as soon as possible after publication).
ACKNOWLEDGMENTS
This work was supported by the MD Anderson Cancer Center Knowledge Gap and Multidisciplinary Research Program funding mechanisms (D.P.K. and S.A.S.). J.R.G.-P. was supported by the MD Anderson Odyssey Fellowship Program and the CFP Foundation for part of the duration of this work as well as by the NIH (1 K01 AI143881-01 from the NIAID). C.B.P. is partially supported by NIH/NCI Cancer Center support grant P30CA016672 (Biostatistics Resource Group). D.P.K. is supported by the Texas 4000 Distinguished Professorship for Cancer Research. This material is based upon work partially supported by the National Science Foundation Graduate Research Fellowship Program under grant no. 1842494 to S.R.
Any opinions, findings, and conclusions or recommendations expressed in this material are ours and do not necessarily reflect the views of the National Science Foundation.
We have no relevant disclosures with respect to conflicts of interest.
REFERENCES
- 1.Shono Y, van den Brink M. 2018. Gut microbiota injury in allogeneic haematopoietic stem cell transplantation. Nat Rev Cancer 18:283–295. doi: 10.1038/nrc.2018.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galloway-Pena JR, Shi Y, Peterson CB, Sahasrabhojane P, Gopalakrishnan V, Brumlow CE, Daver NG, Alfayez M, Boddu PC, Khan MAW, Wargo JA, Do KA, Jenq RR, Kontoyiannis DP, Shelburne SA. 22 August 2019, posting date Gut microbiome signatures are predictive of infectious risk following induction therapy for acute myeloid leukemia. Clin Infect Dis doi: 10.1093/cid/ciz777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hakim H, Dallas R, Wolf J, Tang L, Schultz-Cherry S, Darling V, Johnson C, Karlsson EA, Chang TC, Jeha S, Pui CH, Sun Y, Pounds S, Hayden RT, Tuomanen E, Rosch JW. 2018. Gut microbiome composition predicts infection risk during chemotherapy in children with acute lymphoblastic leukemia. Clin Infect Dis 67:541–548. doi: 10.1093/cid/ciy153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peled JU, Devlin SM, Staffas A, Lumish M, Khanin R, Littmann ER, Ling L, Kosuri S, Maloy M, Slingerland JB, Ahr KF, Porosnicu Rodriguez KA, Shono Y, Slingerland AE, Docampo MD, Sung AD, Weber D, Alousi AM, Gyurkocza B, Ponce DM, Barker JN, Perales MA, Giralt SA, Taur Y, Pamer EG, Jenq RR, van den Brink M. 2017. Intestinal microbiota and relapse after hematopoietic-cell transplantation. J Clin Oncol 35:1650–1659. doi: 10.1200/JCO.2016.70.3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taur Y, Jenq RR, Perales MA, Littmann ER, Morjaria S, Ling L, No D, Gobourne A, Viale A, Dahi PB, Ponce DM, Barker JN, Giralt S, van den Brink M, Pamer EG. 2014. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood 124:1174–1182. doi: 10.1182/blood-2014-02-554725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.El-Jurdi N, Ghannoum MA. 2017. The mycobiome: impact on health and disease states. Microbiol Spectr 5:(3). doi: 10.1128/microbiolspec.FUNK-0045-2016. [DOI] [PubMed] [Google Scholar]
- 7.van der Velden WJ, Netea MG, de Haan AF, Huls GA, Donnelly JP, Blijlevens NM. 2013. Role of the mycobiome in human acute graft-versus-host disease. Biol Blood Marrow Transplant 19:329–332. doi: 10.1016/j.bbmt.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 8.Shelburne SA, Ajami NJ, Chibucos MC, Beird HC, Tarrand J, Galloway-Pena J, Albert N, Chemaly RF, Ghantoji SS, Marsh L, Pemmaraju N, Andreeff M, Shpall EJ, Wargo JA, Rezvani K, Alousi A, Bruno VM, Futreal PA, Petrosino JF, Kontoyiannis DP. 2015. Implementation of a pan-genomic approach to investigate holobiont-infecting microbe interaction: a case report of a leukemic patient with invasive mucormycosis. PLoS One 10:e0139851. doi: 10.1371/journal.pone.0139851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diaz PI, Hong BY, Dupuy AK, Choquette L, Thompson A, Salner AL, Schauer PK, Hegde U, Burleson JA, Strausbaugh LD, Peterson DE, Dongari-Bagtzoglou A. 13 June 2019, posting date Integrated analysis of clinical and microbiome risk factors associated with the development of oral candidiasis during cancer chemotherapy. J Fungi (Basel) doi: 10.3390/jof5020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El Jurdi N, Filali-Mouhim A, Salem I, Retuerto M, Dambrosio NM, Baer L, Lazarus HM, Caimi P, Cooper B, Tomlinson B, Metheny L, Malek E, Otegbeye F, Sekaly RP, Ghannoum M, de Lima M. 2019. Gastrointestinal microbiome and mycobiome changes during autologous transplantation for multiple myeloma: results of a prospective pilot study. Biol Blood Marrow Transplant 25:1511–1519. doi: 10.1016/j.bbmt.2019.04.007. [DOI] [PubMed] [Google Scholar]
- 11.Montassier E, Gastinne T, Vangay P, Al-Ghalith GA, Bruley Des Varannes S, Massart S, Moreau P, Potel G, de La Cochetière MF, Batard E, Knights D. 2015. Chemotherapy-driven dysbiosis in the intestinal microbiome. Aliment Pharmacol Ther 42:515–528. doi: 10.1111/apt.13302. [DOI] [PubMed] [Google Scholar]
- 12.Chen SC, Lewis RE, Kontoyiannis DP. 2011. Direct effects of non-antifungal agents used in cancer chemotherapy and organ transplantation on the development and virulence of Candida and Aspergillus species. Virulence 2:280–295. doi: 10.4161/viru.2.4.16764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hong BY, Sobue T, Choquette L, Dupuy AK, Thompson A, Burleson JA, Salner AL, Schauer PK, Joshi P, Fox E, Shin DG, Weinstock GM, Strausbaugh LD, Dongari-Bagtzoglou A, Peterson DE, Diaz PI. 2019. Chemotherapy-induced oral mucositis is associated with detrimental bacterial dysbiosis. Microbiome 7:66. doi: 10.1186/s40168-019-0679-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bertolini M, Ranjan A, Thompson A, Diaz PI, Sobue T, Maas K, Dongari-Bagtzoglou A. 2019. Candida albicans induces mucosal bacterial dysbiosis that promotes invasive infection. PLoS Pathog 15:e1007717. doi: 10.1371/journal.ppat.1007717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ben-Ami R, Olshtain-Pops K, Krieger M, Oren I, Bishara J, Dan M, Wiener-Well Y, Weinberger M, Zimhony O, Chowers M, Weber G, Potasman I, Chazan B, Kassis I, Shalit I, Block C, Keller N, Kontoyiannis DP, Giladi M, Israeli Candidemia Study Group. 2012. Antibiotic exposure as a risk factor for fluconazole-resistant Candida bloodstream infection. Antimicrob Agents Chemother 56:2518–2523. doi: 10.1128/AAC.05947-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Azevedo MM, Teixeira-Santos R, Silva AP, Cruz L, Ricardo E, Pina-Vaz C, Rodrigues AG. 2015. The effect of antibacterial and non-antibacterial compounds alone or associated with antifungals upon fungi. Front Microbiol 6:669. doi: 10.3389/fmicb.2015.00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Charles PE, Dalle F, Aube H, Doise JM, Quenot JP, Aho LS, Chavanet P, Blettery B. 2005. Candida spp. colonization significance in critically ill medical patients: a prospective study. Intensive Care Med 31:393–400. doi: 10.1007/s00134-005-2571-y. [DOI] [PubMed] [Google Scholar]
- 18.Samonis G, Galanakis E, Ntaoukakis M, Sarchianaki E, Spathopoulou T, Dimopoulou D, Kofteridis DP, Maraki S. 2013. Effects of carbapenems and their combination with amikacin on murine gut colonisation by Candida albicans. Mycoses 56:105–109. doi: 10.1111/j.1439-0507.2012.02212.x. [DOI] [PubMed] [Google Scholar]
- 19.Cui L, Morris A, Ghedin E. 2013. The human mycobiome in health and disease. Genome Med 5:63. doi: 10.1186/gm467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bandara H, Panduwawala CP, Samaranayake LP. 2019. Biodiversity of the human oral mycobiome in health and disease. Oral Dis 25:363–371. doi: 10.1111/odi.12899. [DOI] [PubMed] [Google Scholar]
- 21.Garcia JB, Lei X, Wierda W, Cortes JE, Dickey BF, Evans SE, Ost DE. 2013. Pneumonia during remission induction chemotherapy in patients with acute leukemia. Annals ATS 10:432–440. doi: 10.1513/AnnalsATS.201304-097OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dupuy AK, David MS, Li L, Heider TN, Peterson JD, Montano EA, Dongari-Bagtzoglou A, Diaz PI, Strausbaugh LD. 2014. Redefining the human oral mycobiome with improved practices in amplicon-based taxonomy: discovery of Malassezia as a prominent commensal. PLoS One 9:e90899. doi: 10.1371/journal.pone.0090899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghannoum MA, Jurevic RJ, Mukherjee PK, Cui F, Sikaroodi M, Naqvi A, Gillevet PM. 2010. Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog 6:e1000713. doi: 10.1371/journal.ppat.1000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pagano L, Caira M, Candoni A, Offidani M, Fianchi L, Martino B, Pastore D, Picardi M, Bonini A, Chierichini A, Fanci R, Caramatti C, Invernizzi R, Mattei D, Mitra ME, Melillo L, Aversa F, Van Lint MT, Falcucci P, Valentini CG, Girmenia C, Nosari A. 2006. The epidemiology of fungal infections in patients with hematologic malignancies: the SEIFEM-2004 study. Haematologica 91:1068–1075. [PubMed] [Google Scholar]
- 25.Monteiro-da-Silva F, Araujo R, Sampaio-Maia B. 2014. Interindividual variability and intraindividual stability of oral fungal microbiota over time. Med Mycol 52:498–505. doi: 10.1093/mmy/myu027. [DOI] [PubMed] [Google Scholar]
- 26.Callahan BJ, Sankaran K, Fukuyama JA, McMurdie PJ, Holmes SP. 2016. Bioconductor workflow for microbiome data analysis: from raw reads to community analyses. F1000Res 5:1492. doi: 10.12688/f1000research.8986.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taj-Aldeen SJ, Salah H, Al-Hatmi AMS, Hamed M, Theelen B, van Diepeningen AD, Boekhout T, Lass-Flörl C. 2016. In vitro resistance of clinical Fusarium species to amphotericin B and voriconazole using the EUCAST antifungal susceptibility method. Diagn Microbiol Infect Dis 85:438–443. doi: 10.1016/j.diagmicrobio.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 28.Arvanitis M, Mylonakis E. 2015. Fungal-bacterial interactions and their relevance in health. Cell Microbiol 17:1442–1446. doi: 10.1111/cmi.12493. [DOI] [PubMed] [Google Scholar]
- 29.Gaitanis G, Velegraki A, Frangoulis E, Mitroussia A, Tsigonia A, Tzimogianni A, Katsambas A, Legakis NJ. 2002. Identification of Malassezia species from patient skin scales by PCR-RFLP. Clin Microbiol Infect 8:162–173. doi: 10.1046/j.1469-0691.2002.00383.x. [DOI] [PubMed] [Google Scholar]
- 30.Prohic A, Kuskunovic-Vlahovljak S, Sadikovic TJ, Cavaljuga S. 2015. The prevalence and species composition of Malassezia yeasts in patients with clinically suspected onychomycosis. Med Arch 69:81–84. doi: 10.5455/medarh.2015.69.81-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mukherjee PK, Chandra J, Retuerto M, Sikaroodi M, Brown RE, Jurevic R, Salata RA, Lederman MM, Gillevet PM, Ghannoum MA. 2014. Oral mycobiome analysis of HIV-infected patients: identification of Pichia as an antagonist of opportunistic fungi. PLoS Pathog 10:e1003996. doi: 10.1371/journal.ppat.1003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mussano F, Ferrocino I, Gavrilova N, Genova T, Dell'Acqua A, Cocolin L, Carossa S. 2018. Apical periodontitis: preliminary assessment of microbiota by 16S rRNA high throughput amplicon target sequencing. BMC Oral Health 18:55. doi: 10.1186/s12903-018-0520-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ren W, Zhang Q, Liu X, Zheng S, Ma L, Chen F, Xu T, Xu B. 2016. Supragingival plaque microbial community analysis of children with halitosis. J Microbiol Biotechnol 26:2141–2147. doi: 10.4014/jmb.1605.05012. [DOI] [PubMed] [Google Scholar]
- 34.Kruger W, Vielreicher S, Kapitan M, Jacobsen ID, Niemiec MJ. 2019. Fungal-bacterial interactions in health and disease. Pathogens 8:70. doi: 10.3390/pathogens8020070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Freifeld AG, Bow EJ, Sepkowitz KA, Boeckh MJ, Ito JI, Mullen CA, Raad II, Rolston KV, Young JA, Wingard JR, Infectious Diseases Society of America. 2011. Clinical practice guideline for the use of antimicrobial agents in neutropenic patients with cancer: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis 52:e56–e93. doi: 10.1093/cid/cir073. [DOI] [PubMed] [Google Scholar]
- 36.Nash AK, Auchtung TA, Wong MC, Smith DP, Gesell JR, Ross MC, Stewart CJ, Metcalf GA, Muzny DM, Gibbs RA, Ajami NJ, Petrosino JF. 2017. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 5:153. doi: 10.1186/s40168-017-0373-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 38.Galloway-Pena JR, Smith DP, Sahasrabhojane P, Ajami NJ, Wadsworth WD, Daver NG, Chemaly RF, Marsh L, Ghantoji SS, Pemmaraju N, Garcia-Manero G, Rezvani K, Alousi AM, Wargo JA, Shpall EJ, Futreal PA, Guindani M, Petrosino JF, Kontoyiannis DP, Shelburne SA. 2016. The role of the gastrointestinal microbiome in infectious complications during induction chemotherapy for acute myeloid leukemia. Cancer 122:2186–2196. doi: 10.1002/cncr.30039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galloway-Pena JR, Smith DP, Sahasrabhojane P, Wadsworth WD, Fellman BM, Ajami NJ, Shpall EJ, Daver N, Guindani M, Petrosino JF, Kontoyiannis DP, Shelburne SA. 2017. Characterization of oral and gut microbiome temporal variability in hospitalized cancer patients. Genome Med 9:21. doi: 10.1186/s13073-017-0409-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 42.Edgar RC. 2013. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 43.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koljalg U, Larsson KH, Abarenkov K, Nilsson RH, Alexander IJ, Eberhardt U, Erland S, Hoiland K, Kjoller R, Larsson E, Pennanen T, Sen R, Taylor AF, Tedersoo L, Vralstad T, Ursing BM. 2005. UNITE: a database providing Web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol 166:1063–1068. doi: 10.1111/j.1469-8137.2005.01376.x. [DOI] [PubMed] [Google Scholar]
- 45.McMurdie PJ, Holmes S. 2013. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurtz ZD, Muller CL, Miraldi ER, Littman DR, Blaser MJ, Bonneau RA. 2015. Sparse and compositionally robust inference of microbial ecological networks. PLoS Comput Biol 11:e1004226. doi: 10.1371/journal.pcbi.1004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Differences in mycobiome α-diversity (T6) based on chemotherapy intensity. A box plot compares observed OTUs and Shannon diversity at T6 between those who received high-intensity and low-intensity chemotherapy regimens. The P values are derived from a Mann-Whitney test. Download FIG S1, JPG file, 0.1 MB (129.6KB, jpg) .
Copyright © 2020 Robinson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Taxon abundance differences by chemotherapy subtype. P values represent results of Kruskal-Wallis test performed with data from each time point. Listed are the top 5 taxa by abundance across all 6 time points. Download Table S1, DOCX file, 0.02 MB (17.5KB, docx) .
Copyright © 2020 Robinson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Taxon abundance differences at T6 by receipt of piperacillin-tazobactam. P values represent results of Mann-Whitney test comparing the relative abundances of designated taxa between those who did and did not receive piperacillin-tazobactam. Listed are the top 5 taxa by abundance at T6; however, all genera were tested. Download Table S2, DOCX file, 0.01 MB (12KB, docx) .
Copyright © 2020 Robinson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Taxon abundance differences at T6 by receipt of carbapenem. P values represent results of Mann-Whitney test comparing the relative abundances of designated taxa between those who did and did not receive a carbapenem. Listed are the top 5 taxa by abundance at T6; however, all genera were tested. Download Table S3, DOCX file, 0.01 MB (11.9KB, docx) .
Copyright © 2020 Robinson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Taxon abundance differences at T6 by receipt of cephalosporin. P values represent results of Mann-Whitney test comparing the relative abundances of designated taxa between those who did and did not receive a cephalosporin. Listed are the top 5 taxa by abundance at T6; however, all genera were tested. Download Table S4, DOCX file, 0.01 MB (11.9KB, docx) .
Copyright © 2020 Robinson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Taxon abundance differences at T6 by receipt of echinocandin. P values represent results of Mann-Whitney test comparing the relative abundances of designated taxa between those who did and did not receive an echinocandin. Listed are the top 5 taxa by abundance at T6; however, all genera were tested. Download Table S5, DOCX file, 0.01 MB (12KB, docx) .
Copyright © 2020 Robinson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Taxon abundance differences at T6 by receipt of amphotericin B. P values represent results of Mann-Whitney test comparing the relative abundances of designated taxa between those who did and did not receive amphotericin B. Listed are the top 5 taxa by abundance at T6; however, all genera were tested. Download Table S6, DOCX file, 0.01 MB (12KB, docx) .
Copyright © 2020 Robinson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Taxon abundance differences at T6 by receipt of triazole. P values represent results of Mann-Whitney test comparing the relative abundances of designated taxa between those who did and did not receive a triazole. Listed are the top 5 taxa by abundance at T6; however, all genera were tested. Download Table S7, DOCX file, 0.01 MB (12KB, docx) .
Copyright © 2020 Robinson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Differential abundance of specific taxa at T3 for acute leukemia patients who had infectious complications during induction chemotherapy. The LefSe (linear discriminant analysis effect size) algorithm was used to determine which taxonomic units were most differentially abundant among acute leukemia patients who had infectious complications during induction chemotherapy (“1”) and those who did not experience infection (“0”) at T3. Download FIG S2, JPG file, 0.04 MB (46.9KB, jpg) .
Copyright © 2020 Robinson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Differential abundance of specific taxa at T6 for acute leukemia patients who had infectious complications during induction chemotherapy. The LefSe (linear discriminant analysis effect size) algorithm was used to determine which taxonomic units were most differentially abundant among acute leukemia patients who had infectious complications during induction chemotherapy (“1”) and those who did not experience infection (“0”) at T6. Download FIG S3, JPG file, 0.04 MB (46.3KB, jpg) .
Copyright © 2020 Robinson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
All 16S rRNA gene sequences were deposited in the NCBI Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) under BioProject identifiers (IDs) PRJNA352060 and PRJNA526551, whereas ITS2 sequences were submitted under the BioProject ID PRJNA599151 (the authors could not make this BioProject record available at the time of this paper’s publication due to circumstances related to the COVID-19 pandemic, but it will be made accessible as soon as possible after publication).