

Abstract
Glycine riboswitches utilize both single- and tandem-aptamer architectures. In the tandem system, the relative contribution of each aptamer toward gene regulation is not well understood. To dissect these contributions, the effects of 684 single mutants of a tandem ON switch from Bacillus subtilis were characterized for the wild-type construct and binding site mutations that selectively restrict ligand binding to either the first or second aptamer. Despite the structural symmetry of tandem aptamers, the response to these mutations was frequently asymmetrical. Mutations in the first aptamer often significantly weakened the K1/2, while several mutations in the second aptamer improved the amplitude. These results demonstrate that this ON switch favors ligand binding to the first aptamer. This is in contrast to the tandem OFF switch variant from Vibrio cholerae, which was previously shown to have preferential binding to its second aptamer. A bioinformatic analysis of tandem glycine riboswitches revealed that the two binding pockets are differentially conserved between ON and OFF switches. Altogether, this indicates that tandem ON switch variants preferentially utilize binding to the first aptamer to promote helical switching, while OFF switch variants favor binding to the second aptamer. The data set also revealed a cooperative glycine response when both binding pockets were maximally stabilized with three GC base pairs. This indicates a cooperative response may sometimes be obfuscated by a difference in the affinities of the two aptamers. This conditional cooperativity provides an additional layer of tunability to tandem glycine riboswitches that adds to their versatility as genetic switches.
Keywords: riboswitch, glycine, tandem, transcription termination, cooperativity
INTRODUCTION
Riboswitches are noncoding RNA elements typically found in the 5′-untranslated region of bacterial mRNA. They contain structured aptamer domains that, upon ligand binding, typically alter gene expression by stabilizing one of two or more competing helices in a downstream expression platform. To date, over 40 different classes of riboswitches have been identified (McCown et al. 2017). Most of these utilize a single aptamer to bind their ligand. However, a few riboswitch classes (glycine, glutamine, and guanidine-II) have been identified that use multiple aptamers, with distinct binding sites, followed by a single expression platform to regulate gene expression (Mandal et al. 2004; Ames and Breaker 2011; Sherlock et al. 2017). Similar to these two aptamer systems, two riboswitch classes (tetrahydrofolate—THF and cyclic di-adenosine monophosphate—c-di-AMP) are capable of binding two ligand molecules across distinct binding sites within a single aptamer domain (Trausch et al. 2011; Gao and Serganov 2014; Ren and Patel 2014).
Why do some riboswitches use multiple binding sites when the majority do not? In the case of the guanidine-II riboswitch, all known examples contain two consecutive aptamers that each bind a single molecule of guanidine (Sherlock et al. 2017). In-line-probing experiments indicate that ligand binding is cooperative in this system (Sherlock et al. 2017). This cooperativity may provide a competitive advantage over a single aptamer system as it provides a more “digital” response curve. Alternatively, and more likely given the architecture of the RNA, guanidine-II riboswitches may simply require the folding of both aptamers to achieve ligand binding and modulation of gene expression.
For the glycine riboswitch, the answer to this question is more complicated. Although the majority of riboswitches in this class utilize two consecutive aptamers with distinct binding sites to control a single expression platform, a significant fraction (about one-third) only have one aptamer (Fig. 1A–C; Torgerson et al. 2018). These single-aptamer versions are sometimes referred to as singlets and are subdivided into two types based on whether the aptamer is followed (type-1) or preceded (type-2) by a short hairpin that is required for ligand binding (Ruff et al. 2016). Both the singlet and tandem versions of the glycine riboswitch have been experimentally validated to bind ligand and modulate gene expression in vitro (Mandal et al. 2004; Ruff et al. 2016; Torgerson et al. 2018). Thus, the presence of both aptamers clearly is not a requirement for functional regulation of gene expression in this system.
FIGURE 1.

Glycine riboswitch types and scheme for gene regulation by a transcriptional ON tandem glycine riboswitch. Cartoon representation of the tandem (A), type-1 singlet (B), and type-2 singlet (C) variants of the glycine riboswitch. Two A-minor interactions (labeled as α and β) that occur between adjacent P1 and P3 helices are conserved among tandem systems. Singlet systems each retain one of these interactions. Glycine (orange) binds within the P3 helix of each aptamer domain. (D) Cartoon schematic of ligand-induced helical switching by a transcriptional ON tandem glycine riboswitch. Ligand binding stabilizes the P1 helix of aptamer 2, which acts as an antiterminator and promotes the production of full-length mRNA.
Early studies of the tandem glycine riboswitch indicated that its two aptamers bind glycine in a cooperative manner, similar to guanidine-II riboswitches (Mandal et al. 2004; Lipfert et al. 2007; Kwon and Strobel 2008; Erion and Strobel 2011). The more “digital” response curve this provides has been presented as an explanation for the prevalent tandem architecture of this system (Mandal et al. 2004). However, in 2012, a conserved leader sequence was identified upstream of the first aptamer domain that base pairs with the linker region between the two aptamers to form a “P0” helix. This helix often comprises one side of a canonical kink-turn motif (Kladwang et al. 2012). Glycine riboswitches containing the leader sequence show improved binding affinities, but no longer display cooperative glycine binding (Sherman et al. 2012; Baird and Ferré-D'Amaré 2013; Ruff and Strobel 2014). This loss of cooperativity reopened the debate as to why the tandem architecture is so prevalent among glycine riboswitch systems.
Structural studies of the tandem glycine riboswitch revealed that the two aptamers form a semisymmetric dimer that is mediated by an extensive network of long-range tertiary interactions (Huang et al. 2010; Butler et al. 2011). These interactions include two A-minor motifs, which occur between adenines in the P3 helix of either aptamer and the P1 helix of the adjacent aptamer (denoted as α and β in Fig. 1A). There is also a semiconserved Hoogsteen base pair between nucleotides in the J3a/3b junction of each aptamer (denoted as the γ interaction; not depicted in Fig. 1A). The binding pocket associated with each aptamer is nested within each P3 helix adjacent to these long-range interactions. Both pockets are comprised of approximately five base pairs, starting with a conserved base-triple toward the start of the P3 helices. For the other four base pairs, three are conserved as canonical GC base pairs, while the fourth is typically a purine-U pair (Barrick and Breaker 2007). Within each pocket, glycine binds to a bulged uracil located between the second and third of these five base pairs, and additionally forms direct contacts to a magnesium ion (Huang et al. 2010; Butler et al. 2011). Previous experiments have shown that ligand binding to either aptamer does not require occupation of the binding site in the other aptamer (Ruff and Strobel 2014). However, these experiments also showed that, in the context of the tandem OFF switch from Vibrio cholerae, ligand binding to the first aptamer does depend heavily on the formation of the dimer interface (the α, β, and γ interactions). Ligand binding in the second aptamer was much less dependent on these interactions for this OFF switch.
Here, we present a bioinformatic analysis of tandem glycine riboswitches based on the genetic context of each RNA. This analysis reveals several differences in conservation between glycine riboswitches that are predicted to function as either ON or OFF switches. Based on these results, we set out to explore the sequence requirements for gene regulation by a tandem ON switch. We compare this to previous observations reported for a tandem OFF switch from V. cholerae (Ruff and Strobel 2014). We applied a sequencing-based high-throughput technique termed SMARTT (Sequencing-based Mutational Analysis of RNA Transcription Termination; Torgerson et al. 2018) to generate the in vitro transcription termination profiles of 684 single-point mutants within the aptamer and expression-platform domains of the wild-type (WT) tandem ON switch from Bacillus subtilis. We also collected the profiles of these 684 mutants in the background of two binding-site mutations that each selectively limit ligand binding to either the first or second aptamer. Despite their structural symmetry, our results demonstrate that the two aptamers of tandem systems provide asymmetric contributions toward helical switching in a manner that is dependent on whether the RNA is utilized as an ON or OFF switch. The data also provide insights into the cooperative nature of these tandem glycine riboswitch systems.
RESULTS
Bioinformatic analysis of tandem glycine riboswitches
We updated the consensus sequence of the tandem variant of glycine riboswitches to include new structural motifs that were identified subsequent to the most recent consensus alignment (Barrick and Breaker 2007; Kladwang et al. 2012; Sherman et al. 2012). This was accomplished using a randomly selected subset of 500 out of the 11,626 (4%) unique tandem glycine riboswitch sequences that were identified during a previous bioinformatic search (Fig. 2A; Supplemental Fig. S1; Torgerson et al. 2018). This sample size was chosen because it is expected to be sufficiently large to provide a reasonable statistical measure of covariation and sequence conservation, but is also small enough to allow for manual alignment of the kink-turn and pseudoknot motif regions (see below).
FIGURE 2.

Glycine riboswitch consensus alignments and gene associations. (A) Updated consensus sequence made using 500 randomly selected tandem glycine riboswitch sequences. (B) Gene associations of genes controlled by tandem glycine riboswitches. Consensus alignments are also shown for tandem glycine riboswitches found immediately upstream of genes encoding aminomethyltransferases (C), glycine dehydrogenases (D), and sodium:alanine symporters (E).
We first analyzed all of the sequences in this sample as a group before subdividing the sequences based on their predicted function as an ON or OFF switch as inferred based on the genetic context of the riboswitch (discussed below). The revised consensus sequence for the group retains the core elements identified in previous reports with the addition of two partially conserved features—a pseudoknot and a kink-turn motif (Mandal et al. 2004; Barrick and Breaker 2007). The pseudoknot was observed in 79% of these sequences and is denoted as P5 in Figure 2. It occurs between the P3b stem–loop in aptamer 1 and the region immediately downstream from the P1 helix in aptamer 2. This interaction was first described by Kladwang and coworkers as a possible motif in 2012, but has yet to be experimentally validated in the tandem system (Kladwang et al. 2012). The motif most commonly occurs as a polycytidine tract within the P3b stem–loop that pairs with a polyguanine tract located immediately following the second aptamer. When present, the pseudoknot is usually mutually exclusive of a portion of the predicted alternative helix in the expression platform, suggesting it plays a role in helical switching. However, this pseudoknot does not appear to be absolutely required for gene regulation given that it is only semiconserved.
Kladwang and coworkers also identified a canonical kink-turn motif in 28% of the sequences in their data set (Kladwang et al. 2012). This interaction was shown to improve the binding affinities of each aptamer, while also abolishing the previously observed cooperative binding response (Sherman et al. 2012). The kink-turn is present in all three of the most commonly characterized versions of the tandem glycine riboswitch (i.e., the V. cholerae, B. subtillis, and Fusobacterium nucleatum glycine riboswitches) and appears to aid in preforming long-range inter-aptamer tertiary contacts for these riboswitches (Baird and Ferré-D'Amaré 2013). A canonical kink-turn motif was observed in a similar fraction of the present data set (39%). As with the Kladwang data set, a P0 helix could often be identified, even when a canonical kink-turn could not. It is possible that the intervening region between the P0 and aptamer 1 P1 helices of the remaining ∼60% of tandem sequences produce a fold similar to a kink-turn. However, no alternative conserved sequences were identified for these constructs.
The genes that are controlled by a riboswitch signal whether the riboswitch is likely to up-regulate or down-regulate gene expression upon ligand binding. Of the sequences with annotated downstream genes, 81% are involved in glycine catabolism (or are otherwise related to amino acid metabolism) and another 15% regulate genes annotated as amino acid transporters (Fig. 2B). The majority of the final 4% of sequences were located upstream of genes encoding proteins predicted to be associated with the cell membrane, indicating that many of these may function as transporters as well. Glycine riboswitches located upstream of glycine cleavage system genes have been shown to activate gene expression under high glycine conditions (Mandal et al. 2004; Tezuka and Ohnishi 2014; Khani et al. 2018). In contrast, a glycine riboswitch located upstream of a sodium:alanine symporter in Streptococcus pyogenes was recently reported to repress gene expression under high glycine conditions (Khani et al. 2018). This suggests that >80% of tandem glycine riboswitches function as ON switches and <20% function as OFF switches.
To determine if there were any differences in conservation between the ON and OFF switch versions of tandem glycine riboswitches, we binned the manually aligned 500 sequences based on the identity of the downstream genes and generated individual consensus sequences for the three most abundant gene groups: aminomethyltransferases (ONAMT), glycine dehydrogenases (ONGD), and sodium:alanine symporters (OFFSAS; Fig. 2C–E). These three gene groups account for 81% of glycine riboswitches located upstream of glycine catabolism genes and 79% of amino acid transporters and thereby represent the majority of ON and OFF switch variants. However, the original consensus sequences generated for the ONGD and OFFSAS riboswitch groups (obtained by subdividing the 500 manually aligned sequences) were each based on fewer than 100 sequences. To improve the statistical significance of these alignments, the ONGD and OFFSAS riboswitch groups were expanded to include 250 and 574 sequences, respectively (see Materials and Methods for more details).
In general, the largest differences within the two aptamer domains were observed when comparing the ONAMT and ONGD switches to the OFFSAS switches. These two sets of ON switches show high conservation for the P5 pseudoknot helix, while the OFFSAS switches have a highly conserved P4 stem in aptamer 1 and display significantly higher conservation for the P0 kink-turn motif. The P5 pseudoknot is especially well-conserved among ONGD riboswitches. Over 99% have a polycytidine tract in the P3b stem–loop that is complementary with a polyguanine tract following aptamer 2. Eighty-seven percent of ONAMT riboswitches also display a pseudoknot, but only 66% of all ONAMT riboswitches contain a polyC/polyG pseudoknot. No pseudoknots were identified for any of the OFFSAS riboswitches and most did not contain the P3b stem–loop. In contrast, the OFFSAS riboswitches contained a highly conserved kink-turn motif that was only observed in approximately one-third of ONAMT and ONGD riboswitches. Ninety percent of OFFSAS riboswitches also contained a highly conserved P4 helix in aptamer 1 that was observed in <1% of ONAMT and ONGD riboswitches.
Another striking difference between the three groups was the conservation of the two binding pockets. ONAMT riboswitches display high conservation within the first aptamer binding pocket with 99% containing three GC base pairs in Binding Site 1 compared to just 66% in Binding Site 2. ONGD riboswitches display high conservation within both aptamer binding pockets with 99% containing three GC base pairs in both binding sites. In contrast, OFFSAS riboswitches display high conservation in the second aptamer binding site and lower conservation in the first. While 98% contain three GC base pairs in Binding Site 2, only 35% have three GC pairs in Binding Site 1. These differences in conservation are intriguing as they correlate well with the observations that type-1 singlets act as ON switches, while type-2 singlets act as OFF switches (Torgerson et al. 2018). Put more simply, ONAMT riboswitches appear to conserve the first aptamer binding pocket, which structurally resembles a type-1 singlet, more than the second binding site. In contrast, OFFSAS riboswitches seem to conserve the second aptamer, which resembles the architecture of a type-2 singlet, more than the first. This indicates that there may be differences in the functional requirements of the aptamer domains for gene regulation by the ON and OFF switch versions of the glycine riboswitch. It also suggests that there may be an evolutionary link between tandem ON switches and type-1 singlets, as well as between tandem OFF switches and type-2 singlets as was recently proposed elsewhere (Crum et al. 2019).
Tandem glycine riboswitch aptamers provide differing contributions to helical switching
We set out to assess the functional importance of the structural contacts formed by the aptamer and expression-platform domains of tandem glycine riboswitch ON switch systems for comparison with the observations previously reported for a tandem OFF switch (Ruff and Strobel 2014). To accomplish this, we used SMARTT, a sequencing-based high-throughput assay that we developed to generate large numbers of ligand-dependent in vitro transcription termination profiles (Torgerson et al. 2018). SMARTT uses RNA sequencing to monitor the termination efficiencies of transcriptional riboswitches as a function of ligand concentration (Fig. 3A). We focused our efforts on the tandem ON switch from B. subtilis, a well-characterized transcriptional ONAMT switch that is located within the gcv operon of the B. subtilis genome (Mandal et al. 2004; Sherman et al. 2012; Baird and Ferré-D'Amaré 2013; Babina et al. 2017). Under high glycine conditions, the P1 helix of the second aptamer becomes stabilized in this system and functions as an antiterminator helix, preventing the formation of a downstream terminator hairpin (Fig. 1D). As a result, the production of full-length mRNA encoding glycine-cleavage-system proteins increases.
FIGURE 3.

SMARTT schematic. (A) Schematic of the strategy used for Sequencing-based Mutational Analysis of RNA Transcription Termination (SMARTT). Mutant libraries were produced through error-prone PCR (mutations are depicted by yellow boxes) and used as template DNA in transcription reactions containing varying amounts of ligand. The resulting RNA was prepared for high-throughput sequencing (Illumina adapters are shown in cyan). Each sequence was computationally analyzed to produce response profiles for all variants with zero or one mutations between positions 8 and 235 of the parent construct simultaneously. Representative response profiles are shown for mutants that alter the fit parameters Ymin (B), Ymax (C), amplitude (D), and K1/2 (E). Error bars represent the standard deviation across two replicates at each data point. Figure and legend are adapted from Torgerson et al. (2018).
Using SMARTT, complete ligand-dependent in vitro transcription termination profiles were simultaneously obtained for WT and 684 single-point mutants across positions 8 through 235 of the B. subtilis tandem ON switch. To accomplish this, a mutant library was generated by error-prone PCR and used as template for in vitro transcription reactions with varying concentrations of glycine (Supplemental Tables S1, S2). The full-length and truncated RNAs produced for each construct in the library were then prepared and sent for high-throughput sequencing. Full response profiles were generated by identifying any mutations present and counting the number of full-length and truncated sequences observed at each glycine concentration for each variant with zero or one mutations (read coverages are provided for these variants in a Supplemental Excel file). The data associated with each profile was then individually fit to a modified version of the binding equation (Equation 1):
where Ymin and Ymax are the fraction of full-length RNA produced in the presence of zero ligand and saturating ligand concentrations, respectively, X is the ligand concentration, and K1/2 is the point of half-maximal termination. The fit values generated for each parameter were consistent across two replicates (Supplemental Fig. S1). Three constructs were also tested using a low-throughput gel-based method; similar profiles and parameter values were obtained with both assays (Supplemental Figs. S3, S4; Supplemental Table S3).
Changes to the transcription termination profile for each mutation inform how the energetic properties of the system have been altered. When compared to WT, many of the 684 single-point mutants exhibited altered values for one or more of the fit parameters (Ymin, Ymax, and K1/2; Fig. 3B–E). These parameters are defined by the energetics of the system. The values generated for Ymin and Ymax are determined by the relative stabilities of the terminator and antiterminator helices in the absence or presence of ligand. K1/2 is related to the aptamer binding affinity. Changes to this parameter typically indicate that the binding affinity has been altered. Finally, in some cases, there was no change in termination efficiency over the range of ligand concentrations tested. This is most likely due to mutations that cause large defects in ligand binding or disrupt the communication pathway between the aptamer and expression platform domains. In those cases, K1/2 could not be accurately determined.
To better visualize the effects induced by the mutations assayed, the mean value observed at each position for Ymin, Ymax, amplitude (Ymax − Ymin), and K1/2 was mapped onto the predicted RNA secondary structure (Fig. 4A–D). A full list of the values obtained for each parameter for each mutation is provided in the Supplemental Material. The majority of the mutations that significantly altered Ymin were located in the expression platform (Fig. 4A). In contrast, mutations in the aptamer domains generally did not significantly affect this parameter. As a result, the aptamer domains depicted in the heat maps associated with Ymax and amplitude appear very similar overall (Fig. 4B,C).
FIGURE 4.

Heat maps associated with the WT construct. The mean Ymin (A), Ymax (B), amplitude (C), and K1/2 (D) values for the three single-point mutants at each position are mapped onto the predicted RNA secondary structure. Mutations with error values larger than the generated parameter value for amplitude or K1/2 were excluded from the mean values plotted. Mutants were also filtered if the amplitude value generated was <5% or >100%. Positions with one to two mutants excluded are denoted by a red border. Positions where all three mutant constructs were excluded are grayed out. Heat map scales are based on the parameter values generated for the WT construct. White-filled circles denote that the parameter value is approximately equal to the WT value. Red-filled circles denote that the parameter value has decreased relative to WT, and blue-filled circles denote that the value has increased relative to WT. For K1/2, the apparent change in free energy (ΔΔG) is mapped. This was done as cumulative effects associated with this parameter are expected to be multiplicative rather than additive given the relationship between K1/2 and binding affinity. As such, negative values (which are associated with a lower K1/2 value) are depicted in blue and positive values (associated with a higher K1/2 value) are depicted in red.
The heat maps associated with Ymax/amplitude and K1/2 reveal an intriguing asymmetry between the two aptamer domains. Mutations within the first aptamer, particularly near the binding pocket (located in the P3 helix), produced much more significant effects on K1/2 compared to analogous mutations in the second aptamer (Fig. 4D). Meanwhile, several mutations near the binding pocket in the second aptamer and within the set of purine-purine pairs that stack on top of the P1 helix of the second aptamer resulted in significantly increased Ymax and amplitude values compared to WT. Almost none of the mutations in the first aptamer result in any significant increase to these parameters. Together, these results indicate that binding to the second aptamer is suboptimal in the WT construct. This is in contrast to the tandem glycine OFF switch from Vibrio cholerae, which was previously shown to have preferential binding to its second aptamer (Ruff and Strobel 2014).
Single ligand occupancy is sufficient for helical switching
Given this result, we set out to dissect the individual contributions that each aptamer binding pocket provides to helical switching. Previously, Ruff and Strobel demonstrated that mutating a highly conserved uracil to an adenine in either of the two aptamer binding pockets of the tandem OFF switch from Vibrio cholerae prevents ligand binding by that aptamer, but does not significantly alter the binding affinity of the other aptamer (Ruff and Strobel 2014). These uracil nucleotides each form two hydrogen bonds with glycine (Fig. 5A; Butler et al. 2011). Therefore, substituting either uracil with a larger base appears to sterically occlude glycine from entering the binding site and disrupts the formation of these hydrogen bonds.
FIGURE 5.

Characterization of ligand response for WT and potential single-occupancy constructs. (A) Top-down view of the aptamer 1 binding pocket from the crystal structure of the F. nucleatum tandem glycine riboswitch (PDB ID: 3P49; Butler et al. 2011). Two hydrogen bonds are made between glycine and U81. (B) SMARTT-generated transcription termination response profiles for WT and all four uracil-to-purine binding site mutations. (C) Binding curves generated by equilibrium dialysis for WT and the binding site mutants U81G, U179G, and U81G/U179G. Error bars represent the standard deviation across three replicates at each data point.
We analyzed the individual SMARTT-generated profiles from our data set that were associated with analogous binding site mutations to determine their effects on helical switching (Table 1; Fig. 5B). In the B. subtilis construct tested here, the two uracil nucleotides that form direct contacts with glycine are located at positions 81 and 179. We focus here on U-to-G, rather than U-to-A mutations, as one of the U-to-A binding site mutations (U179A) behaves poorly in this in vitro system, possibly due to misfolding (see Supplemental Text). Mutating the uracil at position 81 to guanine (U81G) resulted in an eightfold decrease to the K1/2 (356 ± 6 µM) and slightly decreased the observed amplitude (26 ± 2%) relative to WT (K1/2: 43 ± 4 µM; Amplitude: 33 ± 1%). This curve and these values are nearly identical to those produced by U81A. In contrast, mutating the uracil located in the second aptamer binding pocket to guanine (U179G) resulted in a construct with a K1/2 (30 ± 10 µM) that was roughly equal WT and had a similar amplitude (20 ± 1%) to U81G. Although each of the two binding site mutants produced a small increase to Ymin (WT: 20 ± 4%; U81G: 29 ± 8%; U179G: 34 ± 6%), their profiles match the WT fit well overall.
TABLE 1.
SMARTT-generated fit parameters for WT and binding site mutants

For additional comparison, the binding affinities of WT, U81G, U179G, and the double mutant U81G/U179G were also determined (Table 2; Fig. 5C). Consistent with previous reports (Sherman et al. 2012; Baird and Ferré-D'Amaré 2013; Ruff and Strobel 2014), the WT construct displayed a Kd of 14 ± 1 µM when fit to a one-site binding equation and had a Hill coefficient near unity (1.08 ± 0.01) when fit to the Hill equation. Under the experimental conditions tested, only the affinity of the strongest binding site is generally observable unless both sites have near equal affinities. Consistent with this, U81G and U179G displayed affinities of 220 ± 40 µM (16-fold weaker than the WT) and 11 ± 3 µM (roughly equivalent to WT), respectively. These differences in affinity relative to WT are comparable to the differences in K1/2 observed for these constructs. The double mutant U81G/U179G did not display any ligand binding up to the highest RNA concentration assayed (400 µM), consistent with both binding sites being inactive.
TABLE 2.
Binding affinities of WT and the U-to-G binding site mutants

To verify that these U-to-G mutations selectively prevent glycine from binding in their associated aptamer, the binding stoichiometries of these constructs were determined (Table 3). As expected, the WT construct binds up to two equivalents of glycine, the single-point mutants each bind up to one equivalent, and the double mutant binds zero equivalents. Combined with the transcription termination analyses, this demonstrates that a single binding event in either aptamer is sufficient to promote helical switching by tandem glycine riboswitch systems, consistent with previous reports (Babina et al. 2017).
TABLE 3.
Binding stoichiometries of WT and the U-to-G binding site mutants

Ligand binding in aptamer 2 depends on the formation of the dimer interface between the two aptamers
The crystal structure of the F. nucleatum tandem glycine riboswitch revealed a network of long-range tertiary interactions that connect the two tandem aptamers in the ligand bound state. These interactions include two A-minor motifs (α and β) and a semiconserved Hoogsteen base pair between nucleotides in the two P3 helices (γ). Previous work demonstrated that disrupting these inter-aptamer tertiary interactions had a significant impact on the binding affinity of the first aptamer of the tandem OFF switch from V. cholerae, but minimal impact on the affinity of the second aptamer binding site (Fig. 6A,B; Ruff and Strobel 2014). This observation indicates that the fold of the first aptamer may be suboptimal in the WT version of this OFF switch and that ligand binding in the second aptamer provides a scaffold for the folding of the first aptamer.
FIGURE 6.
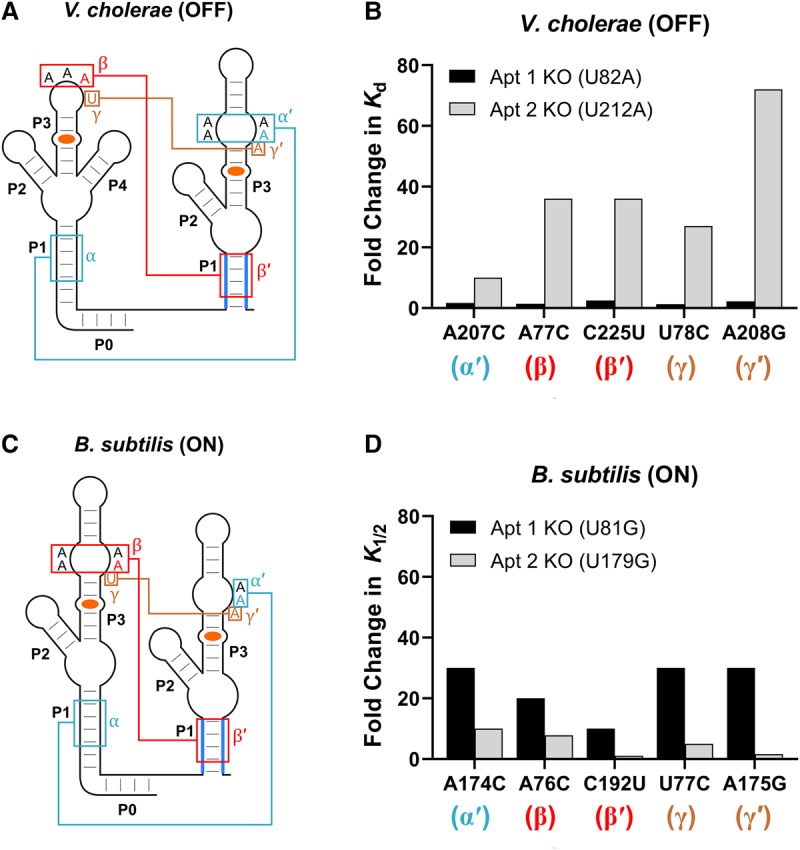
Effects of aptamer interface mutations on ligand binding. (A) Secondary structure of the V. cholerae tandem OFF glycine riboswitch. Nucleotides expected to be involved in long-range tertiary interactions between the two aptamers are labeled α/α′, β/β′, and γ/γ′. (B) Bar graph depicting the fold change in the Kd measured for individual binding sites relative to the parent construct as a result of mutations expected to disrupt the α, β, and γ interactions of the V. cholerae tandem glycine riboswitch. These data were collected by Ruff and Strobel (2014) through equilibrium dialysis. (C) Secondary structure of the B. subtilis tandem ON glycine riboswitch. Nucleotides expected to be involved in long-range tertiary interactions between the two aptamers are again labeled α/α′, β/β′, and γ/γ′. (D) Bar graph depicting the fold change in the K1/2 measured for individual binding sites relative to the parent construct as a result of mutations expected to disrupt the α, β, and γ interactions of the B. subtilis tandem glycine riboswitch. K1/2 values were generated through SMARTT.
To test if this is also observed in the context of a tandem ON switch, we generated complete ligand-induced in vitro transcription termination profiles for the 684 single point mutants between positions 8 and 235 of the B. subtilis ON switch in the background of either U81G or U179G using SMARTT (Supplemental Figs. S5–S8). We then compared the effects of the analogous mutations on K1/2 for each of the two binding sites of the B. subtilis glycine riboswitch. Intriguingly, these analogous mutations, which alter the α, β, and γ tertiary interactions, have a significantly larger impact on ligand binding by the second aptamer versus the first aptamer in this ON switch (Fig. 6C,D; Supplemental Fig. S9)—the opposite trend as was observed for the V. cholerae OFF switch. This suggests that ligand binding to the first aptamer of this ON switch may act as a scaffold for the folding of the second aptamer. We note that the trends are imperfectly reversed between the B. subtilis ON and V. cholerae OFF switches as there are slight differences in the magnitude of the effects caused by the analogous mutations of the adjacent aptamers. However, we expect that these differences are likely due to the distinct sequence contexts of the mutants across these two riboswitches and their aptamers.
A mutation in binding site 2 reveals a cooperative ligand response
Early studies of the glycine riboswitch showed that ligand binding by the two aptamer domains occurs cooperatively. However, these studies were performed using a truncated version of the riboswitch. In 2012, a conserved 5′-leader sequence was identified that often forms part of a kink-turn motif. When this leader sequence was included, cooperative ligand binding was no longer observed for the three RNAs tested (the B. subtilis ON switch, V. cholerae OFF switch, and the F. nucleatum OFF switch).
To determine if any of the mutants in our data set created a cooperative ligand response for the B. subtilis construct, we calculated Hill coefficients for all 684 single point mutants in the WT data set. Sixty-three of the mutants in the data set displayed a Hill coefficient greater than 1.2. However, one mutant stood out from the rest. In addition to displaying a Hill coefficient of 1.28 ± 0.01, U178C also exhibited an amplitude (69 ± 1%) that was roughly double the value observed for the WT construct (33 ± 1%) and had a slightly improved K1/2 (33 ± 5 µM) compared to WT (43 ± 4 µM; Fig. 7; Table 4). This mutation is located within the P3a stem of the second aptamer. It converts the base pair that closes off the top of the binding pocket in the second aptamer from a GU wobble pair to a canonical GC base pair. This is the region that displays lower conservation of GC base pairs in tandem ON switches.
FIGURE 7.
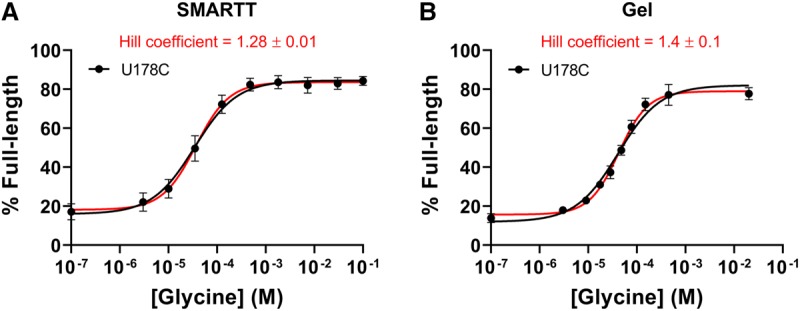
Transcription termination profile of U178C. The transcription termination profile of U178C obtained by SMARTT (A) or the conventional gel-based assay (B). Fits to the one-site equation are shown in black. Fits to the Hill equation are shown in red. Error bars represent the standard deviation across two (SMARTT) or three (gel) replicates at each data point.
TABLE 4.
Parameters for additional binding site mutants

The Hill coefficient observed by SMARTT for U178C only has a few data points around the inflection point. Because of this, we verified the observed cooperativity using the conventional gel-based assay and increased the density of concentrations sampled near the inflection point. We obtained a Hill coefficient of 1.4 ± 0.1 with this method (Fig. 7B).
To determine how U178C affects each aptamer individually and to gain insight as to why the system displays cooperative ligand binding, we examined the effects of the mutation in the background of the two binding site mutations (U81G and U179G; Table 4; Supplemental Fig. S10). When U178C was introduced in the background of U81G (the aptamer 1 binding site mutation), it resulted in a similar improvement to the amplitude (50 ± 1%) compared to the parent construct (25 ± 1%), as well as a 4.6-fold improvement to the K1/2. In contrast, U178C did not significantly alter the K1/2 or amplitude when performed in the background of U179G (the aptamer 2 binding site mutant). This indicates that the increase in amplitude observed for this substitution is due to improvements in ligand binding and folding of the second aptamer.
We then examined the effects of converting the opposite mutation at the analogous position in aptamer 1 to determine if the change in affinity is consistent across both aptamers (Table 4; Supplemental Fig. S10). C80U produces a GC-to-GU base pair conversion at the analogous position in the first aptamer. This substitution produces an approximately threefold weaker K1/2 in the context of the WT construct and a fourfold weaker K1/2 in the context of the second aptamer binding site mutation U179G. No change in K1/2 was observed in the context of the first aptamer binding site mutation U81G. In all three contexts, the amplitude produced is comparable to the value observed for the parent construct. Altogether, this indicates that a GC base pair at this position provides a three- to fourfold improvement in K1/2 compared to a GU base pair in either aptamer. Given that the amplitude was not altered by C80U (versus in aptamer 2 where the difference between a GC or GU pair at this position produced approximately a twofold change in amplitude), these results also indicate that factors beyond the presence of a GC or GU mutation at this position contribute to determining the extent to which ligand binding by each aptamer stabilizes the P1 helix of the expression platform.
DISCUSSION
The tandem architecture used by the majority of glycine riboswitches is a rare feature among the known riboswitch classes. Although single-aptamer versions of this riboswitch exist, the high conservation of its dual-aptamer system implies that it provides some additional fitness benefit within the glycine riboswitch class. The in vitro transcription termination data presented here provide new insights into the mechanism and versatility of gene regulation by the tandem system.
There is a clear asymmetry between the two aptamers of the tandem ON switch from B. subtilis. Mutations within the first aptamer of this riboswitch often require a significantly higher ligand concentration to reach the point of half-maximal termination (K1/2), which is likely due to its tighter binding affinity relative to the second aptamer. Meanwhile, several mutations at key positions in the second aptamer significantly improve the overall amplitude of response. This suggests that the fold of the second aptamer is suboptimal in the WT system as the maximum readthrough efficiency (Ymax) is lower in the WT system compared to these mutants. This means that less free energy is available to stabilize the alternate conformation of the expression platform from ligand binding to the second aptamer. Instead, ligand binding to the first aptamer appears to contribute most of the free energy for helical switching for the WT version of this ON switch.
We also observed an asymmetry in the dependence of each aptamer on long-range tertiary interactions. Mutations disrupting these interactions at the interface between the two aptamers (the α, β, and γ interactions) produced significant effects on the affinity of the second aptamer, but had a much smaller effect on the affinity of the first aptamer. This suggests that the ability of the second aptamer to bind ligand is strongly dependent on the formation of the dimer interface, whereas the ability of the first aptamer is not. Intriguingly, a related asymmetry was observed previously for the tandem OFF switch from V. cholerae, but with the opposite trend—the affinity of the first aptamer was shown to be dependent on the formation of the dimer interface between the two aptamer domains, while the second aptamer was not (Ruff and Strobel 2014).
It is notable that in both cases, the binding pocket that was less dependent on these long-range contacts contains three GC base pairs (the maximum number typically observed), whereas the binding site that was strongly dependent on the formation of these contacts only contains two GC pairs. The bioinformatics presented here indicates that tandem ON switches have a higher conservation of full GC base-pairing in the binding pocket of the first aptamer, while tandem OFF switches conserve full GC base-pairing for the second aptamer binding pocket. We demonstrated here that the affinity of each binding site of the B. subtilis ON switch improves as the number of GC base pairs increases. If this correlation holds in the sequence context of all tandem glycine riboswitches, this implies that most tandem ON switches rely on ligand binding to the first aptamer for functional modulation of gene expression, while most tandem OFF switches rely on ligand binding to the second aptamer. This is also consistent with the observation that type-1 singlets, which are structurally similar to the first aptamer of a tandem system, are predominantly ON switches. Meanwhile, type-2 singlets, which are structurally similar to the second aptamer of a tandem system, are almost exclusively OFF switches.
The observation that a single binding event in either aptamer is sufficient to promote a change in gene expression also implies that the first binding event would preorganize the adjacent unoccupied binding site in the WT system (see Fig. 8). This is based on the concept of reciprocity, which states that if A stabilizes B then B must stabilize A (Ferrell 2009). In other words, if binding in either site can stabilize the antiterminator helix (the P1 helix of aptamer 2), then the reverse must also be true—stabilization of the antiterminator helix must preorganize and stabilize both binding sites. Thus, binding in site A will stabilize the antiterminator helix, which would in turn preorganize site B. This would be expected to improve the affinity of that site and, by definition, means that the system should be cooperative.
FIGURE 8.

Thermodynamic model for ligand binding by the B. subtilis ON switch. Equilibrium arrows depicted in this model have been approximated assuming a glycine concentration between the observed K1/2 values for the two aptamer binding sites. In the apo state, formation of a terminator hairpin is favored. Based on the K1/2 values, binding of glycine (orange) to the first aptamer is favored over binding to the second aptamer. For both pathways depicted, the first binding event is sufficient to stabilize the antiterminator helix (P1 of aptamer 2) and should stabilize the dimer interface (the α and β interactions) and preform the binding pocket of the adjacent aptamer. This, in turn, will increase the probability of glycine binding to the remaining aptamer (i.e., improve the affinity of that aptamer).
The simplest and most common measurement of cooperativity is the Hill coefficient. This has been the measurement used in all studies of the glycine riboswitch. Systems with Hill coefficients greater than 1 produce sigmoidal response curves and are said to be cooperative or display positive cooperativity. Meanwhile, a value of 1 is usually interpreted as “noncooperative.” Cooperative systems produce sigmoidal binding curves because the first binding event induces a conformational change that preorganizes the second binding site. This improves the affinity of that second site to a value that is lower than the affinity of the original binding site for these systems. As a result, the second binding event is now more likely to occur than the first event, which leads to a steeper binding curve. However, a Hill coefficient greater than 1 has not been observed for any of the full-length WT tandem systems that have been tested to date. Why?
The most likely explanation is that the Hill equation, which is used to generate the Hill coefficient, assumes that the two binding sites start with equal affinities. However, the binding affinity of the second aptamer of the B. subtilis ON switch is ∼15-fold weaker than the first aptamer. Thus, as the concentration of glycine increases, ligand binding is expected to predominately proceed through a binding event in aptamer 1 followed by a binding event in aptamer 2 for the B. subtilis ON switch (Pathway 1 of Fig. 8). Yet, binding in the first aptamer may not improve the affinity of the second aptamer domain to the point where it is lower than the affinity of the first binding site. Assuming this is true, a significant fraction of the population will populate the state with a single molecule of glycine bound (upper right of Fig. 8) rather than proceeding to the state with two molecules bound (lower right of Fig. 8) while under conditions where the glycine concentration is between the two binding affinities. This could explain why a Hill coefficient of 1 is observed for the WT response curve of this riboswitch even though the system is expected to be cooperative. It could also explain why the U178C mutation, which improved the affinity of the second aptamer binding site by approximately fivefold, produced a Hill coefficient of 1.4 ± 0.1.
The two other most studied versions of the tandem glycine riboswitch are the tandem OFF switches from V. cholerae and F. nucleatum. As with the tandem ON switch from B. subtilis that was analyzed here, cooperative ligand binding was abolished for these systems upon the addition of the 5′ leader sequence (Sherman et al. 2012; Baird and Ferré-D'Amaré 2013; Ruff and Strobel 2014). Similar to the B. subtilis ON switch, both of these systems only contain three GC base pairs in one of their aptamer binding pockets (the second aptamer) and have two GC base pairs in the adjacent binding pocket. As a result, the first aptamer in these systems is likely to have a reduced binding affinity compared to the second aptamer and may not contribute as much to helical switching. This may explain why the full-length versions of these systems do not exhibit cooperative ligand binding in equilibrium-based binding assays even though ligand binding in the dominant aptamer is likely to preorganize the binding pocket of the adjacent aptamer.
Based on the bioinformatic analysis provided here, approximately two-thirds of all tandem glycine riboswitches contain two maximally stabilized binding pockets (three GC base pairs), with the three most well-studied systems to date being among the exceptions. Thus, a large fraction of glycine riboswitches may naturally display a sigmoidal ligand response curve (Hill coefficient >1). For those that do not, the presence of a second binding domain (even one with a weaker affinity) may still provide a fitness benefit by allowing for additional energy to be contributed to the expression platform upon ligand binding. With more energy available, tandem glycine riboswitches would have increased tunability over singlet systems. Not only can these systems tune the affinities of their aptamer domains to meet different physiological requirements, but also the total amplitude of response upon ligand binding, and even the extent of observable cooperativity. This tunability makes these systems incredibly versatile and allows them to meet a wide variety of biological needs.
MATERIALS AND METHODS
Bioinformatic analysis of RNA sequences
A previous bioinformatic search identified 11,626 unique tandem glycine riboswitch sequences (Torgerson et al. 2018). A pseudo-random subset of 500 of these 11,626 sequences was selected using a custom python script via the random.sample() function. Additional alignment was performed on these 500 sequences manually, with specific attention paid to the kink-turn and pseudoknot regions. A consensus sequence figure based on this alignment was created with R2R and edited with Inkscape.
The downstream gene associations were provided by the Breaker Laboratory for these sequences and were identified as previously described (Weinberg et al. 2015; Torgerson et al. 2018). The corresponding gene-association pie chart was created with Adobe Illustrator and further edited with Inkscape. Consensus sequences for the riboswitches located upstream of the top three gene groups (Aminomethyltransferases, Glycine Dehydrogenases, and Sodium:Alanine Symporters) were made using R2R and Inkscape based on a sampling of the riboswitches controlling these genes. For the aminomethyltransferase group, the sequences annotated as aminomethyltransferases within the group of 500 manually curated sequences (described above; >100 sequences) were used. For the other two gene groups, the 11,626 unique sequences were binned separately if the gene immediately following each sequence was labeled as either “glycine dehydrogenase” or “AlsT” (the most common sodium:alanine symporter annotation; 95%). A total of 250 of the sequences that regulate Glycine Dehydrogenases were then pseudo-randomly selected and additional alignment was performed manually. For the “AlsT” sequences, additional alignment was performed manually on the entire group since these sequences retained a highly conserved canonical kink-turn motif and did not contain the pseudoknot.
Design of plasmids and template DNA for gel-based in vitro transcription termination assays
The WT B. subtilis riboswitch was cloned into a pUC19 plasmid using EcoRI and BamHI restriction digest sites that were added flanking the region of interest. This included 30 nt upstream of the WT promoter through ∼30 nt downstream from its terminator (NC_000964.3/2549731-2549344). This region, and an additional ∼50 nt of the pUC19 plasmid downstream, was PCR amplified using Phusion HF DNA Polymerase (NEB or Thermo Fisher Scientific) and utilized as template DNA in the gel-based in vitro transcription termination assays. Mutant constructs (U81G, U179G, U81G/U179G) were made by QuikChange Mutagenesis. The analogous regions of these constructs were PCR amplified using Phusion HF DNA Polymerase (NEB or Thermo Fisher Scientific) for use in the gel-based in vitro transcription termination assays (see Supplemental Table S4 for the specific sequences used).
Preparation of mutagenized DNA libraries
Mutagenized libraries were created by error-prone PCR using the GeneMorph II Random Mutagenesis Kit from Agilent Technologies. Primers were chosen to flank the aptamer and expression-platform domains of the three B. subtilis constructs (WT, U81G, U179G), and mutations were randomly introduced to each of the constructs through 12 cycles of error-prone PCR. Additional DNA was subsequently added upstream of the riboswitch through a two-step PCR reaction using two forward primers with large overhangs (∼80 nt) on the 5′ ends and Phusion HF DNA Polymerase (NEB or Thermo Fisher Scientific). This was done to make the upstream region of the libraries equivalent to the DNA template used in the gel-based in vitro transcription termination assays. The region downstream from the terminator hairpin was significantly shorter than the template used in the gel-based assays (∼20 nt instead of ∼80 nt) to facilitate analysis by high-throughput sequencing. Following the two-step PCR reaction, each library was gel purified. A final PCR amplification was performed for the three libraries using Phusion HF DNA Polymerase (NEB or Thermo Fisher Scientific) to create sufficient quantities of DNA template for use in the SMARTT transcription termination reactions (see Supplemental Table S4 for the specific sequence used).
Gel-based in vitro transcription termination assays
The gel-based in vitro transcription reactions were performed as described previously (Torgerson et al. 2018). Briefly, reaction mixtures (40 mM Tris-HCl [pH 7.5], 10 mM MgCl2, 150 mM KCl, 1 mM DTT, 10 μg/mL BSA, 1% Glycerol, 25 nM DNA template, 50 μM NTPs, approximately 2.5 μCi [α-32P] GTP, 0.5 units E. Coli RNA Polymerase Holoenzyme [NEB], and varying concentrations of ligand [glycine or alanine]) were set up on ice, incubated at 37°C for 1 h, placed back on ice, and combined with 2 volumes of stop/loading buffer (25 mM EDTA, <0.1% Xylene Cyanol, <0.1% Bromophenol, ∼95% formamide). Full-length and truncated RNA transcripts produced in individual reactions were separated by 6% PAGE, visualized using a Typhoon FLA 9500 (GE Healthcare), and quantified with ImageQuant (GE Healthcare). Quantified band intensities were normalized according to the expected number of guanosine nucleotides contained in each band. Data obtained for each titration was fit to Equation 1:
where Ymin and Ymax are the fractions of full-length RNA produced in the presence of zero ligand and saturating ligand concentrations, respectively, X is the ligand concentration, and K1/2 is the point of half-maximal termination. Each titration was performed in triplicate.
Preparation of RNA for high-throughput sequencing
Preparation of RNA for high-throughput sequencing was carried out as previously described (Torgerson et al. 2018) with only minor changes, such as the sourcing of RNase H. Once prepared for sequencing, the relative concentrations of the final DNA products associated with each sample in a given titration were estimated by qPCR. Samples were combined at approximately 1:1 (for glycine-containing samples) or 0.6:1 (alanine-containing control samples:glycine-containing samples) ratios before being sent to the Yale Center for Genome Analysis (YCGA) for high-throughput sequencing. All samples of a titration were provided a unique i7 index within the Illumina adaptor. For one of the WT library titrations, only i7 indices were used. For all other titrations, samples were split into groups of no more than four samples and each group was provided a unique i5 index to prevent against potential index hopping. Titrations for each mutagenized starting library (WT, U81G, U179G) were performed in duplicate.
High-throughput sequencing
Sequencing was performed at the YCGA on either an Illumina HiSeq 4000 (2 × 150; for one of the WT library replicates) or on an Illumina NovaSeq S4 (2 × 150; for the remaining libraries). Samples were combined and mixed with high sequence diversity samples by the YCGA to prevent sequencing issues stemming from the low sequence diversity of the SMARTT samples. Approximately 9% of a lane was used per sample for the glycine samples run on the Illumina HiSeq 4000. Approximately 1.3% (WT & U179G libraries) or ∼1.5% (U81G library) of a lane was used per sample for the glycine samples run on the NovaSeq. Demultiplexing was done by the YCGA.
Analysis of sequencing results
Sequencing results were analyzed using the SMARTT–Data Analysis Pipeline as previously described (Torgerson et al. 2018). Only sequences with zero or one mutation were analyzed. Reads were labeled as terminated if the last nucleotide was between positions 233 and 239 (inclusive) and full-length if the last nucleotide fell beyond this region. This window was determined manually based on an observed glycine-dependent increase in the frequency of termination in this region. R was used to perform nonlinear regression with inverse-variance weighting to fit the data obtained for each variant to Equation 1 (above). Mutants from the WT library were additionally fit to Equation 2 to determine their Hill coefficient:
where n is the Hill coefficient.
Heat maps
Heat maps were created using the SMARTT–Heat Map Maker python script (Torgerson et al. 2018). The mean parameter value obtained across two replicates was used as the input. Mutants with an error value for either amplitude or K1/2 that was greater than the associated parameter value were excluded when calculating each mean Ymax, amplitude, and K1/2 input value. Variants were also excluded within the Heat Map Maker python script if the mean amplitude value observed was <5% or >100%, as amplitude values outside of this range typically were caused by a poor fit to Equation 1.
RNA preparation and purification
RNA was transcribed using T7 RNA polymerase via runoff transcription. The DNA template used was made by PCR amplification from the appropriate plasmid with Phusion HF Polymerase (NEB or Thermo Fisher Scientific) to allow for easy incorporation of a T7 promoter upstream of the riboswitch. Each RNA was purified by 6% denaturing PAGE, eluted into 0.3 M Sodium Acetate (pH 5.3), ethanol precipitated, and resuspended in equilibrium dialysis buffer (90 mM Tris-Borate [pH 8.3], 100 mM KCl, 10 mM MgCl2). The RNA was then buffer-exchanged four times with additional buffer and concentrated using Amicon Ultra Centrifugal Filters. Any remaining large impurities were removed by filtering the RNA through Corning Costar Spin-X centrifuge tube filters. RNA concentrations were determined by UV absorbance at 260 nm. The extinction coefficient of the WT RNA was determined by Nuclease P1 digestion, according to established protocols (Cavaluzzi and Borer 2004; Wilson et al. 2014). Briefly, ∼100 pmol of RNA was incubated for 1 h at 50°C with 200 mM sodium acetate (pH 5.2), 10 mM zinc acetate, 5 mM EDTA, and 0.5 units of Nuclease P1 enzyme (USBiological). The extinction coefficient of the fully digested sample was calculated to be 2.320 × 106 M−1 cm−1, based on the extinction coefficients of the individual nucleotides, and was used to determine the extinction coefficient of the undigested RNA sample (1.513 × 106 M−1 cm−1). The extinction coefficient of the U81G, U179G, and U81G/U179G RNA samples were assumed to be within error of the value determined for the WT RNA, which was used when calculating the concentrations of these RNA samples.
Determination of RNA binding affinities by equilibrium dialysis
Varying concentrations of RNA were combined with trace 14C-labeled glycine in dialysis buffer (90 mM Tris-Borate [pH 8.3], 100 mM KCl, 10 mM MgCl2). Mixtures were heated to 70°C for 3 min and allowed to slow cool to ∼30°C for 1 h. Samples were then loaded onto one side of a Dispo Equilibrium Dialyzer (Harvard Apparatus) with a molecular weight cutoff of either 5 or 10 kDa. An equal volume of dialysis buffer was loaded onto the opposite side of the cassette and samples were equilibrated at room temperature overnight. For the highest RNA concentrations, osmosis effects resulted in slightly elevated volumes of liquid on the RNA side of the cassette. However, these changes in volume were typically minor and were neglected. For equilibrated samples, the amount of 14C-labeled glycine on each side of the cassette was determined by scintillation counting in Ultima Gold on a PerkinElmer Tri-Carb 4910TR scintillation counter. The fraction bound was determined as follows:
Binding affinities were determined by fitting the data to Equation 4 through nonlinear least squares regression with Graphpad Prism 7:
where Y is the fraction bound, Bmax is the total amplitude, X is the RNA concentration, Kd is the binding affinity, and Y0 is the background number of counts in the absence of RNA. Data collected for the WT construct was also fit to the Hill equation (Equation 5):
where Y is the fraction bound, Bmax is the total amplitude, X is the concentration of RNA, Kd is the binding affinity, Y0 is the background number of counts in the absence of RNA, and n is the Hill coefficient.
Determination of binding stoichiometry
Samples of 200 µM or 400 µM RNA were combined with a threefold excess of cold glycine and trace 14C-labeled glycine in dialysis buffer (90 mM Tris-Borate [pH 8.3], 100 mM KCl, 10 mM MgCl2). RNA samples were refolded, loaded onto Dispo Equilibrium Dialyzer cassettes (Harvard Apparatus; 10 kDa cutoff), and equilibrated against dialysis buffer as described above. The amount of 14C-labeled glycine on each side of the cassette was again determined by scintillation counting. The number of equivalents bound was calculated as follows:
The expected equivalents bound was approximated using the quadratic binding equation:
where θ is the fractional occupancy of the binding site, R is the concentration of RNA added, L is the concentration of ligand on the RNA side of the dialysis cassette (half the amount added), and Kd is the affinity of the binding site. For WT, Equation 7 was solved twice. First, it was solved for the binding site with the stronger affinity. Then, the concentration of free ligand was updated by solving Equation 8:
Equation 7 was then solved again using the updated ligand concentration and the affinity of the weaker binding site. Adding together the fractional occupancies for the two sites provided the expected equivalents bound. This process assumes that ligand binding to the weaker binding site does not affect the fractional occupancy of the stronger site. To verify that this is the case, Equation 7 was solved again using the stronger binding affinity and an updated ligand concentration that did not include the ligand bound to the weaker site. This resulted in minimal impact to the fractional occupancy of the stronger site, indicating the assumption is valid.
DNA oligonucleotides and chemicals
DNA oligos were ordered from the W.M. Keck Oligonucleotide Synthesis Facility at Yale University unless specified otherwise. Glycine and alanine were obtained from Sigma.
Code availability
Custom Python and R scripts used in the SMARTT analyses and for the generation of heat maps are available on GitHub at https://github.com/strobel-lab/SMARTT. All other scripts related to this work are available from the corresponding author upon request.
DATA DEPOSITION
Raw sequencing data used for the analyses presented in this manuscript have been deposited in the Sequence Read Archive under Bioproject PRJNA566051. A table containing the mean fit values for all single point mutants, as well as the number of reads observed at each ligand concentration and the observed percentage of full-length RNA for each replicate, is available in the Supplemental Material. Sequence alignments (in Stockholm format) and the downstream gene associations for the tandem glycine riboswitch constructs identified in the bioinformatic searches are also available in the Supplemental Material. All other data that support the findings in this work are available from the corresponding author upon request.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Caroline Reiss, Caroline Focht, Andrew Knappenberger, and Alec Sexton for thoughtful discussions. We thank Ron Breaker and Shira Stav for providing the genes associated with the tandem glycine riboswitch sequences that were identified in our previous bioinformatic search and analyzed here. We also thank the Yale Center for Genome Analysis and the Yale Center for Research Computing for support and use of their infrastructure. C.D.T. was supported by National Institutes of Health Biophysics Training grant T32GM008283. This work was also supported by NIH grant GM022778 (S.A.S.).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.073577.119.
REFERENCES
- Ames TD, Breaker RR. 2011. Bacterial aptamers that selectively bind glutamine. RNA Biol 8: 82–89. 10.4161/rna.8.1.13864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babina AM, Lea NE, Meyer MM. 2017. In vivo behavior of the tandem glycine riboswitch in Bacillus subtilis. MBio 8: e01602-17 10.1128/mBio.01602-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird NJ, Ferré-D'Amaré AR. 2013. Modulation of quaternary structure and enhancement of ligand binding by the K-turn of tandem glycine riboswitches. RNA 19: 167–176. 10.1261/rna.036269.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick JE, Breaker RR. 2007. The distributions, mechanisms, and structures of metabolite-binding riboswitches. Genome Biol 8: R239 10.1186/gb-2007-8-11-r239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler EB, Xiong Y, Wang J, Strobel SA. 2011. Structural basis of cooperative ligand binding by the glycine riboswitch. Chem Biol 18: 293–298. 10.1016/j.chembiol.2011.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaluzzi MJ, Borer PN. 2004. Revised UV extinction coefficients for nucleoside-5′-monophosphates and unpaired DNA and RNA. Nucleic Acids Res 32: e13 10.1093/nar/gnh015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crum M, Ram-Mohan N, Meyer MM. 2019. Regulatory context drives conservation of glycine riboswitch aptamers. PLoS Comput Biol 15: e1007564 10.1371/journal.pcbi.1007564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erion TV, Strobel SA. 2011. Identification of a tertiary interaction important for cooperative ligand binding by the glycine riboswitch. RNA 17: 74–84. 10.1261/rna.2271511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell JE. 2009. Q&A: cooperativity. J Biol 8: 53 10.1186/jbiol157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao A, Serganov A. 2014. Structural insights into recognition of c-di-AMP by the ydaO riboswitch. Nat Chem Biol 10: 787–792. 10.1038/nchembio.1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Serganov A, Patel DJ. 2010. Structural insights into ligand recognition by a sensing domain of the cooperative glycine riboswitch. Mol Cell 40: 774–786. 10.1016/j.molcel.2010.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khani A, Popp N, Kreikemeyer B, Patenge N. 2018. A glycine riboswitch in Streptococcus pyogenes controls expression of a sodium:alanine symporter family protein gene. Front Microbiol 9: 200 10.3389/fmicb.2018.00200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kladwang W, Chou F-C, Das R. 2012. Automated RNA structure prediction uncovers a kink-turn linker in double glycine riboswitches. J Am Chem Soc 134: 1404–1407. 10.1021/ja2093508 [DOI] [PubMed] [Google Scholar]
- Kwon M, Strobel SA. 2008. Chemical basis of glycine riboswitch cooperativity. RNA 14: 25–34. 10.1261/rna.771608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipfert J, Das R, Chu VB, Kudaravalli M, Boyd N, Herschlag D, Doniach S. 2007. Structural transitions and thermodynamics of a glycine-dependent riboswitch from Vibrio cholerae. J Mol Biol 365: 1393–1406. 10.1016/j.jmb.2006.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal M, Lee M, Barrick JE, Weinberg Z, Emilsson GM, Ruzzo WL, Breaker RR. 2004. A glycine-dependent riboswitch that uses cooperative binding to control gene expression. Science 306: 275–279. 10.1126/science.1100829 [DOI] [PubMed] [Google Scholar]
- McCown PJ, Corbino KA, Stav S, Sherlock ME, Breaker RR. 2017. Riboswitch diversity and distribution. RNA 23: 995–1011. 10.1261/rna.061234.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren A, Patel DJ. 2014. c-di-AMP binds the ydaO riboswitch in two pseudo-symmetry–related pockets. Nat Chem Biol 10: 780–786. 10.1038/nchembio.1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff KM, Strobel SA. 2014. Ligand binding by the tandem glycine riboswitch depends on aptamer dimerization but not double ligand occupancy. RNA 20: 1775–1788. 10.1261/rna.047266.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff KM, Muhammad A, McCown PJ, Breaker RR, Strobel SA. 2016. Singlet glycine riboswitches bind ligand as well as tandem riboswitches. RNA 22: 1728–1738. 10.1261/rna.057935.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherlock ME, Malkowski SN, Breaker RR. 2017. Biochemical validation of a second guanidine riboswitch class in bacteria. Biochemistry 56: 352–358. 10.1021/acs.biochem.6b01270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman EM, Esquiaqui J, Elsayed G, Ye J-D. 2012. An energetically beneficial leader–linker interaction abolishes ligand-binding cooperativity in glycine riboswitches. RNA 18: 496–507. 10.1261/rna.031286.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezuka T, Ohnishi Y. 2014. Two glycine riboswitches activate the glycine cleavage system essential for glycine detoxification in Streptomyces griseus. J Bacteriol 196: 1369–1376. 10.1128/JB.01480-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torgerson CD, Hiller DA, Stav S, Strobel SA. 2018. Gene regulation by a glycine riboswitch singlet uses a finely tuned energetic landscape for helical switching. RNA 24: 1813–1827. 10.1261/rna.067884.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trausch JJ, Ceres P, Reyes FE, Batey RT. 2011. The structure of a tetrahydrofolate-sensing riboswitch reveals two ligand binding sites in a single aptamer. Structure 19: 1413–1423. 10.1016/j.str.2011.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg Z, Kim PB, Chen TH, Li S, Harris KA, Lünse CE, Breaker RR. 2015. New classes of self-cleaving ribozymes revealed by comparative genomics analysis. Nat Chem Biol 11: 606–610. 10.1038/nchembio.1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SC, Cohen DT, Wang XC, Hammond MC. 2014. A neutral pH thermal hydrolysis method for quantification of structured RNAs. RNA 20: 1153–1160. 10.1261/rna.045856.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.