

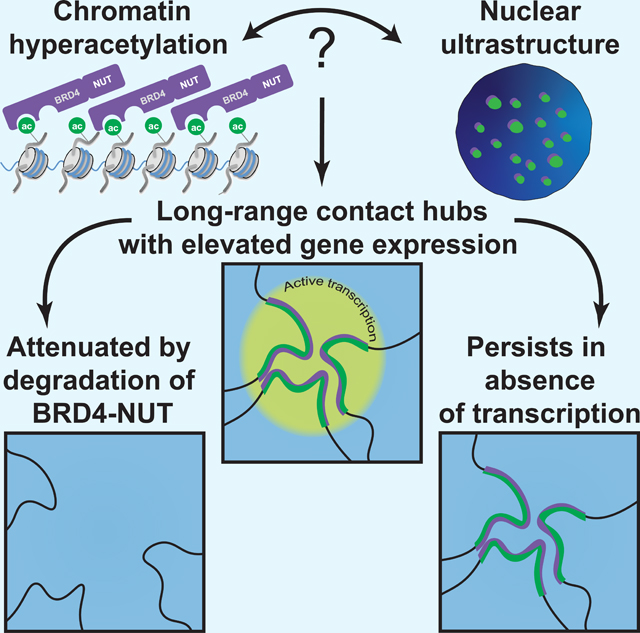
SUMMARY
Delineating how chromosomes fold at length scales beyond one megabase remains obscure relative to smaller-scale folding into TADs, loops, and nucleosomes. We find that rather than simply unfolding chromatin, histone hyperacetylation results in interactions between distant genomic loci separated by tens to hundreds of megabases, even in the absence of transcription. These hyperacetylated “megadomains” are formed by the BRD4-NUT fusion oncoprotein, interact both within and between chromosomes, and form a specific nuclear subcompartment that has elevated gene activity with respect to other subcompartments. Pharmacological degradation of BRD4-NUT results in collapse of megadomains and attenuation of the interactions between them. In contrast, these interactions persist and contacts between newly acetylated regions are formed after inhibiting RNA polymerase II initiation. Our structure-function approach thus reveals that broad chromatin domains of identical biochemical composition, independent of transcription, form nuclear subcompartments, and also indicates the potential of altering chromosome structure for treating human disease.
eTOC Blurb
Rosencrance et al. find that hyperacetylated chromatin domains formed by the BRD4-NUT fusion oncoprotein interact over hundreds of megabases and even between chromosomes resulting in formation of a specific nuclear subcompartment. Subcompartment interactions are lost after small-molecule degradation of BRD4-NUT, but persist when RNA polymerase II initiation is inhibited.
Graphical Abstract
INTRODUCTION
Chromosomes are one of the most mysterious cellular structures. At multiple length scales chromosomes exhibit different folding patterns, most of which lack a molecular basis. Active and inactive genes are differently folded and often located in different regions of the nucleus, but the biochemical basis and functional significance of this relationship remains to be fully determined (Bickmore, 2013; Bickmore and van Steensel, 2013).
Chromosome folding in situ can be detected by Hi-C (Lieberman-Aiden et al., 2009), which combines chemical cross-linking, proximity ligation, and high-throughput DNA sequencing. At a scale of several to hundreds of kilobases, Hi-C has revealed both pairs of loci as well as stretches of chromatin in close physical proximity that form DNA loops (Rao et al., 2014) and that are referred to as topologically associating domains or TADs (also referred to as physical domains, topological domains, or contact domains), respectively (Dixon et al., 2012; Nora et al., 2012; Rao et al., 2014; Sexton et al., 2012). At longer length scales, extending to entire nuclei, groups of loci preferentially associate thereby segregating into a euchromatic A compartment or heterochromatic B compartment (Lieberman-Aiden et al., 2009), both of which can be divided into nuclear subcompartments with distinct patterns of histone post-translational modifications (Rao et al., 2014).
Many TADs coincide with chromatin loops forming loop domains, whose boundaries or anchors are bound by CTCF and cohesin (Rao et al., 2014). Cohesin and CTCF are required for the formation of loops and loop domains (Gassler et al., 2017; Haarhuis et al., 2017; Nora et al., 2017; Rao et al., 2017; Schwarzer et al., 2017; Wutz et al., 2017), consistent with a model of chromatin folding by processive enlargement of DNA loops, referred to as loop extrusion (Fudenberg et al., 2016; Nasmyth, 2001; Sanborn et al., 2015), but the function of cohesin and CTCF cannot account for all chromosome folding patterns detected by Hi-C. Segregation of chromatin into A and B compartments was independent of CTCF or cohesin (Gassler et al., 2017; Haarhuis et al., 2017; Nora et al., 2017; Rao et al., 2017; Schwarzer et al., 2017; Wutz et al., 2017), and, in the case of cohesin removal, chromosome compartmentation became more apparent (Gassler et al., 2017; Haarhuis et al., 2017; Rao et al., 2017; Schwarzer et al., 2017; Wutz et al., 2017). Large-scale chromosome folding over many megabases must reflect an undetermined, cohesin-independent folding mechanism (Eagen, 2018; Falk et al., 2019; Nuebler et al., 2018; Rao et al., 2017; Schwarzer et al., 2017). Furthermore, how megabase-scale chromatin folding contributes to gene regulation or is specifically altered in human disease remains unknown.
BRD4-NUT is a fusion oncoprotein that drives NUT carcinoma, a highly aggressive squamous cell carcinoma (French, 2018). The BRD4 portion binds acetylated chromatin; the NUT portion binds the p300 histone acetyltransferase (French, 2018). This dual function of BRD4-NUT results in exceptionally broad (100 kb to 2 Mb), hyperacetylated chromatin domains, previously defined as megadomains on the basis of chromatin immunoprecipitation with high-throughput sequencing (ChIP-seq) analysis (Alekseyenko et al., 2015), that were identified by liquid chromatography-mass spectrometry based proteomics to be composed of hyperacetylated histones (Zee et al., 2016). BRD4-NUT also forms nuclear foci visible by light microscopy (French et al., 2008; Reynoird et al., 2010; Yan et al., 2011), but the function and significance of these foci with respect to chromosome structure is unknown. These dramatic biochemical and cytological properties prompted us to exploit the genomic changes caused by BRD4-NUT to investigate large-scale chromosome structure and nuclear compartmentation. Based on Hi-C and ChIP-seq analysis, we observe human disease-associated changes in chromosome folding between distant genomic loci that can be pharmacologically modulated. We also relate chromosome folding on the scale of multiple megabases to nuclear subcompartment formation and gene regulation.
RESULTS
Long-Range Interactions Between Broad, Hyperacetylated Chromatin Domains
To examine the relationship between nuclear foci and megabase-scale chromosome folding in a biologically relevant context where BRD4-NUT is endogenously expressed, we turned to a NUT carcinoma patient-derived cell line, TC-797 (Toretsky et al., 2003) (Figure S1A) that exhibits BRD4-NUT nuclear foci colocalized with H3K27ac (Figure 2A). We performed ChIP-seq for BRD4-NUT and for histone H3K27ac with spike-in normalization (Bonhoure et al., 2014; Hu et al., 2015; Orlando et al., 2014) to determine the location of 164 megadomains (Figure S1B) in TC-797 cells. We also performed in situ Hi-C (Rao et al., 2014) to identify 5,407,548,894 chromosomal contacts (read pairs that remain after removal of duplicates, unligated fragments, and reads that align poorly with the reference genome), achieving the densest human Hi-C contact map to date, with a resolution of 850 bp (Table S1).
Figure 2. A Small-molecule PROTAC Modulates Megadomain-Megadomain Interactions.

(A) Immunofluorescence for BRD4-NUT (magenta) and histone H3K27ac (green) from TC-797 cells treated with DMSO, MZ1 (4 hours), or MZ1 (4 hours) followed by drug washout (24 hours) counterstained with DAPI (blue) indicates loss of nuclear foci after treatment with MZ1 that recover after drug washout. Scale bars, 5 μm.
(B) Mean BRD4-NUT (purple) and histone H3K27ac (green) ChIP-seq profiles for megadomains (upper) and heatmap of the ChIP-seq signal for each megadomain (lower) from TC-797 cells treated with DMSO, MZ1, or MZ1 followed by drug washout indicate loss or reduction of BRD4-NUT or H3K27ac, respectively, within megadomains after treatment with MZ1 that partially recover after drug washout. Megadomains were normalized to the same length and 50 kb of flanking DNA is shown next to each normalized megadomain.
(C) Off-diagonal slices of Hi-C contact maps at 100 kb resolution from TC-797 cells treated with DMSO, MZ1, or MZ1 followed by drug washout demonstrating reduced megadomain-megadomain interactions after MZ1 treatment that partially recovered after drug washout. BRD4-NUT (magenta) and histone H3K27ac (green) ChIP-seq profiles are aligned with the maps. Black bars indicate megadomains in DMSO treated cells.
(D) Intrachromosomal, genome-wide aggregate analysis of megadomain-megadomain interactions from TC-797 cells treated with DMSO, MZ1, or MZ1 followed by drug washout demonstrating genome-wide reduction in intrachromosomal megadomain-megadomain interactions after MZ1 treatment that partially recovered after drug washout. Regions around each megadomain-megadomain interaction were extracted from 5 kb resolution Hi-C contact maps, normalized to the same size, and the resulting submatrices then averaged. Domain-domain interaction scores are given at bottom right.
See also Figure S2.
Visual inspection of Hi-C contact maps from TC-797 cells revealed extensive megadomain-megadomain interactions (Figure 1A). We enumerated all possible pairwise interactions and identified, in a manner similar to that used to annotate chromatin loops (Rao et al., 2014) (Figure S1C), 289 intrachromosomal megadomain-megadomain interactions enriched relative to local background. Each interaction was much stronger than the genome-wide average for pairs of loci separated by the same genomic distance (Figure 1B). The average number of contacts between interacting megadomains was 5.09-fold enriched (median 4.02-fold enriched, range of 1.27- to 25.9-fold enrichment) relative to contacts between all pairs of loci separated by the same genomic distance.
Figure 1. Distant Interactions Between Broad, Hyperacetylated Chromatin Domains.

(A) Hi-C contact maps at 100 kb resolution for the patient-derived TC-797 cell line endogenously expressing BRD4-NUT. BRD4-NUT (magenta) and histone H3K27ac (green) ChIP-seq profiles are aligned with the maps. Black bars indicate megadomains. Megadomain-megadomain interactions are indicated by green circles above the diagonal in the leftmost map; below the diagonal is unannotated for clarity. The rightmost two maps are additional examples displayed as off-diagonal slices of the Hi-C contact map.
(B) Intrachromosomal contacts for loci separated by the distance given on the abscissa. The genome-wide average is indicated as a gray line. Each megadomain-megadomain interaction is displayed as a red dot indicating that interactions between megadomains are stronger than the genome-wide average for loci separated by the same distance.
(C) Percent of megadomain-megadomain interactions (green) separated by the distance given on the abscissa and percent of chromatin loops (yellow) of a given size. Distances between interacting megadomains are orders of magnitude larger than chromatin loops. Sizes/distances are logarithmically binned.
Since all megadomain-megadomain interactions were, like chromatin loops, enriched relative to local background, we asked how megadomain-megadomain interactions might differ from loops. We annotated 8,029 chromatin loops (Rao et al., 2014) and compared the distribution of distances separating interacting megadomains to the distribution of chromatin loop sizes. This revealed two distinct distributions: interacting megadomains were separated by 10–100 Mb whereas chromatin loops spanned 100 kb-1 Mb (Figure 1C).
To evaluate if long-range interactions due to BRD4-NUT could be detected in non-NUT carcinoma cells, we transgenically expressed BRD4-NUT under an inducible promoter in human 293TRex cells (Figure S1D), performed immunofluorescence (Figure S1E) and ChIP-seq for BRD4-NUT and histone H3K27ac with spike-in normalization (Bonhoure et al., 2014; Hu et al., 2015; Orlando et al., 2014) to determine the location of megadomains (Figures S1F and S1G), as well as in situ Hi-C to identify chromosomal contacts (Figure S1H and Table S1). Upon induction with tetracycline, we detected off-diagonal interactions between megadomains separated by tens of megabases distinctively in cells expressing BRD4-NUT (Figure S1I), indicating that this fusion protein initiates specific long-range interactions between distant genomic loci.
Pharmacological Modulation of Megadomain-Megadomain Interactions
As a further test of altered chromosome folding at the megabase scale through formation of hyperacetylated megadomains by BRD4-NUT, we asked whether megadomain-megadomain interactions could be pharmacologically attenuated. Proteolysis Targeted Chimeras (PROTACs) are small molecules that induce targeted protein degradation by recruiting a protein of interest to an E3 ubiquitin ligase for proteasome-mediated proteolysis (Coleman and Crews, 2018). Several PROTACs have been developed that target the bromo- and extraterminal (BET) domain family of proteins, which includes BRD2, BRD3, BRD4, and BRDT (Lu et al., 2015; Winter et al., 2015; Zengerle et al., 2015). Since the BRD4 portion of BRD4-NUT contains the bromodomains targeted by BET PROTACs, we treated TC-797 cells for 4 hours with 100 nM of the PROTAC MZ1 (Zengerle et al., 2015) followed by washout of the drug for 24 hours. MZ1 treatment for 4 hours resulted in nearly complete loss of BRD4-NUT by Western blot as well as loss of BRD4-NUT foci by immunofluorescence that recovered 24 hours after washout (Figures 2A and S2A). Global H3K27ac levels, as detected by Western blot, remained unchanged after 4 hours of MZ1 treatment, but H3K27ac nuclear foci were greatly diminished, recovering 24 hours after washout (Figures 2A and S2A).
Next, we performed ChIP-seq for both BRD4-NUT and histone H3K27ac, and in situ HiC after MZ1 treatment and washout, identifying 924,609,952 and 1,195,025,122 chromosomal contacts, respectively (Figure S2B and Table S1). BRD4-NUT binding within megadomains was virtually undetectable upon MZ1 treatment and partially recovered after washout (Figure 2B). H3K27ac within megadomains was largely reduced after MZ1 treatment and also partially recovered after washout (Figure 2B). Consistent with ChIP-seq analysis, interactions between megadomains detected by Hi-C were greatly reduced upon MZ1 treatment and partially recovered after washout (Figure 2C). We substantiated this analysis across all chromosomes by extracting each megadomain-megadomain interaction plus flanking DNA from the Hi-C contact map, re-scaling these extracted regions to the same dimensions, and then averaging the Hi-C signal, pixel-by-pixel, across all such regions. This aggregate analysis demonstrated reduction in megadomain-megadomain interactions upon MZ1 treatment that partially recovered after washout (Figures 2D and S2C), thus confirming alterations of chromosome folding over many megabases through formation of hyperacetylated chromatin domains by BRD4-NUT.
Extensive Interchromosomal Interactions Between Megadomains
Our Hi-C analysis enabled us to interrogate interchromosomal contacts at 100 kb resolution. Similar to intrachromosomal megadomain-megadomain interactions, we also observed interactions between megadomains on different chromosomes that coincided with BRD4-NUT and H3K27ac localization (Figure 3A). Frequently, a single megadomain would contact many other megadomains spread across different chromosomes, thus forming a network of interchromosomal megadomain-megadomain interactions (Figure 3A).
Figure 3. Interchromosomal Interactions Between Acetylated Loci.

(A) Hi-C contact maps at 100 kb resolution from TC-797 cells demonstrating interchromosomal interactions between twelve example megadomains. Interactions between all pairs of megadomains were reduced after MZ1 treatment and partially recovered after drug washout. Each contact map is a 2 × 2 Mb region centered on a single megadomain. BRD4-NUT (magenta) and histone H3K27ac (green) ChIP-seq profiles are aligned with the maps. Black bars indicate megadomains in DMSO treated cells.
(B) Interchromosomal, genome-wide aggregate analysis of megadomain-megadomain interactions from TC-797 cells treated with DMSO, MZ1, or MZ1 followed by drug washout demonstrating genome-wide reduction in interchromosomal megadomain-megadomain interactions after MZ1 treatment that partially recovered after drug washout. Regions around each megadomain-megadomain interaction were extracted from 5 kb resolution Hi-C contact maps, normalized to the same size, and the resulting submatrices then averaged. Domain-domain interaction scores are given at bottom right.
(C) Domain-domain interaction scores for interchromosomal megadomain-megadomain (red), megadomain-super-enhancer (green), and super-enhancer-super-enhancer interactions. Unlike megadomain-megadomain interactions, super-enhancer-super-enhancer interactions are not sensitive to MZ1.
After enumerating all possible pairwise interchromosomal megadomain-megadomain interactions, we identified 2,100 interactions enriched relative to local background. These interactions were also sensitive to MZ1 treatment and partially recovered after drug washout (Figures 3A and 3B).
Since super-enhancers, like megadomains, are broad, highly enriched for H3K27ac (Whyte et al., 2013), and interact with one another over long genomic distances (Beagrie et al., 2017; Rao et al., 2017), we asked if we could detect interchromosomal interactions between megadomains and super-enhancers. Megadomain–super-enhancer interactions were weaker than megadomain-megadomain interactions and were mildly sensitive to MZ1 treatment (Figure 3C). Though detectable, super-enhancer–super-enhancer interactions were not sensitive to MZ1 treatment (Figure 3C).
A Megadomain-Specific Nuclear Subcompartment
Combining the intrachromosomal and interchromosomal analysis identified that a single megadomain could interact with up to 103 other megadomains. The number of interacting partners for a single megadomain was correlated with both H3K27ac (Spearman’s ρ = 0.75) and BRD4-NUT (Spearman’s ρ = 0.69) ChIP-seq enrichment (Figure 4A), but not with megadomain size (Figure S3A). H3K27ac and BRD4-NUT ChIP-seq enrichment was also not correlated with pairwise intrachromosomal or interchromosomal contact strength (Figure S3B and S3C).
Figure 4. A Megadomain-Specific Nuclear Subcompartment.

(A) For each megadomain, the number of genome-wide (sum of intra- and interchromosomal) megadomain-megadomain interactions versus the average histone H3K27ac (green) or BRD4-NUT (purple) ChIP-seq enrichment within that megadomain. ChIP-seq enrichment was normalized for differences in megadomain size.
(B) Normalized counts of triple interactions between three distinct megadomains in TC-797 cells treated with DMSO, MZ1, or MZ1 followed by drug washout demonstrating reduction in triple interactions after MZ1 treatment that partially recovered after drug washout. The observed megadomain triple interactions (left) are compared to a random shuffled control set of interactions (right).
(C) Three-dimensional plot of triple interactions between different megadomains in TC-797 cells treated with DMSO, MZ1, or MZ1 followed by drug washout. The coordinate along each axis represents a distinct megadomain. The maximum value of the colorbar for each condition differs to highlight that the density of distinct triples interactions is reduced after MZ1 treatment and partially recovers after drug washout.
(D) Subcompartment scores and percent of inter-chromosomal Hi-C contacts to megadomains for every 100 kb bin on chromosome 5. Megadomains that strongly interact with megadomains on other chromosomes coincide with very positive subcompartment scores for subcompartment M (gray shading). Inter-chromosomal Hi-C contacts to megadomains at these loci is reduced after MZ1 treatment and partially recovers after drug washout. Colored bars (topmost row) indicate subcompartment invervals in DMSO treated cells; black bars (bottommost row) indicate megadomains.
(E) Contact enrichment between subcompartments on the even and odd chromosomes relative to a random shuffle control of subcompartments indicates that subcompartment M is distinct from the other five subcompartments.
(F) For each subcompartment, the fraction of that subcompartment that overlaps with megadomains compared to a random shuffled control set of megadomains indicates that subcompartment M is specific for megadomains.
See also Figures S3 and S4 and Table S2.
The integrated ChIP-seq and Hi-C analysis, together with directly observable hyperacetylated foci suggests cooperativity in megadomain-megadomain interactions and that multiple megadomains may physically cluster within the nucleus. To search for evidence of megadomain clustering, we examined our Hi-C data for simultaneous contacts between three or more loci, specifically focusing on triple interactions between distinct megadomains (see Methods). We identified four times more triple interactions between megadomains (255 triples) than between a random shuffle control set of megadomains (54 triples) in TC-797 cells (Figures 4B and S3D and Table S2). Clustering of three different megadomains was also observed by FISH (Figure S3F). Triple interactions were reduced after MZ1 treatment (Figures 4B, 4C, S3D, and S3E; p-value < 0.0001; permutation test) and partially recovered after drug washout (Figures 4B, 4C, S3D, and S3E; p-value = 0.0016; permutation test).
Physical clustering of megadomains could indicate they are preferentially associating with each other to form a nuclear compartment or subcompartment (Lieberman-Aiden et al., 2009; Rao et al., 2014). Our exceptionally deeply sequenced Hi-C dataset enabled us to use machine learning to classify inter-chromosomal contact patterns (Rao et al., 2014), thus revealing six subcompartments in TC-797 cells (Figures S4A-F), five of which corresponded to those in GM12878 lymphoblastoid cell lines (Rao et al., 2014) (see Methods). For each subcompartment, this classification, based on a hidden Markov model, parameterized every locus in the genome with a mean value based on a Gaussian distribution. This mean value, which we refer to as the subcompartment score, reflects how likely every locus is to be in each subcompartment, with positive subcompartment scores indicating a high likelihood of being in the subcompartment compared with negative values. One subcompartment, which we refer to as subcompartment M, had exceptionally positive subcompartment scores at megadomains that made strong interchromosomal contacts with other megadomains (Figure 4D and S4G). Scores for the other subcompartments at megadomains was much weaker than that for subcompartment M, indicating that these megadomains were not in the other subcompartments (Figure 4D).
Interactions between loci within subcompartment M were much more enriched than interactions between loci within any of the five other subcompartments, including the active A1L and A2L subcompartments (Figure 4E). Chromatin within subcompartment M was depleted for contacts with chromatin in each of the five other subcompartments, including the other two active subcompartments A1L and A2L (Figure 4E). Subcompartment M is therefore not a subset of another active subcompartment. Subcompartment M overlapped almost exclusively with megadomains, thus revealing a megadomain-specific nuclear subcompartment (Figure 4F). A megadomain-specific subcompartment is also supported by a loss of strong interchromosomal contacts between megadomains within subcompartment M after MZ1 treatment that partially recovered after drug washout (Figure 4D).
Megadomain Formation Does Not Alter Local Chromatin Structure
Changes in interchromosomal or long-range intrachromosomal interactions resulting in subcompartment M formation could reflect re-organization of chromatin structure at the sub-megabase scale. At 5 kb resolution, megadomains in both TC-797 cells and 293TRex cells often spanned many chromatin loops, visible in Hi-C contact maps as radially symmetric focal peaks of contact enrichment, and were co-incident with TADs, apparent in contact maps as on-diagonal boxes of enriched contact frequency (Figures 5A and S5A). Local loop and TAD organization was unaltered after de novo formation of megadomains in 293TRex cells or after MZ1-mediated removal of BRD4-NUT in TC-797 cells (Figures 5A and S5A).
Figure 5. Local Chromatin Organization is Unchanged After Removal of BRD4-NUT.

(A) Hi-C contact maps at 5 kb resolution of DMSO (above the diagonal) and MZ1 (below the diagonal) treated TC-797 cells. BRD4-NUT (magenta) and histone H3K27ac (green) ChIP-seq profiles are aligned with the maps. Black bars indicate megadomains identified in induced and DMSO cells. The Hi-C contact map from DMSO treated cells was at a comparable sequencing depth to that from MZ1 treated cells.
(B) Genome-wide, aggregate analysis at 5 kb resolution of Hi-C signal from DMSO, MZ1, and MZ1 followed by drug washout treated TC-797 cells centered on contact domains, loop domains, loop-independent contact domains, megadomains, and random shuffled control megadomains. Megadomains and contact domains remained unchanged after treatment with MZ1 or after treatment with MZ1 followed by drug washout. Contact domain annotations and megadomain locations are from DMSO treated cells. Domains were re-scaled to the same size and observed submatrices around each megadomain-megadomain interaction were averaged.
(C) Genome-wide, aggregate peak analysis (APA) at 5 kb resolution of Hi-C signal from DMSO, MZ1, and MZ1 followed by drug washout treated TC-797 cells centered on loops, contact domain corners, loop domain corners, loop-independent contact domain corners, megadomain corners, and random shuffled control megadomain corners. Loops, megadomains, and contact domains remain largely unchanged after treatment with MZ1 or after treatment with MZ1 followed by drug washout. Loop and contact domain annotations and megadomain locations are from DMSO treated cells. APA scores are given at bottom right.
See also Figure S5.
We performed genome-wide analysis to more extensively assess if megadomain formation alters local domain structure. To examine intradomain contacts, each domain plus flanking DNA was extracted from the Hi-C contact map and re-scaled to the same dimensions. The Hi-C signal was then averaged, pixel-by-pixel, across all such extracted regions. We observed not change in signal after removal or expression of BRD4-NUT, indicating that domain organization appears largely unchanged after megadomain removal or formation (Figures 5B and S5B-D).
Similarly, we performed genome-wide aggregate peak analysis (APA) (Rao et al., 2014), in which a single pixel of interest (e.g. peak pixel of a chromatin loop) plus a constant amount of flanking DNA is extracted from the Hi-C contact map and then all extracted regions are averaged, to assess if megadomain formation alters chromatin loops. We also divided contact domains into those that have a loop at their corner, referred to as loop domains (Rao et al., 2014), that reflect TAD formation (Haarhuis et al., 2017; Nora et al., 2017; Rao et al., 2017; Schwarzer et al., 2017), and those that do not, referred to as loop-independent contact domains, to examine if megadomains were related to loop-like structures. This analysis indicated that loop and domain organization appear largely unchanged after megadomain formation or removal, and also indicated the equivalence of megadomains with loop-independent contact domains, but not with chromatin loops (Figures 5B, 5C, S5B-E). Since chromatin looping is distinct from compartmentation (Eagen, 2018; Rao et al., 2017; Schwarzer et al., 2017), many loop-independent contact domains seem to correspond to subcompartment intervals, also known as compartment domains (Eagen, 2018; Rao et al., 2017; Rowley et al., 2017). That loci within subcompartment intervals exhibit the same pattern of histone modifications (Rao et al., 2014), that megadomains are broad, hyperacetylated regions of chromatin (Alekseyenko et al., 2015), and that we have identified a nuclear subcompartment consisting nearly entirely of megadomains indicate that megadomains are equivalent to large, loop-independent contact domains or compartment domains.
Elevated Gene Expression Within the Megadomain-Specific Subcompartment
Genes residing in megadomains are important for NUT carcinoma growth (Alekseyenko et al., 2015). A megadomain is found within the Myc locus in numerous NUT carcinoma samples, including upstream of Myc in TC-797 cells (Alekseyenko et al., 2015), and Myc blocks differentiation and maintains cell growth in this cancer (Grayson et al., 2014). Hi-C and ChIP-seq from TC-797 cells indicated that the megadomain within the upstream regulatory region for Myc lies within subcompartment M, (Figure 6A) which also contains Nrg1, another gene important for NUT carcinoma growth (Alekseyenko et al., 2015). We assessed nascent transcription by performing transient transcriptome sequencing (TT-seq) (Schwalb et al., 2016), revealing elevated gene activity at these loci (Figure 6A).
Figure 6. Elevated Gene Expression in the Megadomain-Specific Subcompartment.

(A) Off-diagonal slices of Hi-C contact maps at 100 kb resolution from TC-797 cells treated with DMSO, MZ1, or MZ1 followed by drug washout. BRD4-NUT (magenta) and histone H3K27ac (green) ChIP-seq profiles are aligned with the maps. Purple bars indicate subcompartment M intervals and black bars indicate megadomains in DMSO treated cells. Zoomed areas show nascent transcription profiles analyzed by TT-seq (sense transcripts in blue, anti-sense transcripts in red).
(B) Violin plots overlaid with Tukey box plots (white bars show medians; boxes indicate interquartile range; whiskers extend 1.5 times the interquartile range; black dots are outliers) indicating that expression levels for genes within subcompartment M are greater than that for genes in the other subcompartments. Value in parenthesis indicate the number of genes in each subcompartment. p-value is from one-sided Mann-Whitney U tests.
(C) Volcano plots (significance versus fold change) of differentially expressed genes show that after MZ1 treatment genes within subcompartment M (purple dots) are downregulated (left) whereas after drug washout genes within subcompartment M (purple dots) are upregulated (right) in TC-797 cells. Horizontal dashed line indicates a Benjamini-Hochberg adjusted p-value of 0.01; vertical dashed lines indicate 2-fold change in expression.
To extend this analysis genome-wide, we examined gene expression within subcompartments. Genes within subcompartment M were more highly expressed than genes within other subcompartments (Figure 6B). Although megadomains are bound by BRD4-NUT and are hyperacetylated, expression levels of genes within all megadomains were not correlated with H3K27ac or BRD4-NUT ChIP-seq enrichment (Figure S6A). It seems that residence in a particular region of the nucleus, for instance subcompartment M, is associated with enhanced gene activity.
Expression of genes within megadomains that interact with the Myc locus was greatly reduced after MZ1 treatment and partially recovered after drug washout (Figure 6A), and differential gene expression analysis over the course of PROTAC treatment and withdrawal also revealed a relationship between nuclear subcompartments and gene regulation. Of the 7,610 differentially expressed genes (≥ 2-fold change in expression and Benjamini-Hochberg adjusted p-value < 0.01) after MZ1 treatment, downregulated genes within subcompartment M (107 of 5,932 all downregulated genes) were 5.04-fold enriched relative to upregulated genes within subcompartment M (6 of 1,678 all upregulated genes) (Figure 6C). Of the 8,438 differentially expressed genes after MZ1 washout, upregulated genes within subcompartment M (111 of 7,003 all upregulated genes) were 4.55-fold enriched relative to downregulated genes within subcompartment M (5 of 1,435 all downregulated genes) (Figure 6C). 98.1% (105 genes) of the genes downregulated after MZ1 treatment were subsequently upregulated after drug washout (Table S3). Such specificity was not observed across the other subcompartments (Figure S6B).
Subcompartment Persistence in the Absence of Transcription
As MZ1 treatment results in loss of BRD4-NUT, H3K27ac (Figure 2B), and transcription (Figure 6A) within megadomains, we asked whether we could decouple gene expression from chromatin composition by inhibiting RNA polymerase II initiation with triptolide (Jonkers et al., 2014; Titov et al., 2011; Y. Wang et al., 2011). Treating TC-797 cells for 4 hours with 500 nM of triptolide resulted in clearance of RNA polymerase II from promoters, gene bodies (Figure 7A) and megadomains (Figure S7A) as well as genome-wide loss of transcription (Figure 7B). However, BRD4-NUT or H3K27ac nuclear foci persisted in the absence of transcription (Figure 7C). H3K27ac increased genome-wide, including at megadomains (Figure 7D), indicating that chromatin hyperacetylation did not depend on RNA polymerase II initiation or transcription.
Figure 7. Subcompartment Persistence in the Absence of Transcription.

(A) Mean RNA polymerase II ChIP-seq profiles for expressed genes (FPKM ≥ 0.5) (upper) and heatmap of the ChIP-seq signal for each expressed gene (lower) from TC-797 cells treated with DMSO or triptolide indicate clearance of RNA polymerase II from promoters and gene bodies after triptolide treatment.
(B) Heatmaps of TT-seq signal (nascent transcription) for expressed genes (FPKM ≥ 0.5) from TC-797 cells treated with DMSO or triptolide (4 hours) indicate inhibition of nascent transcription after triptolide treatment.
(C) Immunofluorescence for BRD4-NUT (magenta) and histone H3K27ac (green) from TC-797 cells treated with DMSO or triptolide counterstained with DAPI (blue) indicates that nuclear foci persist in the absence of transcription. Scale bars, 5 μm.
(D) Heatmaps of H3K27ac ChIP-seq signal for each H3K27ac enriched region from TC-797 cells treated with DMSO or triptolide indicate an increase in H3K27ac after inhibiting transcription. H3K27ac enriched regions were normalized to the same length and 50 kb of flanking DNA is shown next to each normalized region.
(E) Hi-C contact maps at 50 kb resolution from TC-797 cells treated with DMSO or triptolide demonstrating persistence of old and formation of new contacts between hyperacetylated loci in the absence of transcription. Histone H3K27ac ChIP-seq (green) and TT-seq profiles (sense transcripts in blue, anti-sense transcripts in red) are aligned with the maps.
(F) Genome-wide aggregate analysis of megadomain-megadomain interactions from TC-797 cells treated with DMSO or triptolide demonstrating no genome-wide change in average megadomain-megadomain interactions after inhibiting transcription. Regions around each megadomain-megadomain interaction were extracted from 5 kb resolution Hi-C contact maps, normalized to the same size, and the resulting submatrices then averaged. Aggregate scores are given at bottom right.
(G) Loci (100 kb bins) within subcompartment M (ovals) are largely preserved after TC-797 cells are treated with triptolide, indicating persistence of the subcompartment in the absence of transcription. 47 of 63 (75%) of subcompartment M intervals remained after triptolide treatment. Colors alternate for successive chromosomes.
See also Figure S7
Megadomain-megadomain interactions identified by in situ Hi-C (2,201,862,149 contacts; Table S1) persisted in the absence of transcription, and additional interactions were also formed between newly hyperacetylated regions that were previously highly transcribed and occupied by RNA polymerase II (Figure 7E). On average, megadomain-megadomain interactions were unchanged genome-wide in the absence of transcription (Figure 7F), consistent with megadomain-megadomain interaction strength not being correlated with H3K27ac ChIP-seq enrichment (Figures S3B and S3C). Triple megadomain interactions also remained unaltered (Figures S7B-D) and the vast majority of subcompartment M persisted in the absence of transcription (Figures 7G and S7E).
DISCUSSION
The chromosome folding pattern reported here is notable for several reasons: 1) megadomains interact over hundreds of megabases and even between chromosomes to form a novel nuclear subcompartment, 2) altered chromosome folding by a small-molecule PROTAC, 3) PROTAC-induced specific changes in expression only for genes within this subcompartment, and 4) subcompartment formation in the absence of transcription. Long-range megadomain-megadomain interactions are significant for delineating large-scale chromosome folding that arises due to interactions between distant genomic loci with similar composition of the chromatin fiber but without reorganization of local structure. Unaltered local structure was surprising because unfolding or re-organization of pre-existing structures might have been expected due to histone hyperacetylation (Shogren-Knaak et al., 2006). Rather, after changing the local composition of the chromatin fiber, loci now interact over long genomic distances within and between chromosomes to form a nuclear subcompartment.
Sequential imaging of multiple loci suggested a polarized arrangement of A and B compartments (S. Wang et al., 2016), but the physical organization of subcompartments within the nucleus remains unexplored. The most parsimonious interpretation of the HiC, ChIP-seq, and imaging results presented here is that BRD4-NUT nuclear foci identified by immunofluorescence physically represent subcompartment M. Approximately 100 foci are observed in single NUT carcinoma cells (French et al., 2008; Reynoird et al., 2010; Yan et al., 2011), suggesting that every megadomain-megadomain interaction detected in a cell population by Hi-C does not occur in each cell, and that a single subcompartment identified on the basis of genomics is composed of many distinct physical regions of the nucleus.
Persistence of megadomain-megadomain interactions after inhibiting RNA polymerase II initiation importantly demonstrates that a nuclear subcompartment can be maintained in the absence of transcription. We have also identified newly hyperacetylated regions and interactions between them after inhibiting transcription suggesting, in certain cases, that transcription may actually inhibit these interactions. It seems likely that the local composition of the chromatin fiber holds the subcompartment together and is even perhaps instructive for forming new contacts independent of transcription, suggesting that histone post-translational modifications and/or proteins that bind to modified histones establish nuclear compartmentation.
Genome-wide approaches have identified physical proximity of highly transcribed or regulatory loci that tend to be important for determining cell fate and that are separated by large genomic distances or even located on different chromosomes (Beagrie et al., 2017; Bonev et al., 2017; Monahan et al., 2019; Rao et al., 2017). Here, we have altered folding and related these changes to human disease. Modifying structure begins to delineate how these interactions arise, identifying roles for BRD4-NUT and H3K27ac. By co-opting normal cellular machinery, BRD4-NUT alters megabase-scale chromosome folding, which can be pharmacologically modulated through removal of the oncogenic fusion protein. Large-scale chromosome folding into nuclear subcompartments thus depends on histone post-translational modifications and/or proteins that bind to modified histones. Specific interactions we report here between chromosomes due to an oncogenic fusion protein reveal that the inter-chromosomal interactions identified in mouse olfactory neurons (Monahan et al., 2019) are not restricted to cellular differentiation, but also occur in the context of human disease. In olfactory neurons (Monahan et al., 2019), inter-chromosomal interactions are attributed to LHX2, a transcription factor, and LDB1, an adaptor protein, which both bind to specific genomic loci, whereas subcompartment M arises due to spreading of BRD4NUT across large stretches of chromatin.
Megadomain locations, like those of super-enhancers, are cell-lineage specific, but megadomains do not arise from preexisting super-enhancers (Alekseyenko et al., 2015). Since we can detect interactions between megadomains and super-enhancers, and since both are broad and highly enriched for H3K27ac (Whyte et al., 2013), it is likely that these interactions are due to local composition of the chromatin fiber. Unlike super-enhancer–super-enhancer interactions, which are observed in the absence of cohesin (Rao et al., 2017), megadomains interact in the presence of cohesin, suggesting that cohesin does not necessarily restrict subcompartmentation. Super-enhancer–super-enhancer interactions are also not sensitive to MZ1 treatment, whereas megadomain-megadomain interactions are sensitive.
The identification of a megadomain-specific subcompartment indicates aberrant nuclear compartmentation in NUT carcinoma. Rather than TAD dysfunction through boundary misregulation, as in congenital limb malformations (Franke et al., 2016; Lupiáñez et al., 2015), glioma (Flavahan et al., 2016), and T-cell acute lymphoblastic leukemia (Hnisz et al., 2016), oncogenic BRD4-NUT results in pathological long-range interactions within a novel nuclear subcompartment. Results presented here identify an additional mechanism by which chromosome folding is aberrantly regulated in human disease.
BRD4-NUT and H3K27ac localization patterns from many NUT carcinoma samples led to a feed-forward model of megadomain formation (Alekseyenko et al., 2015). BRD4 bromodomains bind acetylated histones and NUT recruits p300 leading to spreading of BRD4-NUT and histone acetylation across long stretches of chromatin (Alekseyenko et al., 2015). Conceptualized in terms of chemical composition of a one-dimensional chromatin fiber, our findings now place this model in the context of three-dimensional chromosome structure. Megadomains, particularly those harboring genes important for NUT carcinoma pathogenesis, are not physically isolated within the nucleus, but extensively interact with one another in a nuclear subcompartment. At present, it is unclear if megadomain formation and megadomain-megadomain interactions arise simultaneously, or if megadomains need to be established prior to the formation of the megadomain-specfic subcompartment.
Loop extrusion, a leading model for the formation of many TADs and chromatin loops (Fudenberg et al., 2016; Sanborn et al., 2015), apparently is inconsistent with the megadomain-megadomain and subcompartment interactions reported here. Loop sizes, which likely reflect processivity of the loop extrusion machinery (Haarhuis et al., 2017; Wutz et al., 2017), are orders of magnitude shorter than distances separating interacting megadomains (Figure 1C). The megadomain-specific subcompartment is most apparent as megadomain-megadomain interactions between chromosomes (Figures 3 and 4); loop extrusion acts along a single chromatin fiber. BRD4-NUT and interacting proteins (Alekseyenko et al., 2017) contain intrinsically disordered regions, which stabilize many weak, multivalent interactions throughout the cell (Banani et al., 2017; Shin and Brangwynne, 2017) including interactions established by recombinant BRD4 on in vitro reconstituted, acetylated chromatin fibers (Gibson et al., 2019). If weak, multivalent interactions, which have been linked to heterochromatin formation (Larson et al., 2017; Strom et al., 2017) and transcriptional regulation (Boija et al., 2018; Cho et al., 2018; Chong et al., 2018; Sabari et al., 2018), do promote megadomain-megadomain interactions, apparent as hyperacetylated, BRD4-NUT nuclear foci, then these molecular interactions now seem to be important for chromosome structure, particularly for maintaining contacts between loci separated by many megabases and for nuclear compartmentation.
Genetic manipulations (de Wit et al., 2015; Guo et al., 2015; Haarhuis et al., 2017; Sanborn et al., 2015; Schwarzer et al., 2017; Wutz et al., 2017), including those coupled with inducible protein degradation (Nora et al., 2017; Rao et al., 2017; Wutz et al., 2017) have been used to study chromatin folding. We have used BRD4-NUT and a small-molecule BET PROTAC as tools to study chromosome structure. BET inhibitors have been used to treat NUT carcinoma (Filippakopoulos et al., 2010; French, 2018) and BET degraders act on proteins that regulate transcription (Winter et al., 2017). Our results suggest that these transcriptional defects may be due, in part, to altered chromosome folding. Furthermore, the therapeutic potential of PROTACs (Coleman and Crews, 2018) indicates that pharmacological modulation of chromosome structure may be one option for the treatment of human disease.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kyle P. Eagen (eagen@northwestern.edu). Distribution of the 293TRex-Flag-BRD4-NUT-HA and TC-797 cell lines are restricted due to Material Transfer Agreements with Brigham and Women’s Hospital and Georgetown University, respectively.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
293TRex-Flag-BRD4-NUT-HA cells were a kind gift from Christopher A. French (Brigham and Women’s Hospital, Boston, MA). Cells were grown in DMEM with 4.5 g/L glucose and sodium pyruvate without L-glutamine (Corning #15013CV) + 10% Fetal Bovine Serum (Atlanta Biologicals #S10350) + 1% GlutaMAX (Gibco #35050061) + 1% Penicillin-Streptomycin (Gibco #15140122) + 15 μg/mL blasticidin (Gibco #A1113903) + 100 μg/mL hygromycin (Corning #30240CR) at 37° C wi th 5% CO2. TC-797 cells were a kind gift from Jeffrey A. Toretsky (Georgetown University, Washington, DC). Cells were grown in DMEM with 4.5 g/L glucose and sodium pyruvate without L-glutamine (Corning #15013CV) + 10% Fetal Bovine Serum (Atlanta Biologicals #S10350) + 1% GlutaMAX (Gibco #35050061) + 1% Penicillin-Streptomycin (Gibco #15140122) at 37° C with 5% CO2. 293TRex-Flag-BRD4-NUT-HA cell line was validated by immunofluorescence before and after induction of BRD4-NUT. Sex of the TC-797 cell line is male and this cell line was validated by karyotyping and immunofluorescence for BRD4-NUT expression. Cell lines regularly tested negative for mycoplasma with the Universal Mycoplasma Detection Kit (ATCC #30–1012K).
Kc167 cells were obtained from the Drosophila Genomics Resource Center (#1; RRID:CVCL_Z834; Bloomington, IN). Cells were grown in CCM3 Media (HyClone #SH30062.02) at 25° C.
Tetracycline hydrochloride (Sigma-Aldrich #T7660) was solubilized in water. 293TRexFlag-BRD4-NUT-HA were induced to express BRD4-NUT with 1 μg/mL tetracycline for 8 hours.
MZ1 (Cayman Chemical #21622) was solubilized in DMSO. TC-797 cells were treated with 100 nM MZ1 (0.01% final DMSO) for 4 hours. For washout experiments, TC-797 cells were treated with 100 nM MZ1 (0.01% final DMSO) for 4 hours, washed three times with PBS, and then returned to normal culture media (as described above) for 24 hours.
Triptolide (TPL; Cayman Chemical #11973) was solubilized in DMSO. TC-797 cells were treated with 500 nM TPL (0.01% final DMSO) for 4 hours.
METHOD DETAILS
Immunofluorescence
Immunofluorescence was performed on 293TRex-Flag-BRD4-NUT-HA and TC-797 cells with DAPI counterstaining. Primary antibodies used were NUT rabbit monoclonal antibody (Cell Signaling Technologies #3625, lot 4, RRID: AB_2066833) and H3K27ac mouse monoclonal antibody (Active Motif #39685, RRID: AB_2722569). Secondary antibodies included Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (Thermo Fisher #A-11008, RRID: AB_143165) and Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 (Thermo Fisher #A-21236, RRID: AB_2535805).
High-precision cover glass (Bioscience Tools #CSHP-No1.5–22×22) was cleaned by submerging the cover glass in 1 M KOH and sonicating for 15 minutes in an ultrasonic cleaner. Cover glasses were washed 5 times with ultrapure water, once with 70% ethanol, and then air dried in a tissue culture hood. One cover glass was placed in each well of a 6-well culture dish. Cover glasses were treated for 5 minutes with 0.01% poly-lysine (Sigma #P4707–50ML) while at room temperature. Cover glasses were washed three times with sterile water, air dried, and sterilized by UV light overnight in the tissue culture hood.
Cells were seeded in wells 2–24 hours before fixation, adjusting for optimal density. Cells were washed with 5 mL of PBS per well, and fixed with 5 mL of 4% EM-grade paraformaldehyde (Electron Microscopy Sciences #15714) in PBS for 5 minutes at room temperature while rocking. Cells were then permeabilized with 5 mL of 4% PFA, 0.1% Triton X-100 in PBS while rocking for 1 minute at room temperature. Cells were washed three times with 5 mL of PBS, and then blocked for 30 minutes at room temperature with blocking buffer (5% normal goat serum (Cell Signaling Technologies #5425S), 0.1% Triton X-100 in PBS) while rocking. Primary antibodies were diluted 1:1000 using antibody dilution buffer (1% BSA IgG-free, Protease-free (Jackson ImmunoResearch #001–000-161), 0.1% Triton X-100 in PBS) and 0.5 mL of diluted primary antibody was added to each well and then incubated with the fixed, permeabilized cells with rocking for 2 hours at room temperature. Cells were washed three times with 5 mL of PBS. Secondary antibodies, diluted 1:1000 with antibody dilution buffer, were added after the last wash and incubated with rocking for 1 hour at room temperature protected from light. While maintaining protection from light, cells were washed with 5 mL of PBS followed by counterstaining with 300 nM DAPI dilactate (Fisher #D3571) in PBS for 10 minutes at room temperature with rocking. Three final rounds of washing were done with 5 mL of PBS and the cover glass was mounted on microscope slides using ProLong Diamond (Thermo Fisher #P36965). Slides were left to cure overnight protected from light.
Slides were imaged on a ZEISS LSM 800 confocal microscope using a Zeiss Plan-Apochromat 63x/1.40 NA oil DIC M27 objective and images acquired with Zeiss Zen 2.3 (blue edition) software at an image bit depth of 8 bits. Images were collected as Z-stacks on a GaAsP detector, one for each imaging channel, with a 65 nm pixel size. Appropriate filters for fluorochromes Alexa Fluor 488, Alexa Fluor 647, and DAPI were used. Images were projected along the Z-axis taking the maximum intensity, the BRD4-NUT channel was pseudocolored magenta, the histone H3K27ac channel was pseudocolored green, and the DAPI channel was pseudocolored blue each using a linear LUT that covered the full range of the data, the channels merged, and the image converted to TIFF format using Zeiss Zen 2.3 lite (blue edition) software.
DNA FISH
DNA FISH was performed on TC-797 cells using Oligopaint FISH probes (Beliveau et al., 2012). A pool of 959 probes per megadomain (Table S4) was selected from the previously designed “balanced” set of Oligopaint probes against human genome build 38 (Beliveau et al., 2018). A 20 bp constant reverse transcriptase priming sequence was added to the 5’ end of each probe. Each set of probes corresponding to one megadomain was then flanked with unique 20 bp priming sequences on both the 5’ and 3’ ends. The resulting oligo pool was ordered from CustomArray (Bothell, WA). Probe sets were amplified from the pool using limited cycle PCR with unique flanking primers, further amplified via T7 in vitro transcription, and then reverse transcribed to ssDNA containing a unique 5’ fluorophore per megadomain as previously described (Beliveau et al., 2017).
Cells were seeded on high-precision cover glass (prepared as described above) in wells 2–24 hours before fixation, adjusting for optimal density. In situ hybridization was performed as previously described (Bintu et al., 2018), with minor modifications. Cells were washed with 5 mL of PBS per well, and fixed with 5 mL of 4% EM-grade paraformaldehyde (Electron Microscopy Sciences #15714) in PBS for 10 minutes at room temperature while rocking. Cells were washed three times with 5 mL of PBS and then treated with 5 mL of freshly prepared 1 mg/mL sodium borohydride for 7 minutes at room temperature. Cells were washed three times with 5 mL of PBS and then permeabilized with 5 mL 0.5% v/v Triton X-100 in PBS while rocking for 10 minutes at room temperature. Cells were washed three times with 5 mL of PBS and then permeabilized a second time with 5 mL of 0.1 M HCl while rocking for 5 minutes at room temperature. Cells were washed three times with 5 mL of PBS and then endogenous RNA was digested by incubating the cells in 1 mL 0.1 mg/mL RNase A in PBS for 45 min at 37° C. Cells were washed three t imes with 5 mL of 2xSSC (300 mM NaCl, 30 mM sodium citrate, pH 7.0) per well each time and then incubated in 1 mL prehybridization buffer (2xSSC, 0.1% v/v Tween 20, 50% formamide) for 30 minutes at room temperature. Cells on coverslips were inverted onto 25 μl of hybridization mix (2x SSC, 0.1% v/v Tween 20, 50% formamide, 10% dextran sulfate, 25 pmol of each probe) deposited on a glass slide. The sample was denatured at 78° C for 3 minutes in a Slide Denaturation & Hybridization System (Biotang #TDH-500).
The sample was hybridized overnight at 37° C in the Slide Denaturation & Hybridization System. Coverslips were washed with 2xSSC + 0.1% v/v Tween 20 twice at 60° C and then twice at room temperature for 10 minutes each time while protected from light. Coverslips were washed with 2xSSC at room temperature and then counterstained with 300 nM of DAPI dilactate in 2xSSC for 10 minutes at room temperature while protected from light. Three final rounds of washing were done with 5 mL of 2xSSC and the cover glass was mounted on microscope slides using ProLong Diamond (Thermo Fisher #P36965).
Slides were imaged as above, with appropriate filters for fluorochromes Alexa Fluor 488, Alexa Fluor 565, Alexa Fluor 647, and DAPI, and maximum intensity Z-projections were also taken as above.
Cell Cross-linking for Hi-C and ChIP-seq
Mammalian cells were grown to a density of 1×106 cells/mL and then washed twice with 10 mL of media without serum while attached to the dish. 2.5 mL of TrypLE Express (Gibco #12605010) was added to the dish and incubated for 5–10 minutes to detach adherent cells. TrypLE Express was quenched by adding media without serum. Kc167 cells were detached from the plate by blowing medium at the cells using a serological pipette. Kc167 cells were then centrifuged at 500 × g for 5 minutes and resuspended in mammalian media without serum to reach a density of 1×107 cells/mL. Multiples of 5 million mammalian cells were added to conical tubes, and Kc167 cells were added to the tube to achieve a spike-in ratio of 2:1 (mammalian cells:fly cells). Cells were then centrifuged at 300 × g for 5 minutes and resuspended at a density of 1×106 cells/mL in media without serum three times to remove traces of serum. Cell suspensions were crosslinked by adding 32% EM-grade paraformaldehyde (Electron Microscopy Sciences #15714) to a final concentration of 1%, and incubated at room temperature for 10 minutes while constantly rotating end-over-end. Paraformaldehyde was quenched by adding 2.5 M glycine to a final concentration of 0.15 M while constantly rotating end-over-end for 5 minutes at room temperature. Cells were centrifuged at 500 × g for 5 minutes at 4° C, resuspended in 1 mL of ice-cold PB S per 10 million mammalian cells, and subsequently pelleted. The cell pellet was then flash frozen in liquid nitrogen and stored at −80° C.
Immunoblotting
Primary antibodies were NUT (C52B1) Rabbit mAb (Cell Signaling Technologies #3625, lot 4, RRID: AB_2066833) diluted 1:100000, Acetyl-Histone H3 (Lys27) (D5E4) XP Rabbit mAb (Cell Signaling Technology #8173S, lot 6, RRID: AB_10949503) diluted 1:1000. Loading control primary antibodies were beta-tubulin mouse antibody (Developmental Studies Hybridoma Bank #E7, RRID: AB_528499) diluted 1:1000, and histone H3 rabbit antibody (Shilatifard Lab #42) diluted 1:5000. Secondary antibodies were goat anti-rabbit IgG–HRP conjugate (Sigma #A6154, RRID: AB_258284) diluted 1:5000, and goat anti-mouse IgG-HRP conjugate (Sigma #A4416, RRID: AB_258167) diluted 1:5000.
1 million uncrosslinked cells, collected simultaneously with those cross-linked for ChIP-seq and Hi-C, were collected in a 1.5 mL tube, centrifuged at 300 × g for 5 minutes at 4° C, the supernatant discarded, and the cell pellet resuspended in 1 mL of ice-cold PBS. Cells were then centrifuged at 300 × g for 5 minutes at 4° C, the supernatant discarded, and the cell pellet resuspended in 50 μL of lysis buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% SDS). Cells were lysed at 95° C for 5 minutes, cooled to room temperature, and whole-cell extracts flash frozen in liquid nitrogen and stored at −80° C.
Cell extracts were thawed on ice. Nucleic acid was removed by treated samples with 250 U of Benzonase (Sigma-Aldrich #1014) and incubated at room temperature for 5 minutes. Protein concentration was quantified using a Bio-Rad DC Protein Assay (Bio-Rad #5000112). 10 μg of protein in 1× Laemmli Sample Buffer (Bio-Rad #161–0737) with 2.5% 2-mercaptoethanol (Sigma #M6250) was incubated at 95° C for 2 minutes, and then run on a 4–20% Mini-PROTEAN TGX Stain-Free gel (Bio-Rad #456–8093) at 150 V. Samples were transferred to a 0.2 μM PVDF membrane using a Bio-Rad Trans-Blot Turbo Transfer System and Trans-Blot Turbo Transfer Pack (Bio-Rad #1704156). The membrane was blocked with blocking buffer (TBST + 5% non-fat dry milk) for 30 minutes at room temperature while rocking. Blocking buffer was discarded and primary antibody (diluted in blocking buffer) was added and incubated overnight at 4° C while rocking. Buffer was discarded and the membrane was washed three times for 10 minutes at room temperature with TBST (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% Tween 20) while rocking. Secondary antibody (diluted in blocking buffer) was added, incubated for 1 hour while rocking at room temperature, and then washed three times for 10 minutes at room temperature with TBST while rocking. After removing the TBST, the blot was developed with Clarity Max Western ECL Substrate (Bio-Rad #1705062) and imaged with a Bio-Rad ChemiDoc Imaging System (Bio-Rad, Hercules, CA).
For loading controls, membranes were first treated with blocking buffer + 1 mM sodium azide and incubated overnight at 4° C while rocking . Tubulin loading control primary antibody was added as described above except incubated for 1 hour at room temperature. For total histone H3 loading control, stripping buffer (200 mM glycine pH 2.2, 0.1% SDS, 0.01% Tween 20) was added to the blot, incubated for 10 minutes at room temperature while rocking, washed twice with PBS and twice with TBST. The membrane was then blocked a second time and antibody was added as described above.
ChIP-seq Library Preparation
40 million cross-linked mammalian cells (with Kc167 cell spike-in), thawed on ice, were lysed with 3 mL of buffer I (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.50% Igepal CA-630, 0.25% Triton ×−100, 1 cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail tablet (Sigma-Aldrich #11836170001) added fresh per 10 mL of buffer) and incubated on ice for 10 minutes, occasionally being inverted by hand. The lysate was centrifuged at 1,811 × g for 5 minutes at 4° C and the supernatant discarded. Pellets were resuspended in 3 mL of buffer II (10 mM Tris-HCl pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1 cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail tablet added fresh per 10 mL of buffer) and incubated at room temperature for 10 minutes while slowly rotating end-over-end. The sample was centrifuged at 1,811 × g for 5 minutes at 4° C and the supernatant discarded. Pe llets were resuspended in 1 mL of buffer III (10 mM Tris-HCl pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, 0.5% sarkosyl, 1 cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail tablet added fresh per 10 mL of buffer) corresponding to an initial cell density of 4 × 107 cells/mL. A 10 μL aliquot was removed prior to sonication as an unsonicated control.
For histone H3K27ac and RNA polymerase II ChIP:
For chromatin shearing, samples were transferred to Covaris milliTUBE 1 mL AFA Fiber (Covaris #520135) and sonicated using a Covaris E220 focused-ultrasonicator (Covaris, Woburn, MA) at 20% duty factor, 140 peak intensity power, and 200 cycles per burst for 5 minutes for histone H3K27ac ChIP or at 10% duty factor, 140 peak intensity power, and 200 cycles per burst for 4 minutes for RNA polymerase II ChIP. The sample was transferred to a fresh 1.5 mL low-bind tube, the milliTUBE washed with 200 μl of buffer III, and added to the sample on ice. Triton ×−100 was added to the sonicated chromatin to a final concentration of 1% and incubated at 4° C for 5 minutes while slowly rotating end-over-end. The sample was centrifuged at 15,493 × g for 10 minutes at 4° C to remove insoluble material. The supernatant was transferred to a fresh 1.5 mL low-bind tube, and a 10 μl aliquot was removed as a sonicated control. The chromatin was then aliquoted into 1.5 mL low-bind tubes to obtain a concentration corresponding to an initial cell density of 5 × 106 cells per tube for H3K27ac ChIP or 10 ×106 cells per tube for RNA polymerase II ChIP. 20 ng of each control (unsonicated and sonicated) was run on a 1X TBE 1% analytical agarose gel with Quick-Load 2-log DNA Ladder (NEB #N0550) to verify successful DNA shearing.
50 μl of Protein A/G PLUS-Agarose beads (Santa Cruz Biotechnology #sc-2003) were washed twice with 1 mL of buffer III + 1% Triton ×−100. 50 μl of beads was added to chromatin and rotated end-over-end for 2 hours at 4° C to preclear the chromatin. Beads were centrifuged at 1000 ×g for 5 minutes at 4° C, the supernatant transferred to a fresh 1.5 mL low-bind tube, and a 1% aliquot removed as the input control.
For immunoprecipitation, 5 μl of Acetyl-Histone H3 (Lys27) (D5E4) XP Rabbit mAb (Cell Signaling Technology #8173S, lot 6, RRID: AB_10949503) for H3K27ac ChIP or 10 μL of Rpb1 NTD (D8L4Y) Rabbit mAb (Cell Signaling Technology #14958S, lot 3, RRID: AB_2687876) for RNA polymerase II ChIP was added to the chromatin and incubated overnight at 4° C while slowly rotating end-over-end. 50 μl of beads was added to the antibody/chromatin mix and rotated end-over-end for 2 hours at 4° C. The beads were washed 5 times with 1 mL of ice-cold ChIP washing buffer (50 mM HEPES pH 7.5, 0.5 M LiCl, 1 mM EDTA, 0.7% sodium deoxycholate, 1% Igepal CA-630). For each wash, the sample was rotated for 5 minutes at 4° C, centrifuged at 1,000 ×g for 2 minutes at 4°C, and the supernatant discarded. The beads were washed once with 1 mL of ice-cold 10 mM Tris-HCl pH 8.0, 50 mM NaCl, 1 mM EDTA, rotated for 5 minutes at 4° C, centrifuged at 1,000 × g for 2 minutes at 4° C, and the supernatant discarded.
For BRD4-NUT ChIP:
As NUT is only expressed in post-meiotic spermatids (French, 2018; French et al., 2003), we employed an anti-NUT antibody for BRD4-NUT ChIP, as done previously (Alekseyenko et al., 2015).
For chromatin shearing for BRD4-NUT ChIP, we adapted a combined mild sonication and nuclease digestion approach (Pchelintsev et al., 2016) that can improve the immunoprecipitation of high molecular weight proteins (BRD4-NUT MW: 200.1 kDa). Samples were transferred to Covaris milliTUBE 1 mL AFA Fiber (Covaris #520135) and sonicated using a Covaris E220 focused-ultrasonicator (Covaris, Woburn, MA) at 1% duty factor, 140 peak intensity power, and 200 cycles per burst for 20 minutes. The sample was transferred to a fresh 1.5 mL low-bind tube, the milliTUBE washed with 200 μl of buffer III, and added to the sample on ice. Triton ×−100 was added to the sonicated chromatin to a final concentration of 1%. The sample was centrifuged at 15,493 × g for 10 minutes at 4° C to remove insoluble material. Th e supernatant was transferred to a fresh 1.5 mL low-bind tube, and a 10 μl aliquot was removed as a sonicated control. The chromatin was then aliquoted into 1.5 mL low-bind tubes to obtain a concentration corresponding to an initial cell density of 20 ×106 cells per tube. 100 μl of 10× TBS (10× TBS: 0.5 M Tris pH 8.0, 1.5 M NaCl), 100 μl of 10% Triton X-100, then 10 μl of 100 mM MgCl2 was added for every 1 mL of chromatin. 11.2 units of Benzonase (Sigma-Aldrich #1014) was added to the sample and incubated for 25 minutes at room temperature. Benzonase was inactivated by adding 0.5 M EDTA pH 8.0 to a final concentration of 5 mM, and a 10 μL aliquot was removed as the Benzonase-treated control. 20 ng of each control (unsonicated, sonicated, and Benzonase-treated) was run on a 1× TBE 1% analytical agarose gel with Quick-Load 2-log DNA Ladder (NEB #N0550) to verify successful DNA shearing.
The day before immunoprecipitation, 50 μL of DynaBeads Protein A (Thermo Scientific #10001D) were blocked overnight at 4° C with 1 mL o f buffer III (without protease inhibitors) + 1% Triton ×−100, 0.1% SDS, 1 mg/mL BSA. The beads were then resuspended in 50 μl of buffer III + 0.1% SDS. 10 μL (2.55 μg) of NUT (C52B1) Rabbit mAb (Cell Signaling Technology Signaling #3625S, lot 4 RRID: AB_2066833) was added to the beads and incubated for 8 hours at 4° C. The beads were then collected with a magnet and the supernatant discarded.
A 1% aliquot was removed from the sample as the input control prior to immunoprecipitation and an amount of sample equivalent to 20 × 106 cells was added to the antibody-bound beads and incubated overnight at 4°C while rotating end-over-end. The beads were washed three times with 1 mL of ice-cold ChIP washing buffer (50 mM HEPES pH 7.5, 0.5 M LiCl, 1 mM EDTA, 0.7% sodium deoxycholate, 1% Igepal CA-630). For each wash, the sample was rotated for 5 minutes at 4° C, collected with a magnet, and the supernatant discarded. The beads were washed once with 1 mL of ice-cold 10 mM Tris-HCl pH 8.0, 50 mM NaCl, 1 mM EDTA, rotated for 5 minutes at 4° C, collected with a magnet, and the supernatant discarded.
Proceeded identically for both histone H3K27ac and BRD4-NUT ChIP:
DNA extraction was performed by adding 120 μl of elution buffer (20 mM Tris pH 8.0, 50 mM NaCl, 5 mM EDTA, 1% SDS, 0.1 μg/μl proteinase K) to the beads, resuspending the beads by vortexing at medium speed or tapping the tube, and incubating for at least 2 hours (up to overnight) at 65° C with constant mixing at 750 rpm on a ThermoMixer (Eppendorf). DNA (ChIP, input, and controls) was purified using a GenCatch PCR Cleanup Kit (Epoch Life Science #2360250) following the manufacturer’s instructions, eluting with 20 μl of elution buffer. DNA concentration was determined by Qubit 3.0 fluorometer and Qubit dsDNA HS Assay Kit (Invitrogen #Q32854).
DNA (ChIP and input) was prepared for high-throughput sequencing following the directions for “NEBNext End Prep” and “Adaptor Ligation” in the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs #E7645). After cleanup of adaptor-ligated DNA without size selection, the sample was eluted and reclaimed using 15 μL 10 mM Tris-HCl pH 8.0. DNA concentration was determined by Qubit 3.0 fluorometer and Qubit dsDNA HS Assay Kit (Invitrogen #Q32854). A PCR was set-up using 15 μL adaptor-ligated DNA, 5 μL i7 Index Primer, 5 μL Universal Primer or 5 μL i5 Index Primer, and 25 μL NEBNext Ultra II Q5 Master Mix. The sample was run on a thermocycler at 98° C for 30 seconds followed by 6 cycles of 98° C for 10 seconds, 65° C for 75 seconds, followed by 1 cycle at 65° C for 5 minutes, and finally held at 4° C. 50 μl of 10 mM Tris-HCl pH 8.0 was added to each sample and the sample size selected by first adding 65 μL (0.65x) of SPRIselect mixture (Beckman Coulter #B23318). This was vortexed to mix, incubated for 5 minutes at room temperature, and then the beads were collected in a magnetic stand for 5 minutes. The supernatant was transferred to a new tube, 15 μL (0.8x final) of SPRIselect was added, vortexed to mix, and incubated for 5 minutes at room temperature. Beads were collected with a magnet and washed twice with 200 μL 85% ethanol for 30 seconds. Beads were air dried on the magnet, resuspended in 15 μL of 10 mM Tris-HCl pH 8.0, and then incubated at room temperature for 5 minutes to elute the DNA. Beads were collected with a magnet and 15 μL of the eluate was transferred to a new tube. Sample concentration was determined with a Qubit dsDNA HS Assay. A second round PCR was set-up using 15 μL adaptor-ligated DNA, 5 μL i7 Index Primer, 5 μL Universal Primer or 5 μL i5 Index Primer, and 25 μL NEBNext Ultra II Q5 Master Mix. The sample was run on a thermocycler at 98° C for 30 seconds followed by 9 cycles of 98° C for 10 seconds, 65° C for 75 seconds, followed by 1 cycle at 65° C for 5 minutes, and fin ally held at 4° C. The amplified library was purified twice as in “Cleanup of PCR Amplification” in the NEBNext Ultra II DNA Library Prep Kit for Illumina, using SPRIselect volumes of 0.9x. The DNA was eluted and reclaimed in 15 μL 10mM Tris-HCl pH 8.0.
Final sample concentration was determined with a Qubit dsDNA HS Assay and DNA integrity assessed with a Bioanalyzer High Sensitivity DNA Chip (Agilent, Santa Clara, CA). DNA was either single-read sequenced with 50 sequencing cycles on an Illumina NextSeq 500 instrument or paired-end sequenced, 150 cycles each read, on an Illumina HiSeq × instrument.
ChIP-seq was performed in duplicate, where each biological replicate represents cells from a different passage as well as treated and cross-linked independently.
Hi-C Library Preparation
Hi-C libraries were prepared using minor modifications to the previously described in situ Hi-C protocol (Rao et al., 2014). Five million cross-linked mammalian cells (with Kc167 cell spike-in), thawed on ice, were resuspended in 250 μL of Hi-C lysis buffer (10 mM Tris-HCl pH 8.0, 10 mM NaCl, 0.2% Igepal CA-630) with 50 μL of 100x Protease Inhibitor Cocktail (Sigma #P8340), and incubated on ice for 15 minutes. The lysate was then centrifuged at 2,500 x g for 5 minutes at 4°C and the supernatant discarded. Nuclei were washed with 500 μL of ice-cold Hi-C lysis buffer, centrifuged at 2,500 × g for 5 minutes at 4°C, and the supernatant discarded. The pellet was gently resuspended in 50 μL of 0.5% SDS, incubated at 62° C for 10 minutes, and immediately cooled on ice. SDS was neutralized by adding 145 μL of water and then 25 μL of 10% Triton X-100 to the tube, mixed gently by inversion, and incubated for 15 minutes at 37° C. A 10% aliquot of the sample was taken as an undigested control. 25 μL of 10x NEBuffer 2 (New England Biolabs), followed by 4 μL of 25 U/μL MboI (New England Biolabs #R0147) was added to the sample and then gently inverted to mix. The sample was incubated overnight at 37° C in a ThermoMixer (Eppe ndorf) set to 750 rpm, 15 seconds on, 4 minutes 45 seconds off, to digest the chromatin.
To inactivate MboI, samples were incubated at 62° C for 20 minutes while shaking at 750 rpm, 15 seconds on, 4 minutes 45 seconds off in a ThermoMixer, and immediately cooled on ice. A second 10% aliquot was taken as a digested control. Restriction fragment overhangs were filled in and biotin labeled by adding 50 μL of fill-in master mix (24.75 μL water, 16.5 μL 1 mM Biotin-14-dATP (Axxora #JBS-NU-835-BIO14-L), 1.65 μL 10 mM dTTP, 1.65 μL 10 mM dGTP, 1.65 μL 10 mM dCTP, 8.8 μL 5U/μL DNA Polymerase I, Large (Klenow) Fragment (New England Biolabs #M0210) and incubated at 37° C for 45 minutes while shaking at 750 rpm, 1 5 seconds on, 4 minutes 45 seconds off in a ThermoMixer. 900 μL of ligation master mix (695 μL water, 126 μL 10x T4 DNA Ligase Buffer (New England Biolabs), 105 μL 10% Triton X-100, 12.6 μL 10 mg/mL BSA, 1.05 μL of Hi-C Ligation Control Template (pUC19 digested with SmaI @ 21pg/μL), 5.25 μL 400U/μL T4 DNA ligase (New England Biolabs #M0202)) was added and incubated at room temperature while slowly rotating end-over-end for 4 hours.
DNA extraction was performed by centrifuging the nuclei at 2,500 x g for 5 min at 4° C, discarding the supernatant, and resuspending the pellet in 300 μL of extraction buffer (10 mM Tris-HCl pH 8.0, 0.5 M NaCl, 1% SDS). For the undigested and digested control samples, 275 μL of extraction buffer was added to each control sample. 50 μL of 800 U/mL proteinase K was added and samples were incubated at 55° C for 30 minutes with constant mixing at 750 rpm on a ThermoMixer. The temperature was increased to 68° C and constant mixing was maintained while incubating overnight to reverse the cross-links. Samples were cooled to room temperature and briefly centrifuged. 1 μL of 20 mg/mL glycogen followed by 875 μL of 100% ice-cold ethanol was added to each sample, mixed by inverting the tube, and incubated at −80° C for 20 minutes. Samples were centrifuged at max speed for 30 minutes at 4° C. The pellet was washed twice with 800 μL of 70% ethanol and centrifuging at 20,627 × g for 10 minutes at 4° C. Samples were air-dried on ice for < 5 minutes, and, before completely drying, resuspended in 80 μL of 10 mM Tris-HCl pH 8.0. Samples were incubated at 42° C for 15 minutes mixing constantly at 750 rpm on a ThermoMixer to completely dissolve the DNA. 1 μL of RNase A (Thermo Scientific #EN0531) was added and incubated at 37° C for 30 minutes while constantly mixing at 750 rpm on a ThermoMixer. DNA concentration was determined by Qubit 3.0 fluorometer and Qubit dsDNA HS Assay Kit (Invitrogen #Q32854).
No more than 10 μg of DNA per Covaris microTUBE AFA Fiber (Covaris #520045) was sheared to a target peak of 400 bp using a Covaris E220 focused-ultrasonicator (Covaris, Woburn, MA) at duty factor 10%, peak incident power 140 W, 200 cycles per burst for 60 seconds. 125 μL of sample was transferred to a fresh 1.5 mL low-bind tube, the microTUBE washed with 75 μL of 10 mM Tris-HCl pH 8.0, 1 mM EDTA and added to the sample, and the sample size selected by first adding 130 μL (0.65x) of SPRIselect mixture (Beckman Coulter #B23318). This was vortexed to mix, incubated for 5 minutes at room temperature, and then the beads were collected in a magnetic stand for 5 minutes. The supernatant was transferred to a new tube, 30 μL (0.8× final) of SPRIselect was added, vortexed to mix, and incubated for 5 minutes at room temperature. Beads were collected with a magnet and washed twice with 200 μL 85% ethanol for 30 seconds. Beads were air dried on the magnet, resuspended in 53 μL of 10 mM Tris-HCl pH 8.0, and then incubated at room temperature for 5 minutes to elute the DNA. Beads were collected with a magnet and 51 μL of the eluate was transferred to a new tube. Sample concentration was determined with a Qubit dsDNA HS Assay.
50 μL of Dynabeads MyOne Streptavidin C1 beads (Life Technologies #65001) were washed twice with 100 μL 1× B&W buffer + 0.05% Tween 20 (2x Bind & Wash (B&W) buffer: 10 mM Tris-HCl pH 7.5, 2 M NaCl, 1 mM EDTA) and then resuspended in 50 μL of 2x B&W buffer. DNA was added to the beads and incubated at room temperature for 30 minutes while rotating end-over-end. Beads were washed twice with 100 μL 1x B&W buffer + 0.05% Tween 20, washed twice with 100 μL of TE buffer, and resuspended in 50 μL of 10 mM Tris-HCl pH 8.0. DNA was prepared for high-throughput sequencing following the directions for “NEBNext End Prep” and “Adaptor Ligation” in the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs #E7645) while still bound to the beads. Beads were then collected with a magnet, the supernatant discarded, the beads washed twice with 100 μL 1x B&W buffer + 0.05% Tween 20, and then twice with 100 μL 10 mM Tris-HCl pH 8.0, 1 mM EDTA. The sample was eluted by resuspending the beads in 5 μL of 95% freshly deionized formamide, 10 mM EDTA pH 8.0, and incubating at 90° C for 10 minutes. Beads were collected with a magnet and the supernatant was transferred to a fresh tube containing 16 μL of 10 mM Tris-HCl pH 8.0.
Small-scale qPCR was utilized to determine the optimal number of PCR cycles for library amplification. Reactions were performed in triplicate with 4 μL 100x diluted library DNA, 6 μL primer mix (3.33 μM Universal PCR primer, 3.33 μM Index Primer, 1.66 mM MgCl2, 3.33x SYBR Green I), 10 μL NEBNext Ultra II Q5 Master Mix and thermocycled at 98° C for 30 seconds followed by 35 cycles of 98 ° C for 10 seconds, 65° C for 75 seconds. Linear Rn versus cycle number was plotted to determine the cycle number corresponding to one-third of maximum fluorescent intensity, and, after accounting for 100-fold dilution, the optimal number of PCR cycles needed was determined. Final library amplification was performed in duplicate using 2.5 μL water, 2.5 μL 10 mM MgCl2, 5 μL i5 Index Primer, 5 μL i7 Index Primer, 10 μL eluted DNA (undiluted), 25 μL NEBNext Ultra II Q5 Master Mix. The split sample was run on a thermocycler at 98° C for 30 seconds followed by X cycles (as determined above by qPCR) of 98° C for 10 seconds, 65° C for 75 seconds, followed by 1 cycle at 65° C for 5 minutes, and finally held at 4° C. The amplified library was purified twice as in “Cleanup of PCR Amplification” in the NEBNext Ultra II DNA Library Prep Kit for Illumina, using SPRIselect volumes of 0.7x and 0.8x. The DNA was eluted in 17 μL 10mM Tris-HCl pH 8.0 and 15 μL was collected as the final product.
Final sample concentration was determined with a Qubit dsDNA HS Assay and DNA integrity assessed with a Bioanalyzer High Sensitivity DNA Chip (Agilent, Santa Clara, CA). DNA was paired-end sequenced, 150 cycles each read, on either an Illumina HiSeq × or NovaSeq 6000 instrument.
Hi-C was performed across six replicates for untreated TC-797 cells, in duplicate for uninduced and induced 293TRex cells, and in duplicate for MZ1, MZ1 washout, and TPL treated TC-797 cells, where each biological replicate represents cells from a different passage as well as treated and cross-linked independently. Of the untreated TC-797 cell replicates, four replicates were completely untreated and two replicates were 0.01% DMSO controls.
TT-seq Library Preparation
Media was removed from TC-797 cells growing on a 15 cm plate and replaced with media containing 500 μM 4-thiouridine (Sigma-Aldrich #T4509) prewarmed to 37° C. Cells were incubated for 5 minutes at 37° C with 5% CO2. The media was then removed, 2.5 mL of TRIzol reagent (Thermo Fisher #15596018) was immediately added to lyse the cells and total RNA was isolated following the manufacturer’s instructions, except 1 μL of 20 mg/mL RNase-free glycogen and 1 μL of freshly prepared 1 M DTT (1 mM final) was added during the isopropanol precipitation step and the samples were protected from light. Sample concentration was determined by Qubit 3.0 fluorometer and Qubit RNA BR Assay Kit (Invitrogen #Q10210). A 1 μL aliquot was removed and RNA integrity was confirmed (RIN ≥ 9.0) with a Bioanalyzer RNA 6000 Pico or Nano Chip (Agilent, Santa Clara, CA).
From the yield of total RNA determined above, the amount of total RNA per 10 million cells was determined. 400 ng of 4-thiouridine labelled total Drosophila Kc167 cell RNA was added/spiked-in to an aliquot of total RNA corresponding to 10 million cells. 2 μL 10x NEBNext RNA Fragmentation Buffer (New England Biolabs #E6150S) was added to 18 μL of sample (no more than 50 μg RNA, if more than 50 μg of RNA all volumes were linearly scaled up accordingly). The sample was incubated for exactly 2 minutes at 94° C in a preheated thermal cycler and then immediately transferred to ice followed by addition of 2.5 μL of 10x NEBNext RNA Fragmentation Stop Solution (New England Biolabs #E6150S) and 2.5 μL of 164 mM DTT (1 mM final DTT during RNAClean XP bead-binding below) for every 20 μL reaction volume. If necessary, total sample volume was adjusted to 50 μL using RNase-free (DEPC-treated) water and 1.8 volumes of RNAClean XP mixture (Beckman Coulter #A63987) and 270 μL of 100% isopropanol was added to each sample. The sample was mixed by pipetting up and down 10 times and then incubated at room temperature for 5 minutes. Tubes were placed in a magnetic stand for 5 minutes, or until the supernatant turned completely clear, at room temperature, and then the supernatant discarded. Beads were washed twice with 600 μL of freshly prepared 85% ethanol while the beads are still on the magnetic stand. Beads were incubated in the presence of ethanol for 30 seconds during each wash. Beads were air dried on the magnetic stand until the alcohol had completely evaporated. Beads were resuspended in 52 μL of RNase-free (DEPC-treated) water, mixed by pipetting up and down 10 times, and then incubate at room temperature for 2 minutes. Beads were collected with a magnetic stand for 2 minutes, or until the supernatant turns completely clear, at room temperature and then 50 μL of the supernatant transferred to a fresh microcentrifuge tube.
RNA was denatured by incubating at 65° C for 5 minu tes and then rapidly cooled on ice. RNA was biotinylated by mixing 1.5 μL 1 M HEPES-KOH pH 7.5, 1.5 μL 0.1 M EDTA, fragmented RNA (no more than 50 μg RNA, if more than 50 μg of RNA all volumes were linearly scaled up accordingly), RNase-free (DEPC-treated) water to 120 μL and then 30 μL 0.167 mg/mL MTSEA-biotin-XX (Biotium #90066) dissolved in DMF was added. The sample was incubated at room temperature while slowly rotating end-over-end for at least 30 minutes, but no more than 2 hours. One volume of 24:1 chloroform:isoamyl alcohol was added to each tube, mixed by shaking by hand for 15 seconds and then let sit for 2 minutes. The sample was centrifuged at 12,000 ×g for 5 minutes at 4° C. The aqueous upper phase was trans ferred to a new microcentrifuge tube and the RNA purified using RNAClean XP with supplemental isopropanol, as described above, except the sample was eluted with 55 μL of RNase-free (DEPC-treated) water.
100 μL of Dynabeads MyOne Streptavidin C1 beads (Life Technologies #65001) were washed twice with 100 μL pre-wash buffer 1 (50 mM NaCl, 0.1 M NaOH in DEPC-treated water), once with 100 μL pre-wash buffer 2 (0.1 M NaCl in DEPC-treated water), twice with 1x B&W buffer (2x Bind & Wash (B&W) buffer: 200 mM Tris-HCl pH 7.5, 2 M NaCl, 20 mM EDTA, 0.1% (v/v) Tween 20 in DEPC-treated water), and then blocked with 100 μL 1× B&W buffer + 40 ng/μL RNase-free glycogen by incubating for 1 hour at room temperature. Beads were washed twice with 100 μL 1X B&W buffer and then resuspended in 50 μL of 2X B&W buffer.
RNA was denatured by incubating at 65° C for 5 minutes and then rapidly cooled on ice. A 5 μL aliquot was saved as the input and then 50 μL of the bead suspension was rapidly added to the denatured, labelled RNA, pipetted up and down to mix, and then incubated for 15 minutes at room temperature while slowly rotating end-over-end while protected from light. Beads were washed three times with 100 μL 1x B&W buffer pre-warmed to 65° C. For each wash, the sample was incu bated for 2 minutes at 65° C in a ThermoMixer set to 750 rpm. Beads were then washed three times with 100 μL room-temperature 1x B&W buffer. RNA was eluted twice by incubating the beads in 50 μL of freshly prepared 100 mM DTT for 15 minutes at room temperature while protected from light and then purified using RNAClean XP with supplemental isopropanol, as described above, except the sample was eluted with 15 μL of RNase-free (DEPC-treated) water. Sample concentration was determined with a Qubit RNA HS Assay and RNA fragmentation verified with a Bioanalyzer RNA 6000 Pico Chip (Agilent, Santa Clara, CA).
rRNA was depleted from the sample using a NEBNext rRNA Depletion Kit (New England Biolabs #E6310) following the manufacturer’s instructions, except the RNA was purified using RNAClean XP with supplemental isopropanol, as described above, eluted with 65 μL of RNase-free (DEPC-treated) water, and sample concentration was determined with a Qubit RNA HS Assay. A DNA library (from reverse transcribed RNA) was prepared for high-throughput sequencing following chapter 5 in the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England Biolabs #E7760) with the following minor modifications to account for small insert sizes: During the cleanup after adaptor ligation 96.5 μL (1.0 volume) of SPRIselect beads was used. During the PCR cleanup step 60 μL (1.2 volumes) of SPRIselect beads was used, DNA was eluted and reclaimed using 50 μL 0.1× TE buffer or 10 mM Tris-HCl pH 8.0, and the PCR cleanup step repeated using 60 μL (1.2 volumes) of SPRIselect beads. During the second cleanup, DNA was eluted with 17 μL 10 mM Tris-HCl pH 8.0 and 15 μL reclaimed from the beads as the final product.
Final sample concentration was determined with a Qubit dsDNA HS Assay and DNA integrity assessed with a Bioanalyzer High Sensitivity DNA Chip (Agilent, Santa Clara, CA). DNA was paired-end sequenced, 150 cycles each read, on an Illumina HiSeq × instrument.
TT-seq was performed in triplicate for paired DMSO, MZ1, and MZ1 washout treated TC-797 cells and in duplicate for paired DMSO and TPL treated TC-797 cells, where each biological replicate represents cells from a different passage as well as treated and subsequently labelled with 4-thiouridine independently.
QUANTIFICATION AND STATISTICAL ANALYSIS
ChIP-seq Data Processing
ChIP-seq Profile Generation
Reads were trimmed for adaptor sequences using trimmomatic v0.36 and then aligned to concatenated hg38/Genome Reference Consortium Human Reference 38 Patch 15 (GRCh38.p15) and dm6/BDGP Release 6 + ISO1 MT assemblies of the human and Drosophila melanogaster genomes, respectively, using bowtie2 v2.3.1 with option --very-sensitive. Only reads with a MAPQ ≥ 30 were retained using samtools v1.3.1. PCR duplicates were removed with picard MarkDuplicates v2.9.2. MACS2 v2.1.4 (Zhang et al., 2008) was used to compute signal tracks by first running macs2 callpeak with parameters -B --SPMR --keep-dup all -g hs -f BAM and then running macs2 bdgcmp with parameter -m FE. Histone H3K27ac ChIP-seq profiles were calibrated using spike-in normalization with D. melanogaster as the calibration genome, as previously described (Bonhoure et al., 2014; Hu et al., 2015; Orlando et al., 2014). As the NUT epitope is not expressed in Drosophila Kc167 cells, BRD4-NUT ChIP-seq profiles were not spike-in normalized.
Megadomain and Super-enhancer Annotation
We used histone H3K27ac ChIP-seq data to identify megadomains, as done previously (Alekseyenko et al., 2015). The epic v0.2.12 implementation of the SICER algorithm (Xu et al., 2014) was used to identify broadly enriched domains from histone H3K27ac ChIP-seq data setting the window size to 2500 bp and gap size to 1 with parameters -w 2500 -g 1, respectively. Input libraries were used as controls for domain annotation. Often, we observed closely spaced enriched regions and to span these regions and capture the entire domain, SICER/epic-identified domains within 25 kb of one another were combined using bedtools v2.27.1 with command bedtools merge. To identify megadomains, the list of called domains was filtered by signal strength and by domain size. Signal strength was determined by subtracting the rpm normalized input reads from rpm normalized sample reads for each called domain and subsequently ranked in increasing order of counts. The ROSE algorithm (Rank Ordering of Super-Enhancers) (Whyte et al., 2013) was applied to the ranked dataset to determine the cutoff threshold for signal strength by sliding a diagonal line and finding the point along the abscissa where the diagonal line was tangent to the rank-ordered curve. The cutoff for feature size was determined in a similar manner by ranking the domain length in base pairs and applying ROSE. Domains that exceeded both the signal cutoff and the size threshold were retained using bedtools intersect with default parameters. The above was performed for each biological replicate, and, then, to create the highest confidence domain list, domains identified in both replicates were merged using bedtools intersect with default parameters. The resulting domains were considered “megadomains”.
To identify super-enhancers, domains that exceeded both the megadomain signal and megadomain size threshold were removed from the list of SICER/epic-identified domains and the above filtering procedure for both signal strength and domain size was repeated. Super-enhancers within 100 kb of one another were combined using bedtools v2.27.1 with command bedtools merge for analysis of megadomain–super-enhancer and super-enhancer–super-enhancer interactions.
Random Shuffle Control Generation
A random shuffled control set of megadomains was determined using bedtools shuffle with parameters -chrom -noOverlapping -excl to ensure that shuffled megadomains are retained on the chromosome they originated from, that shuffled megadomains do not overlap, and that shuffled megadomains are excluded from genome assembly gaps, respectively.
Metaplot and Heatmap Visualization
deepTools v3.1.3 (Ramírez et al., 2016) was used to generate metaplots and heatmaps of BRD4-NUT, H3K27ac, and RNA polymerase II ChIP-seq profiles within megadomains and 50 kb flanking regions.
Hi-C Data Processing
Hi-C Contact Map Generation
Data was processed as previously described using the Juicer pipeline (Durand et al., 2016a; 2016b; Rao et al., 2014). In brief, reads were aligned to concatenated hg38/Genome Reference Consortium Human Reference 38 Patch 15 (GRCh38.p15) and dm6/BDGP Release 6 + ISO1 MT assemblies of the human and Drosophila melanogaster genomes, respectively, using BWA-MEM v0.7.15-r1140. Reads were then assigned to MboI restriction fragments, duplicates removed, reads with a MAPQ ≥ 30 were retained, and intra restriction fragment reads removed. The genome was divided into equally spaced, non-overlapping bins and the number of reads counted in each pair of bins gave the number of contacts, generating a Hi-C contact map.
Hi-C contact maps were normalized by matrix balancing using the method of Knight and Ruiz (KR normalization) using Juicer Tools v1.8.9 with command juicer pre (Durand et al., 2016b; Knight and Ruiz, 2012; Rao et al., 2014). As biological replicates were highly reproducible, including those between completely untreated and 0.01% DMSO control TC-797 cells (referred to simply as DMSO treated TC-797 cells), datasets were merged after filtering, as described above, and the combined contact maps KR normalized. Hi-C contact maps were further normalized for differences in the number of contacts between samples using a normalization factor of contacts per billion (CPB). This is equivalent to reads per million (RPM) normalization for ChIP-seq and RNA-seq data. All figures represent merged, KR normalized, and sequencing depth normalized contact maps.
Pearson correlation matrices of Hi-C contact maps were computed using Juicer Tools (Durand et al., 2016b) v1.8.9 with command juicer pearsons with options KR BP 100000.
Contact Domain and Loop Annotation
Contact domains were annotated at 10 kb resolution using the previously described Arrowhead algorithms (Durand et al., 2016b; Rao et al., 2014) using Juicer Tools v1.8.9 with command juicer arrowhead with option -r 10000. Loops were annotated using the previously described HiCCUPS algorithm (Durand et al., 2016b; Rao et al., 2014) using Juicer Tools v1.9.0 with command juicer hiccups with default parameters.
Contact domains overlapping loops, that is loop domains, were identified by determining for every contact domain corner at location Mi,j was a loop located within distance 0.2*|i-j| (Rao et al., 2014). Loop-independent contact domains were identified as those contact domains that did not meet this criterion.
Aggregate Analysis of Domains and Loops
Aggregate domain analysis (ADA) was performed similarly to that as previously described (Flyamer et al., 2017). For every domain at location Mi,j with size l=|i-j|, a submatrix from [i-l:i+l, j-l:j+l] was extracted from the observed and log2(observed/expected) Hi-C contact maps. The expected number of contacts was determined as previously described (Durand et al., 2016b; Lieberman-Aiden et al., 2009; Rao et al., 2014) and, in the case of TC-797 cells, normalized to the DMSO sample based on the Drosophila spike-in data. Submatrices that extended beyond the ends of the chromosome were excluded from further analysis. This submatrix was then rescaled to 90 × 90 pixels using linear interpolation and block-averaging (Flyamer et al., 2017). The average signal at each pixel was then computed across all submatrices (Figure S5B).
An aggregate domain analysis (ADA) score was implemented to assess the log2(observed/expected) Hi-C contact enrichment at the domain corner relative to outside the domain and adjacent to the boundaries. The domain corner was chosen as loop domains visually have more contact enrichment at the corner than loop-independent contact domains; outside the domain and adjacent to the domain boundaries was chosen as these regions should be depleted of contacts if the domains analyzed are contact domains. From the 90 × 90 pixel observed/expected ADA matrix, the domain corner was defined as the upper triangle of the submatrix from rows 30 to 44 and columns 45 to 59 ([30:45, 45:60] in Python slicing notation). From the 90 × 90 pixel observe/expected ADA matrix, outside the domain and adjacent to the boundaries was defined as two regions: the lower triangle of the submatrix from rows 15 to 29 and columns 30 to 44 ([15:30, 30:45]) and the lower triangle of the submatrix from rows 45 to 59 and columns 60 to 74 ([45:60, 60:75]). The mean of the log2(observed/expected) contacts within these three regions was determined and the ADA score computed as the difference of the mean of the domain corner region and ½ the sum of the means of the outside the domain and adjacent to the boundary regions (Figure S5C).
Aggregate peak analysis (APA) was performed as previously described (Durand et al., 2016b; Rao et al., 2014) using Juicer Tools v1.8.9 with command juicer apa with default parameters, except the aggregate signal was averaged instead of summed.
Contacts as a Function of Genomic Distance
The average number of intrachromosomal contacts as a function of genomic distance was computed by determining the ratio of the number of observed contacts separated by a given genomic distance to the number of pairs of loci separated by that same genomic distance. Genomic distance was logarithmically binned.
Annotating Megadomain-Megadomain Interactions
Robust megadomain-megadomain interactions were identified based on enrichment relative to local background, similar to that used for identifying chromatin loops (Rao et al., 2014).
First, all possible megadomain-megadomain interactions are enumerated. Next, a local background region, defined as a donut surrounding each possible megadomain-megadomain interaction region, is identified. This donut is defined to have a 1-pixel gap (e.g. at 100 kb resolution a 100 kb gap) between the possible megadomain-megadomain interaction region and the inner part of the donut and a width of 3 pixels (e.g. at 100 kb resolution a 300 kb width). The average number of KR normalized (if KR normalization was not available VC normalization (Rao et al., 2014) was used) contacts in the donut is determined. For interchromosomal interactions genome-wide normalizations were used. To account for all donut pixels not being the same distance from the diagonal and for the donut pixels being a different distance from the diagonal than the possible megadomain-megadomain interaction region, this value is normalized by multiplying by the average expected value within the megadomain-megadomain interaction region divided by the average expected value within the donut region. As there is no one-dimensional distance dependence (i.e. no diagonal) for interchromosomal contacts, this step was omitted during identification of interchromosomal megadomain-megadomain interactions. This expected value is KR (or VC) normalized and does not obey Poisson statistics, so this value is multiplied by the average of the KR (or VC) correction factors within the possible megadomain-megadomain interaction region.
Prior to determination of statistical significance and multiple hypothesis testing, two filtering steps are performed. First, possible megadomain-megadomain interaction regions where its respective donut region would overlap with the diagonal are removed to ensure that the donut reflects the true local background and not the symmetric half of the Hi-C contact map. Second, possible megadomain-megadomain interaction regions less than 2-fold enriched for both intrachromosomal and interchromosomal relative to its respective donut region are removed for DMSO treated TC-797 cells and less than 2fold enriched for intrachromosomal interactions or 4-fold enriched for interchromosomal interactions relative to its respective donut region are removed for MZ1 treated, MZ1 washout, and triptolide treated TC-797 cells. This allowed for very stringent identification of megadomain-megadomain interactions, in a manner similar to that for chromatin loop annotation (Rao et al., 2014). Stronger enrichment of interchromosomal interactions was chosen because of the greater sparsity of interchromosomal Hi-C contact maps in MZ1 treated, MZ1 washout, and triptolide treated TC-797 cells.
We then test the hypothesis that the average number of observed raw contacts (rounded to obey Poisson statistics) within the possible megadomain-megadomain interaction region is significantly enriched relative to the donut region, defined above. We then corrected for multiple hypothesis testing using the Benjamini-Hochberg FDR control procedure with an FDR threshold of 0.005. Again, an extremely stringent value is used for robust and conservative identification of megadomain-megadomain interactions.
This process was performed at 100 kb resolution genome-wide for DMSO treated TC-797 cells and at 100 kb resolution for intrachromosomal interactions and 250 kb resolution of interchromosomal interactions in MZ1 treated, MZ1 washout, and triptolide treated TC-797 cells to generate the final annotation of megadomain-megadomain interactions.
Analysis of Megadomain-Megadomain Interactions
Aggregate analysis of megadomain-megadomain interactions was performed as above for aggregate domain analysis (ADA), except observed contacts instead of the log2(observed/expected) contacts were used and since megadomain-megadomain interactions are typically rectangular they were re-scaled to be square during linear interpolation and block-averaging.
The domain-domain interaction score was implemented to assess the observed Hi-C contact enrichment within the interaction relative to that outside this region. This score was computed as the ratio of the mean of the number of Hi-C contacts within the rescaled interaction region, rows 30 to 59 and columns 30 to 59 ([30:60, 30:60] in Python slicing notation) to the mean of the number of Hi-C contacts outside this region, all pixels except those from rows 30 to 59 and columns 30 to 59 ([30:60, 30:60]) (Figure S2C).
Analysis of Triple Interactions
Interactions between three genomic loci (triples) were identified from higher-order contacts identified as DNA concatemers in Hi-C data (Darrow et al., 2016; Rao et al., 2017). Abnormal chimeric read pairs that aligned to two or more loci were identified by the Juicer pipeline (Durand et al., 2016b; 2016a; Rao et al., 2014) and parsed into triple, quadruple, or quintuple contacts. Duplicates were removed analogously to the deduplication procedure implemented by the Juicer pipeline (Durand et al., 2016b; 2016a; Rao et al., 2014). Counts of unique (non-duplicated) higher-order contacts are given in Table S2.
Triple and quadruple interactions that overlapped megadomains were identified using bedtools intersect. Only megadomains separated by greater than 1 Mb were considered, and changing the window size from 0.1 to 2 Mb did not influence the results. Interactions were filtered for those where each locus overlapped a distinct megadomain. Among quadruple interactions, triple megadomain-megadomain-megadomain interactions were extracted where one and only one locus in the quadruple did not overlap a megadomain. Where each locus in the quadruple overlapped a megadomain, the quadruple was collapsed to four distinct triple megadomain-megadomain-megadomain interactions. Differences due to sequencing depth between samples were normalized by dividing by the ratio of total unique higher-order contacts between samples.
We used a bootstrapping approach to account for the sparse number of higher-order interactions in Hi-C datasets (Darrow et al., 2016; Rao et al., 2017), thereby estimating the expected random distribution of megadomain-megadomain-megadomain interactions and evaluating for differences between DMSO treated, MZ1 treated, MZ1 washout, and triptolide treated TC-797 cells. Following the above procedure, we identified the number triple megadomain-megadomain-megadomain interactions using a random shuffle control set of megadomains and then determined the ratio of random shuffle triple interactions between DMSO treated versus MZ1 treated, MZ1 washout versus MZ1 treated, and triptolide versus DMSO treated TC-797 cells. This procedure was repeated 10,000 times generating a new ratio each time. P-values were then determined by comparing the observed ratio to the distribution of shuffled ratios using a random permutation test.
Subcompartment Annotation
Subcompartments were annotated at 100 kb resolution using machine learning to cluster loci based on interchromosomal contact patterns, similar to that as previously described (Rao et al., 2014). An interchromosomal, genome-wide KR normalized contact matrix was constructed with odd chromosomes on the rows and even chromosomes on the columns. Rows and columns with undefined or zero contacts were removed. A row-based z-score of each entry was determined using R’s scale function. Then, an unsupervised Gaussian hidden Markov model (GHMM) with 2–19 components (i.e. putative subcompartments) was trained on the resulting matrix using Python’s hmmlearn.hmm.GaussianHMM.fit function with 1000 iterations to estimate model parameters. Python’s hmmlearn.hmm.GaussianHMM.predict function was then used to infer the states (i.e. putative subcompartments) on the odd chromosomes. The matrix was transposed and the above procedure followed to infer the states on the even chromosomes. Sixteen states (i.e. putative subcompartments) were chosen for further analysis.
To assess both accuracy of clustering and to match clusters from the even chromosomes with those from the odd chromosomes, we calculated the contact enrichment between each pair of states on the even and odd chromosomes. Contact enrichment was determined as the mean number of contacts for each pair of states divided by the number of contacts from the average of 100 random shuffled control set of states. State 14 on even chromosomes preferentially interacted with state 15 on the odd chromosomes, indicating a one-to-one mapping between these states and visual inspection of these states revealed that they overlapped with megadomains. After merging states even 14 and odd 15, we referred to this state as subcompartment M (196 100 kb bins, or 63 merged subcompartment intervals after merging adjacent bins).
However, there was not a clear one-to-one mapping between the other states. An apparent lack of one-to-one mapping could be due to the necessity to fix ab initio the number of components for the GHMM resulting in one subcompartment being split into smaller states. For example, the patterns of contact enrichment (Figure S4A) and the mean parameter estimated by the GHMM (Figure S4B) for states 1 and 8 on the odd chromosomes appear very similar, implying that these states should be merged together. Therefore, to merge states lacking a clear one-to-one mapping, subcompartment M was first removed from further consideration, each row of the contact enrichment matrix was rescaled using R’s scale function, dissimilarity values between the rows were calculated by R’s dist(method = “Euclidean”) function, and the resulting vectors were then clustered using hierarchical clustering by R’s hclust(method = “complete”, members = NULL) function (Figure S4C, left). The same method was applied for the columns, which corresponded to the odd chromosomes (Figure S4C, right). After each round of merging, we computed a new contact enrichment between each pair of merged states on the even and odd chromosomes, as described above. Our final choice of five states (in addition to subcompartment M) was based on the finding of a one-to-one mapping between states on the even and odd chromosomes (Figure S4D).
We next checked if the above procedure resulted in putative subcompartments with properties in agreement with previously defined compartments (Lieberman-Aiden et al., 2009) and subcompartments (Rao et al., 2014). The five states separated into two groups: A1L and A2L in one group, had contact enrichment correlated with each other and anticorrelated with the other states (Figure S3G), and had higher GC content than the other states (Figure S4D). The other group, B1L, B2L, and B3, had contact enrichment correlated with each other and anticorrelated with states A1L and A2L (Figure S3G), and had lower GC content than the other states (Figure S4D). Histone H3K27ac is highly enriched in subcompartment M (Figure S4E). Histone H3K27ac is also enriched in subcompartments A1L and A2L (Figure S4E) and positively correlated with these subcompartments (Figure S4F). We labelled these subcompartments A1L (i.e. A1-Like), A2L (i.e. A2-Like), B1L (i.e. B1-Like), etc. for consistency with previously defined subcompartments (Rao et al., 2014).
A subcompartment score was implemented to assess the how likely each locus is to be in each subcompartment. This score was defined to be the mean parameter from the GHMM for each locus in the genome. For subcompartments derived from merged states, given that the subcompartment score is similar between states (Figure S4B), the average of the mean score from each state was used for the subcompartment score. As this score comes from a parameter from the model, we compared the subcompartment score to a biased inter-chromosomal contact frequency, similar to that as previously described (Monahan et al., 2019). From the genome-wide KR normalized contact map, the number of inter-chromosomal megadomain contacts for every 100 kb bin in the genome was determined and this value was divided by the total interchromosomal contacts for each respective bin to yield the percent of inter-chromosomal Hi-C contacts to megadomains.
Subcompartment M was annotated in triptolide treated TC-797 cells by applying the GHMM model trained on the interchromosomal Hi-C matrices from DMSO treated cells to the interchromosomal Hi-C matrices from triptolide treated cells. First, 100 kb interchromosomal contact matrices from triptolide treated cells were normalized using a row-based z-score, as described above. Next, to match the exact coordinates from DMSO treated cells, undefined rows or columns due to genome-wide KR normalization specifically from the triptolide treated cells were replaced with zeros. Finally, subcompartment M was predicted using hmmlearn.hmm.GaussianHMM.predict by retaining states even 14 and odd 15 after filtering out rows and columns in these states which were all zeros. Hi-C contact maps from MZ1 treated and MZ1 washout TC-797 cells were too sparse for accurate subcompartment annotation.
TT-seq Data Processing
TT-seq Profile Generation
Reads were trimmed for adaptor sequences using trimmomatic v0.36 and then aligned to concatenated hg38/Genome Reference Consortium Human Reference 38 Patch 15 (GRCh38.p15) and dm6/BDGP Release 6 + ISO1 MT assemblies of the human and Drosophila melanogaster genomes, respectively, using STAR (Dobin et al., 2013) v2.5.2b with options --outFilterType BySJout --outFilterMultimapNmax 20 -alignSJoverhangMin 8 --alignSJDBoverhangMin 1 --outFilterMismatchNmax 999 --outFilterMismatchNoverReadLmax 0.04 --alignIntronMin 20 --alignIntronMax 1000000 -alignMatesGapMax 1000000. Duplicate and multimapper reads were marked with STAR with option --bamRemoveDuplicatesType UniqueIdentical and then removed with samtools v1.3.1 with option -F 1024. STAR was used to compute signal tracks with options --outWigType bedGraph --outWigStrand Stranded --outWigNorm RPM. TT-seq profiles were calibrated using spike-in normalization with D. melanogaster as the calibration genome, as previously described (Lugowski et al., 2018).
Heatmap Visualization
deepTools v3.1.3 (Ramírez et al., 2016) was used to generate metaplots and heatmaps of TT-seq profiles within genes.
Differential Gene Expression Analysis
Reads overlapping genes were counted using htseq-count v0.9.1 (Anders et al., 2015) with parameters -f bam -r pos -s reverse -t gene -i gene_id. The gene set used consisted of protein-coding genes and lincRNAs from human GENCODE v24 (gtf column “transcript_type” either “protein_coding” or “lincRNA”) concatenated with those from FlyBase release 6.21. Expression levels (fpkm) and differentially expressed genes were determined using DESeq2 v1.18.1 (Love et al., 2014), excluding genes with less than 10 counts in all samples, using size factors determined from the D. melanogaster gene counts, and calling significance at a Benjamini-Hochberg adjusted p-value < 0.01.
DATA AND CODE AVAILABILITY
Hi-C, ChIP-seq, and TT-seq datasets generated in this study have been deposited in the Gene Expression Omnibus (GEO) and are publicly available under accession numbers GSE133163, GSE133122, and GSE133164, respectively.
Supplementary Material
Table S1. Hi-C Sequencing Statistics and Quality Metrics, Related to Figure 1 For definitions of each metric see Rao et al., 2014.
Table S2. Counts of Unique (Non-Duplicated) Higher-Order Contacts, Related to Figure 3
Table S3. Differentially Expressed Genes Within Subcompartment M, Related to Figure 5
Table S4. Oligopaint FISH Probes, Related to Figure S3
HIGHLIGHTS.
Hyperacetylated chromatin interacts over multiple megabases and between chromosomes
BRD4-NUT oncoprotein drives interactions to form a specific nuclear subcompartment
Pharmacological degradation of BRD4-NUT abolishes subcompartment interactions
Subcompartment persists in the absence of transcription
ACKNOWLEDGMENTS
We thank Christopher A. French for providing the 293TRex-Flag-BRD4-NUT-HA cell line; Jeffrey A. Toretsky for providing the TC-797 cell line; Katrin M. Carlson Leuer and Dianna Dichtl for karyotyping; and Roger D. Kornberg, Jason H. Brickner, Edwin R. Smith, and Kaixiang Cao for critical reading of the manuscript. This research was supported by an NIH Director’s Early Independence Award (DP5OD024587) to K.P.E.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alekseyenko AA, Walsh EM, Wang X, Grayson AR, Hsi PT, Kharchenko PV, Kuroda MI, French CA, 2015. The oncogenic BRD4-NUT chromatin regulator drives aberrant transcription within large topological domains. Genes Dev 29, 1507–1523. doi: 10.1101/gad.267583.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko AA, Walsh EM, Zee BM, Pakozdi T, Hsi P, Lemieux ME, Dal Cin P, Ince TA, Kharchenko PV, Kuroda MI, French CA, 2017. Ectopic protein interactions within BRD4-chromatin complexes drive oncogenic megadomain formation in NUT midline carcinoma. Proceedings of the National Academy of Sciences 114, E4184–E4192. doi: 10.1073/pnas.1702086114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W, 2015. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 66–169. doi: 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banani SF, Lee HO, Hyman AA, Rosen MK, 2017. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 18, 285–298. doi: 10.1038/nrm.2017.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beagrie RA, Scialdone A, Schueler M, Kraemer DCA, Chotalia M, Xie SQ, Barbieri M, de Santiago I, Lavitas L-M, Branco MR, Fraser J, Dostie J, Game L, Dillon N, Edwards PAW, Nicodemi M, Pombo A, 2017. Complex multi-enhancer contacts captured by genome architecture mapping. Nature 543, 519–524. doi: 10.1038/nature21411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beliveau BJ, Boettiger AN, Nir G, Bintu B, Yin P, Zhuang X, Wu C-T, 2017. In Situ Super-Resolution Imaging of Genomic DNA with OligoSTORM and OligoDNA-PAINT. Methods Mol Biol 1663, 231–252. doi: 10.1007/978-1-4939-7265-4_19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beliveau BJ, Joyce EF, Apostolopoulos N, Yilmaz F, Fonseka CY, McCole RB, Chang Y, Li JB, Senaratne TN, Williams BR, Rouillard J-M, Wu C-T, 2012. Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proceedings of the National Academy of Sciences 109, 21301–21306. doi: 10.1073/pnas.1213818110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beliveau BJ, Kishi JY, Nir G, Sasaki HM, Saka SK, Nguyen SC, Wu C-T, Yin P, 2018. OligoMiner provides a rapid, flexible environment for the design of genome-scale oligonucleotide in situ hybridization probes. Proceedings of the National Academy of Sciences 115, E2183–E2192. doi: 10.1073/pnas.1714530115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickmore WA, 2013. The spatial organization of the human genome. Annu Rev Genomics Hum Genet 14, 67–84. doi: 10.1146/annurev-genom-091212-153515 [DOI] [PubMed] [Google Scholar]
- Bickmore WA, van Steensel B, 2013. Genome architecture: domain organization of interphase chromosomes. Cell 152, 1270–1284. doi: 10.1016/j.cell.2013.02.001 [DOI] [PubMed] [Google Scholar]
- Bintu B, Mateo LJ, Su J-H, Sinnott-Armstrong NA, Parker M, Kinrot S, Yamaya K, Boettiger AN, Zhuang X, 2018. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362, eaau1783. doi: 10.1126/science.aau1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boija A, Klein IA, Sabari BR, Dall’Agnese A, Coffey EL, Zamudio AV, Li CH, Shrinivas K, Manteiga JC, Hannett NM, Abraham BJ, Afeyan LK, Guo YE, Rimel JK, Fant CB, Schuijers J, Lee TI, Taatjes DJ, Young RA, 2018. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 175, 1842–1855.e16. doi: 10.1016/j.cell.2018.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot J-P, Tanay A, Cavalli G, 2017. Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 171, 557–572.e24. doi: 10.1016/j.cell.2017.09.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhoure N, Bounova G, Bernasconi D, Praz V, Lammers F, Canella D, Willis IM, Herr W, Hernandez N, Delorenzi M, CycliX Consortium, 2014. Quantifying ChIP-seq data: a spiking method providing an internal reference for sample-to-sample normalization. Genome Res. 24, 1157–1168. doi: 10.1101/gr.168260.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho W-K, Spille J-H, Hecht M, Lee C, Li C, Grube V, Cisse II, 2018. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415. doi: 10.1126/science.aar4199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong S, Dugast-Darzacq C, Liu Z, Dong P, Dailey GM, Cattoglio C, Heckert A, Banala S, Lavis L, Darzacq X, Tjian R, 2018. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 361, eaar2555. doi: 10.1126/science.aar2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman KG, Crews CM, 2018. Proteolysis-Targeting Chimeras: Harnessing the Ubiquitin-Proteasome System to Induce Degradation of Specific Target Proteins. Annual Review of Cancer Biology 2, 41–58. doi: 10.1146/annurev-cancerbio-030617-050430 [DOI] [Google Scholar]
- Darrow EM, Huntley MH, Dudchenko O, Stamenova EK, Durand NC, Sun Z, Huang S-C, Sanborn AL, Machol I, Shamim M, Seberg AP, Lander ES, Chadwick BP, Aiden EL, 2016. Deletion of DXZ4 on the human inactive X chromosome alters higher-order genome architecture. Proceedings of the National Academy of Sciences 201609643. doi: 10.1073/pnas.1609643113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit E, Vos ESM, Holwerda SJB, Valdes-Quezada C, Verstegen MJAM, Teunissen H, Splinter E, Wijchers PJ, Krijger PHL, de Laat W, 2015. CTCF Binding Polarity Determines Chromatin Looping. Mol Cell 60, 676–684. doi: 10.1016/j.molcel.2015.09.023 [DOI] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B, 2012. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. doi: 10.1038/nature11082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, Aiden EL, 2016a. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst 3, 99–101. doi: 10.1016/j.cels.2015.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, Aiden EL, 2016b. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst 3, 95–98. doi: 10.1016/j.cels.2016.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagen KP, 2018. Principles of Chromosome Architecture Revealed by Hi-C. Trends Biochem. Sci. 43, 469–478. doi: 10.1016/j.tibs.2018.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk M, Feodorova Y, Naumova N, Imakaev M, Lajoie BR, Leonhardt H, Joffe B, Dekker J, Fudenberg G, Solovei I, Mirny LA, 2019. Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature 570, 395–399. doi: 10.1038/s41586-019-1275-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE, 2010. Selective inhibition of BET bromodomains. Nature 468, 1067–1073. doi: 10.1038/nature09504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suvà ML, Bernstein BE, 2016. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114. doi: 10.1038/nature16490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyamer IM, Gassler J, Imakaev M, Brandão HB, Ulianov SV, Abdennur N, Razin SV, Mirny LA, Tachibana-Konwalski K, 2017. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 544, 110–114. doi: 10.1038/nature21711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke M, Ibrahim DM, Andrey G, Schwarzer W, Heinrich V, Schöpflin R, Kraft K, Kempfer R, Jerković I, Chan W-L, Spielmann M, Timmermann B, Wittler L, Kurth I, Cambiaso P, Zuffardi O, Houge G, Lambie L, Brancati F, Pombo A, Vingron M, Spitz F, Mundlos S, 2016. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538, 265–269. doi: 10.1038/nature19800 [DOI] [PubMed] [Google Scholar]
- French CA, 2018. NUT Carcinoma: Clinicopathologic features, pathogenesis, and treatment. Pathol. Int. 51, 3327. doi: 10.1111/pin.12727 [DOI] [PubMed] [Google Scholar]
- French CA, Miyoshi I, Kubonishi I, Grier HE, Perez-Atayde AR, Fletcher JA, 2003. BRD4-NUT fusion oncogene: a novel mechanism in aggressive carcinoma. Cancer Res. 63, 304–307. [PubMed] [Google Scholar]
- French CA, Ramirez CL, Kolmakova J, Hickman TT, Cameron MJ, Thyne ME, Kutok JL, Toretsky JA, Tadavarthy AK, Kees UR, Fletcher JA, Aster JC, 2008. BRD-NUT oncoproteins: a family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene 27, 2237–2242. doi: 10.1038/sj.onc.1210852 [DOI] [PubMed] [Google Scholar]
- Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, Mirny LA, 2016. Formation of Chromosomal Domains by Loop Extrusion. Cell Reports 15, 2038–2049. doi: 10.1016/j.celrep.2016.04.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassler J, Brandão HB, Imakaev M, Flyamer IM, Ladstatter S, Bickmore WA, Peters JM, Mirny LA, Tachibana K, 2017. A mechanism of cohesin-dependent loop extrusion organizes zygotic genome architecture. EMBO J e201798083. doi: 10.15252/embj.201798083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, Doolittle LK, Schneider MWG, Jensen LE, Gamarra N, Henry L, Gerlich DW, Redding S, Rosen MK, 2019. Organization of Chromatin by Intrinsic and Regulated Phase Separation. Cell 179, 470–484.e21. doi: 10.1016/j.cell.2019.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson AR, Walsh EM, Cameron MJ, Godec J, Ashworth T, Ambrose JM, Aserlind AB, Wang H, Evan GI, Kluk MJ, Bradner JE, Aster JC, French CA, 2014. MYC, a downstream target of BRD-NUT, is necessary and sufficient for the blockade of differentiation in NUT midline carcinoma. Oncogene 33, 1736–1742. doi: 10.1038/onc.2013.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU, Jung I, Wu H, Zhai Y, Tang Y, Lu Y, Wu Y, Jia Z, Li W, Zhang MQ, Ren B, Krainer AR, Maniatis T, Wu Q, 2015. CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. Cell 162, 900–910. doi: 10.1016/j.cell.2015.07.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarhuis JHI, van der Weide RH, Blomen VA, Yáñez-Cuna JO, Amendola M, van Ruiten MS, Krijger PHL, Teunissen H, Medema RH, van Steensel B, Brummelkamp TR, de Wit E, Rowland BD, 2017. The Cohesin Release Factor WAPL Restricts Chromatin Loop Extension. Cell 169, 693–707.e14. doi: 10.1016/j.cell.2017.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Weintraub AS, Day DS, Valton A-L, Bak RO, Li CH, Goldmann J, Lajoie BR, Fan ZP, Sigova AA, Reddy J, Borges-Rivera D, Lee TI, Jaenisch R, Porteus MH, Dekker J, Young RA, 2016. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 351, 1454–1458. doi: 10.1126/science.aad9024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Petela N, Kurze A, Chan K-L, Chapard C, Nasmyth K, 2015. Biological chromodynamics: a general method for measuring protein occupancy across the genome by calibrating ChIP-seq. Nucleic Acids Res 43, e132. doi: 10.1093/nar/gkv670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers I, Kwak H, Lis JT, 2014. Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. elife 3, e02407. doi: 10.7554/eLife.02407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight PA, Ruiz D, 2012. A fast algorithm for matrix balancing. IMA J Numer Anal 33, drs019–1047. doi: 10.1093/imanum/drs019 [DOI] [Google Scholar]
- Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, Narlikar GJ, 2017. Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 22, 1181. doi: 10.1038/nature22822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J, 2009. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 326, 289–293. doi: 10.1126/science.1181369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S, 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K, Crews CM, 2015. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol 22, 755–763. doi: 10.1016/j.chembiol.2015.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugowski A, Nicholson B, Rissland OS, 2018. DRUID: A pipeline for transcriptome-wide measurements of mRNA stability. RNA rna.062877.117. doi: 10.1261/rna.062877.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, Santos-Simarro F, Gilbert-Dussardier B, Wittler L, Borschiwer M, Haas SA, Osterwalder M, Franke M, Timmermann B, Hecht J, Spielmann M, Visel A, Mundlos S, 2015. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025. doi: 10.1016/j.cell.2015.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan K, Horta A, Lomvardas S, 2019. LHX2- and LDB1-mediated trans interactions regulate olfactory receptor choice. Nature 565, 448–453. doi: 10.1038/s41586-018-0845-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth K, 2001. Disseminating the genome: joining, resolving, and separating sister chromatids during mitosis and meiosis. Annu. Rev. Genet. 35, 673–745. doi: 10.1146/annurev.genet.35.102401.091334 [DOI] [PubMed] [Google Scholar]
- Nora EP, Goloborodko A, Valton A-L, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, Bruneau BG, 2017. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169, 930–944.e22. doi: 10.1016/j.cell.2017.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, Gribnau J, Barillot E, Blüthgen N, Dekker J, Heard E, 2012. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385. doi: 10.1038/nature11049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuebler J, Fudenberg G, Imakaev M, Abdennur N, Mirny LA, 2018. Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proceedings of the National Academy of Sciences 115, E6697–E6706. doi: 10.1073/pnas.1717730115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, Fritz CC, Bradner JE, Guenther MG, 2014. Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Reports 9, 1163–1170. doi: 10.1016/j.celrep.2014.10.018 [DOI] [PubMed] [Google Scholar]
- Paul MR, Markowitz TE, Hochwagen A, Ercan S, 2018. Condensin Depletion Causes Genome Decompaction Without Altering the Level of Global Gene Expression in Saccharomyces cerevisiae. Genetics 210, 31–344. doi: 10.1534/genetics.118.301217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pchelintsev NA, Adams PD, Nelson DM, 2016. Critical Parameters for Efficient Sonication and Improved Chromatin Immunoprecipitation of High Molecular Weight Proteins. PLoS ONE 11, e0148023. doi: 10.1371/journal.pone.0148023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T, 2016. deepTools2: a next generation web server for deepsequencing data analysis. Nucleic Acids Res 44, W160–5. doi: 10.1093/nar/gkw257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huang S-C, Glenn St Hilaire B, Engreitz JM, Perez EM, KiefferKwon K-R, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, Huang X, Shamim MS, Shin J, Turner D, Ye Z, Omer AD, Robinson JT, Schlick T, Bernstein BE, Casellas R, Lander ES, Aiden EL, 2017. Cohesin Loss Eliminates All Loop Domains. Cell 171, 305–320.e24. doi: 10.1016/j.cell.2017.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL, 2014. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. doi: 10.1016/j.cell.2014.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynoird N, Schwartz BE, Delvecchio M, Sadoul K, Meyers D, Mukherjee C, Caron C, Kimura H, Rousseaux S, Cole PA, Panne D, French CA, Khochbin S, 2010. Oncogenesis by sequestration of CBP/p300 in transcriptionally inactive hyperacetylated chromatin domains. EMBO J 29, 2943–2952. doi: 10.1038/emboj.2010.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley MJ, Nichols MH, Lyu X, Ando-Kuri M, Rivera ISM, Hermetz K, Wang P, Ruan Y, Corces VG, 2017. Evolutionarily Conserved Principles Predict 3D Chromatin Organization. Mol Cell 67, 837–852.e7. doi: 10.1016/j.molcel.2017.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, Li CH, Guo YE, Day DS, Schuijers J, Vasile E, Malik S, Hnisz D, Lee TI, Cisse II, Roeder RG, Sharp PA, Chakraborty AK, Young RA, 2018. Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958. doi: 10.1126/science.aar3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn AL, Rao SSP, Huang S-C, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, Geeting KP, Gnirke A, Melnikov A, McKenna D, Stamenova EK, Lander ES, Aiden EL, 2015. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proceedings of the National Academy of Sciences 112, E6456–65. doi: 10.1073/pnas.1518552112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalb B, Michel M, Zacher B, Frühauf K, Demel C, Tresch A, Gagneur J, Cramer P, 2016. TT-seq maps the human transient transcriptome. Science 352, 1225–1228. doi: 10.1126/science.aad9841 [DOI] [PubMed] [Google Scholar]
- Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe-Mie Y, Fonseca NA, Huber W, H Haering C, Mirny L, Spitz F, 2017. Two independent modes of chromatin organization revealed by cohesin removal. Nature 551, 51–56. doi: 10.1038/nature24281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G, 2012. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 148, 458–472. doi: 10.1016/j.cell.2012.01.010 [DOI] [PubMed] [Google Scholar]
- Shin Y, Brangwynne CP, 2017. Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382. doi: 10.1126/science.aaf4382 [DOI] [PubMed] [Google Scholar]
- Shogren-Knaak M, Ishii H, Sun J-M, Pazin MJ, Davie JR, Peterson CL, 2006. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844–847. doi: 10.1126/science.1124000 [DOI] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, Karpen GH, 2017. Phase separation drives heterochromatin domain formation. Nature 144, 732. doi: 10.1038/nature22989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titov DV, Gilman B, He Q-L, Bhat S, Low W-K, Dang Y, Smeaton M, Demain AL, Miller PS, Kugel JF, Goodrich JA, Liu JO, 2011. XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat. Chem. Biol. 7, 182–188. doi: 10.1038/nchembio.522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toretsky JA, Jenson J, Sun C-C, Eskenazi AE, Campbell A, Hunger SP, Caires A, Frantz C, Hill JL, Stamberg J, 2003. Translocation (11;15;19): a highly specific chromosome rearrangement associated with poorly differentiated thymic carcinoma in young patients. Am. J. Clin. Oncol. 26, 300–306. doi: 10.1097/01.COC.0000020960.98562.84 [DOI] [PubMed] [Google Scholar]
- Wang S, Su J-H, Beliveau BJ, Bintu B, Moffitt JR, Wu C-T, Zhuang X, 2016. Spatial organization of chromatin domains and compartments in single chromosomes. Science 353, 598–602. doi: 10.1126/science.aaf8084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lu J-J, He L, Yu Q, 2011. Triptolide (TPL) inhibits global transcription by inducing proteasome-dependent degradation of RNA polymerase II (Pol II). PLoS ONE 6, e23993. doi: 10.1371/journal.pone.0023993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA, 2013. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. doi: 10.1016/j.cell.2013.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE, 2015. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381. doi: 10.1126/science.aab1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Mayer A, Buckley DL, Erb MA, Roderick JE, Vittori S, Reyes JM, di Iulio J, Souza A, Ott CJ, Roberts JM, Zeid R, Scott TG, Paulk J, Lachance K, Olson CM, Dastjerdi S, Bauer S, Lin CY, Gray NS, Kelliher MA, Churchman LS, Bradner JE, 2017. BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol Cell 67, 5–18.e19. doi: 10.1016/j.molcel.2017.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wutz G, Várnai C, Nagasaka K, Cisneros DA, Stocsits RR, Tang W, Schoenfelder S, Jessberger G, Muhar M, Hossain MJ, Walther N, Koch B, Kueblbeck M, Ellenberg J, Zuber J, Fraser P, Peters JM, 2017. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO Journal 36, 3573–3599. doi: 10.15252/embj.201798004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Grullon S, Ge K, Peng W, 2014. Spatial clustering for identification of ChIP-enriched regions (SICER) to map regions of histone methylation patterns in embryonic stem cells. Methods Mol Biol 1150, 97–111. doi: 10.1007/978-1-4939-0512-6_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Diaz J, Jiao J, Wang R, You J, 2011. Perturbation of BRD4 protein function by BRD4-NUT protein abrogates cellular differentiation in NUT midline carcinoma. Journal of Biological Chemistry 286, 27663–27675. doi: 10.1074/jbc.M111.246975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zee BM, Dibona AB, Alekseyenko AA, French CA, Kuroda MI, 2016. The Oncoprotein BRD4-NUT Generates Aberrant Histone Modification Patterns. PLoS ONE 11, e0163820. doi: 10.1371/journal.pone.0163820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M, Chan K-H, Ciulli A, 2015. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 10, 1770–1777. doi: 10.1021/acschembio.5b00216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS, 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. doi: 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Hi-C Sequencing Statistics and Quality Metrics, Related to Figure 1 For definitions of each metric see Rao et al., 2014.
Table S2. Counts of Unique (Non-Duplicated) Higher-Order Contacts, Related to Figure 3
Table S3. Differentially Expressed Genes Within Subcompartment M, Related to Figure 5
Table S4. Oligopaint FISH Probes, Related to Figure S3
Data Availability Statement
Hi-C, ChIP-seq, and TT-seq datasets generated in this study have been deposited in the Gene Expression Omnibus (GEO) and are publicly available under accession numbers GSE133163, GSE133122, and GSE133164, respectively.