

Abstract
Interferon‐γ (IFN‐γ) is a major proinflammatory cytokine, and binding to its nearly ubiquitous receptor induces a wide variety of biological functions. To explore the role(s) of IFN‐γ signaling in astrocytes, transgenic mice (GFAP/IFN‐γR1ΔIC) expressing a dominant‐negative IFN‐γ receptor alpha chain under control of the astrocyte‐specific glial fibrillary acid protein (GFAP) promoter were generated. Transgenic mice developed normally, had normal astrocyte numbers and distribution, and exhibited no clinically overt phenotype. Transgene mRNA expression was detected only in the CNS, and the transgene‐encoded IFN‐γ receptor 1 colocalized with GFAP, which is consistent with astrocyte expression. Astrocytes from transgenic mice exhibited reduced IFN‐γ‐induced signaling as measured by major histocompatibility class II induction. Neither CNS inflammation nor perforin‐mediated clearance of a neurotropic mouse hepatitis virus from astrocytes was impaired following infection. Transgenic mice with impaired astrocyte responsiveness to IFN‐γ provide a model for studying the selective astrocyte‐dependent effects of this critical cytokine in CNS immunopathology. © 2005 Wiley‐Liss, Inc.
Keywords: central nervous system, glia, inflammation
Interferon‐γ (IFN‐γ) is a critical component in immunological defense against a variety of pathogenic agents, acting via induction of antimicrobial activity, macrophage and natural killer cell (NK cell) activation, increased expression of major histocompatibility complex (MHC) antigens, and regulation of B cell function (Schroder et al., 2004). IFN‐γ is produced by predominantly T cells and NK cells (Schroder et al., 2004); however, it has also been suggested that antigen‐presenting cells can secrete IFN‐γ (Frucht et al., 2001). IFN‐γ represents a critical component of immunological defense within the central nervous system (CNS; Benveniste, 1998; Chesler and Reiss, 2002). IFN‐γ‐secreting cells are normally absent from the resting CNS; however, during inflammation, IFN‐γ‐secreting cells are rapidly recruited into the CNS and exert their immunological function locally (Hart and Fabry, 1995; Popko et al., 1997). In addition to its critical role in both peripheral and CNS infections, IFN‐γ plays an important regulatory role in the pathogenesis of the human autoimmune demyelinating disease multiple sclerosis (MS) and its murine model, experimental autoimmune encephalomyelitis (EAE). Elevated IFN‐γ levels correlate with disease severity in both MS and EAE (Olsson, 1992; Popko et al., 1997; Benveniste, 1998). IFN‐γ secretion by peripheral blood mononuclear cells increases prior to MS clinical relapses, and treatment of MS patients with IFN‐γ had deleterious effects (Panitch et al., 1987; Dettke et al., 1997).
Astrocytes, the most abundant CNS glial cells, are essential for maintaining normal brain physiology. In addition to metabolic and trophic functions, they play a crucial role in the maintenance of blood–brain barrier integrity (Rubin and Staddon, 1999). Astrocytes are also a central component of the CNS response to injury and disease (Eng and Ghirnikar, 1994; Minagar et al., 2002). Astrocytic reactivity has been described in Alzheimer's disease, in Parkinson's disease (Minagar et al., 2002; Teismann and Schulz, 2004), and following traumatic injury and ischemia/hypoxia (Eng and Ghirnikar, 1994; Stoll et al., 1998). Astrocytes are also involved in viral and parasitic infections of the CNS and are believed to play important roles in demyelinating disease (Minagar et al., 2002; De Keyser et al., 2003; Wilson and Hunter, 2004). Astrocytes express several immune functions potentially associated with CNS immunopathology, including the ability to serve as antigen‐presenting cells with IFN‐γ‐inducible surface expression of MHC and costimulatory molecules as well as secretion of a variety of immune mediators, including both chemokines and cytokines (Benveniste, 1998; Dong and Benveniste, 2001).
Several neurotropic viruses infect astrocytes; however, none shows exclusive astrocyte tropism (Tyler and Gonzales‐Scarano, 1996). CNS infection with the neurotropic mouse hepatitis virus (MHV) strain JHM (JHMV) results in acute encephalomyelitis (Marten et al., 2001). JHMV initially infects ependymal cells but progresses rapidly into the CNS parenchyma, infecting astrocytes, microglia, oligodendroglia, and occasionally neurons (Wang et al., 1992). Acute infection induces rapid CNS recruitment of mononuclear cells, with elements of both the innate and the adaptive immune response (Marten et al., 2001), regulated in part by astrocyte‐secreted chemokines (Sun et al., 1995; Lane et al., 1998). Infiltration of T lymphocytes is associated with clearance of infectious virus, with both CD4+ and CD8+ T cells contributing to the reduction in virus replication (Williamson and Stohlman, 1990). CD8+ T cells; however, are the major effectors of virus control, utilizing a perforin‐dependent mechanism to limit replication in astrocytes, macrophages, and microglia (Lin et al., 1997; Bergmann et al., 2004). Similarly to its critical role(s) in the control of other CNS viral infections (Weidinger et al., 2001; Chesler and Reiss, 2002), IFN‐γ plays a protective role within the CNS during JHMV infection (Zhang et al., 1997; Parra et al., 1999), specifically by limiting virus replication in oligodendroglia. In addition to being targets of JHMV replication, astrocytes are a major source for immune mediators during JHMV infection (Sun et al., 1995; Lane et al., 1998), and it has been suggested that MHV neurovirulence correlates with the astrocyte proinflammatory response (Li et al., 2004).
Numerous astrocyte‐associated homeostatic repair mechanisms and immune functions occur in response to, or are enhanced by, IFN‐γ. Therefore, transgenic (tg) mice expressing a dominant‐negative IFN‐γ receptor alpha chain under the control of the astrocyte‐specific glial fibrillary acid protein (GFAP) promoter were produced to provide a tool for analyzing the consequences of disrupted IFN‐γ signaling by a prominent CNS resident cell type.
MATERIALS AND METHODS
Generation of GFAPγR1Δ Transgenic Mice
Transgenic GFAP/IFN‐γR1ΔIC (GFAPγR1Δ) mice expressing a IFN‐γR1 with a truncated cytoplasmic domain by astrocytes were generated by using the human GFAP promoter (Besnard et al., 1991). The truncated receptor, designated IFN‐γR1ΔIC (Dighe et al., 1995), has only three amino acids of the intracellular domain while retaining the IFN‐γR1 extracellular and transmembrane domains, abrogating intracellular signaling. The extracellular domain retains ligand binding activity and contains a human c‐myc tag. The IFN‐γR1ΔIC transgene was initially excised from plasmid pSFFV.myc‐mgRΔIC. A 1‐kb EcoR I fragment was first subcloned into pBluescript SK (Stratagene, La Jolla, CA) and a BamH I/Hind III fragment subsequently cloned into pRSGFP‐C1 (BD Biosciences Clontech, Palo Alto, CA). A BamH I/Bgl II fragment was finally subcloned into the gfa2‐lacZ plasmid (Brenner et al., 1994), containing the gfa2 promoter (Fig. 1). The BamH I/Bgl II‐flanked IFN‐γR1ΔIC fragment places the transgene coding region between the gfa2 promoter and a region comprising bp +95 to +625 from the mouse protamine‐1 (mp1) structural gene, which provides an intron and 3′‐untranslated and poly‐A sequences (Fig. 1). An approximately 3.7‐kb Bgl II fragment containing the promoter, transgene, and 3′ regulatory regions was purified from plasmid pGfa2/IFN‐γR1ΔIC, injected into the pronuclei of B6 × CBA F1 fertilized oocytes, which were transferred into pseudopregnant B6 × CBA F1 mice.
Figure 1.
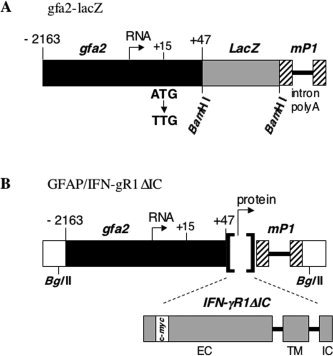
Schematic of the GFAP/IFN‐γR1ΔIC transgene construct. A: Diagram of the gfa2‐lacZ construct containing the gfa2 designated human GFAP promoter, spanning bp –2,163 to +47 relative to the transcriptional start site and showing the initiating ATG at +15 converted to TTG. The construct also contains mouse protamine‐1 gene sequences (mp1, hatched boxes) comprising an intron and the poly‐A signal sequence. In the GFAP/IFN‐γR1ΔIC construct (B), LacZ was replaced by the IFN‐γR1ΔIC sequence (gray boxes; EC, extracellular domain; TM, transmembrane domain; IC, truncated intracellular domain; c‐myc, human c‐myc tag) encoding the cytoplasmically truncated IFN‐γR1 chain. Open boxes represent vector sequences and the black bar the GFAP promoter.
Founder transgenic mice were identified by PCR analysis of genomic DNA from tail biopsies prepared by digestion with proteinase K (250 μg/ml) in 50 mM of Tris buffer pH 8.0, 100 mM EDTA, 100 mM NaCl, and 1% sodium dodecyl sulfate (SDS) at 55°C overnight, followed by two phenol: chloroform (1:1) extractions and a final extraction with chloroform alone. DNA was precipitated with isopropanol and resuspended in 10 mM Tris buffer, pH 8.0. PCR amplification was performed with the primer pairs 5′‐GCA‐GAG‐CCA‐GAG‐CAG‐GAT‐GGA‐GAG‐GAG‐3′ for GFAP and 5′‐GGA‐ATC‐AGT‐CCA‐GGA‐ACC‐CG‐3′ for IFN‐γR1ΔIC. Reaction conditions were as follows: initial denaturation at 95°C for 3 min, 30 cycles of denaturation at 94°C for 45 sec, annealing at 55°C for 1 min 30 sec, and extension at 72°C for 45 sec, followed by a final extension at 72°C for 4 min. Amplification products with an expected length of approximately 400 bp were analyzed by electrophoresis on 1.0% agarose gels. Founders (2 and 3) were back‐crossed to C57BL/6 and BALB/cBy mice (Jackson Laboratories, Bar Harbor, ME), and transmission was monitored via Southern blot by hybridization of genomic DNA with an EcoR I‐derived 1‐kb fragment of pSFFV.myc‐mgRΔIC. At the third back‐cross, transgene‐positive mice were screened for MHC class I expression by flow cytometric analysis of peripheral blood cells. Back‐crossing was continued with transgene and MHC double‐positive mice.
Tissue‐specific transgene mRNA expression was analyzed by using RNA extracted from organs of adult (6–8 week old) transgenic and syngeneic control C57BL/6 and BALB/c mice, obtained from the National Cancer Institute (Frederick, MD), by using Trizol reagent (Gibco BRL, Rockville, MD). RNA was transcribed with oligo(dT) and AMV reverse transcriptase (Promega, Madison, WI). Transgene mRNA was detected with a primer within the human c‐myc sequence (5′‐AGC‐TCA‐TTT‐CTG‐AAG‐AGG‐ACT‐TG‐3′) and a primer within the IFN‐γR1 extracellular domain (5′‐GGA‐ACC‐CGA‐ATA‐CAC‐CTT‐TAC‐C‐3′). HPRT primers 5′‐TTG‐CTC‐GAG‐ATG‐TCA‐TGA‐AGG‐A‐3′ and 5′‐AGC‐AGG‐TCA‐GCA‐AAG‐AAC‐TTA‐TAG‐C‐3′ were used as controls for expression of the housekeeping gene. PCR amplification was performed in 1× PCR buffer containing 0.25 μg primers, 200 μM dNTPs, and 2.5 U HotStar Taq DNA polymerase (Qiagen, Valencia, CA) under the following reaction conditions: 30 cycles of 1 min denaturation at 94°C, 1 min annealing at 60°C, and 1 min extension at 72°C. PCR products were analyzed by electrophoresis on 1.2% agarose gels. All data presented are derived from mice obtained from founder 3 at the sixth or seventh back‐cross to C57BL/6 mice. Mice were housed in an accredited animal facility at the University of Southern California.
Immunohistochemistry
After anesthesia, adult 6–8‐week‐old naive mice were perfused with cold 4% paraformaldehyde (PFA) in phosphate‐buffered saline (PBS), pH 7.2. Brains and spinal cords were removed, postfixed with 4% PFA at 4°C for 24 hr, and then equilibrated with 30% sucrose in PBS (pH 7.4) overnight at 4°C. Frozen (10 μm) sections were incubated with 2% H2O2 in PBS for 20 min, and nonspecific binding was inhibited by incubation in 3% goat serum. Sections were incubated overnight at 4°C with mouse anti‐GFAP cocktail of monoclonal antibody (mAb; BD PharMingen, San Diego CA), washed with PBS, and incubated with biotinylated anti‐mouse IgG secondary antibody (Ab), and antigen–antibody complexes were visualized by using avidin‐biotin peroxidase with diaminobenzidine as chromogen, with OsO4 enhancement (Vectastain ABC kit; Vector Laboratories, Burlingame, CA).
For confocal microscopy, 6‐μm sections were dried and postfixed in ice‐cold methanol/acetone (1:1). All further incubations were performed at room temperature (RT). Sections were permeabilized with 0.2% Triton X‐100, and nonspecific binding was inhibited by incubation in PBS (pH 7.4) containing 1% bovine serum albumin, 1% cold water fish skin gelatin (Sigma‐Aldrich, St. Louis, MO), 1 M glycine, and 3% goat serum. Coexpression of IFNγR1 and GFAP was detected with hamster anti‐IFNγR1 (BD PharMingen) and Texas red‐conjugated anti‐hamster IgG (Vector Laboratories), followed by a mouse anti‐GFAP cocktail (BD PharMingen) and fluorescein isothiocyanate (FITC)‐labeled anti‐mouse IgG (Vector Laboratories). Coexpression of GFAP and c‐myc was detected with the mouse anti‐GFAP cocktail (BD PharMingen) and Texas red‐labeled anti‐mouse IgG (Vector Laboratories), followed by rabbit anti‐c‐myc (clone A‐14; Santa Cruz Biotechnology, Santa Cruz, CA) and FITC‐conjugated anti‐rabbit IgG (Vector Laboratories). Sections were mounted with aqueous mounting medium (Vector Laboratories), and immunofluorescence was analyzed on a laser scanning confocal microscope (LSM 510; Zeiss, Jena, Germany) with a ×40 oil immersion objective.
For analysis of viral antigen, brains and spinal cords were removed, snap frozen, and embedded in O.C.T. compound (American Master Tech Scientific Inc., Lodi, CA). Colocalization of viral antigen and GFAP was detected by immunoperoxidase staining (Vector Laboratories). The distribution of viral antigen was determined by using mAb J.3.3 specific for the C terminus of the viral nucleocapsid protein and horse anti‐mouse secondary Ab (Vector Laboratories). Astrocytes were detected with mouse anti‐GFAP cocktail mAb (BD PharMingen) and anti‐mouse IgG (Vector Laboratories).
For analysis of CNS inflammation, brains and spinal cords were removed, fixed with Clark's solution (75% ethanol and 25% glacial acetic acid), and embedded in paraffin. Sections (6 μm) were stained with hematoxylin and eosin as described elsewhere (Lin et al., 1997; Parra et al., 1999). All sections were scored in a blinded manner. Representative fields were identified based on the average score of all sections in each experimental group.
Mixed Glial Cell Cultures
Brains were isolated from newborn GFAPγR1Δ transgenic and syngeneic control C57BL/6 mice. Meninges and superficial blood vessels were removed prior to digestion of frontal lobes with 0.25% trypsin/EDTA for 20 min at 37°C. After digestion and dissociation by trituration, cells from three to five mice were suspended in Dulbecco's modified minimal essential medium (Irvine Scientific, Santa Ana, CA) supplemented with 10% fetal calf serum (FCS) and penicillin (1,000 U/ml)/streptomycin (1,000 μg/ml) and added to T75 flasks. Medium was replaced after 12–16 hr incubation and at 3‐day intervals. Cells were subcultured once after 7–10 days and used at confluence. To examine IFN‐γ‐mediated signaling, mixed glial cultures were incubated in the presence or absence of recombinant murine IFN‐γ (BD PharMingen). After 48 hr, cells were removed by trypsinization (0.025% trypsin/EDTA) for flow cytometric analysis.
Virus Infection
Age‐matched GFAPγR1Δ tg and syngeneic 6–8‐week‐old C57BL/6 mice were infected with the neutralizing mAb variant 2.2v‐1 of the JHMV (Fleming et al., 1986). Mice were injected in the left hemisphere with 250 plaque forming units diluted in endotoxin‐free Dulbecco's PBS in a 30‐μl volume or with an equal volume of PBS. Virus was propagated and CNS virus titers were determined by plaque assay on monolayers of DBT cells, a murine astrocytoma, as previously described (Fleming et al., 1986; Lin et al., 1997). Briefly, one‐half of the brains were homogenized in RPMI containing 25 mM HEPES, pH 7.2, with TenBroeck tissue homogenizers. After clarification by centrifugation at 500g for 7 min, supernatants were either assayed directly for infectious virus or stored at –70°C.
CNS‐Derived Cells
For analysis of CNS inflammatory cells during JHMV infection, brains from infected PBS‐perfused mice were homogenized in serum‐free PBS with ice‐cold TenBroeck tissue homogenizers as described elsewhere (Bergmann et al., 2004). Cells were collected by centrifugation (400g for 7 min at 4°C), resuspended in RPMI medium containing 25 mM HEPES (pH 7.2), and adjusted to 30% Percoll (Amersham Biosciences, Piscataway, NJ). A 1.0‐ml underlay of 70% Percoll was added prior to centrifugation at 800g for 20 min at 4°C. Cells from the 30/70% interface were collected and washed in RPMI medium supplemented with 1% FCS prior to analysis via flow cytometry.
Flow Cytometry
Nonspecific mAb binding to CNS‐derived cells and mixed glial culture cells was blocked by incubation with anti‐mouse CD16/CD32 mAb (0.5 μg per sample; clone 2.4G2; BD PharMingen) and a 10% mixture of goat, mouse, rat, and horse serum at 4°C for 20 min. Microglia and astrocytes in the mixed glial cultures and infiltrating inflammatory cells in the CNS‐derived cell preparations were separated based on differential CD45 expression (Sedgwick et al., 1991). Surface molecule expression of mixed glial cells was examined by using APC labeled anti‐CD45 (Ly‐5, clone 30‐F11) and PE‐labeled anti‐MHC class II (M5/114.15.2) mAbs (BD PharMingen). FITC‐, PE‐, PerCP‐, and APC‐conjugated mAb were used for four‐color analysis of CD45hi (Sedgwick et al., 1991) virus‐induced CNS inflammatory cells by using the following mAb: CD4 (L3T4, clone GK1.5), CD8a (Ly‐2, clone 53‐6.7), CD45 (Ly‐5, clone 30‐F11), NK1.1 (PK136; all BD PharMingen), and F4/80 (Serotec, Raleigh, NC). Populations identified were CD4+ T cells (CD45hiCD4+), CD8+ T cells (CD45hiCD8+), NK cells (CD45hiNK1.1+), and macrophages (CD45hiF4/80+). Cells were stained at 4°C for 30 min in PBS containing 0.1% bovine serum albumin, fixed in 2% PFA, and analyzed on a FACSCalibur flow cytometer with CellQuest Pro software (BD Biosciences, Mountain View, CA).
Immunization and Peripheral T Cell Response
OVA323–339 (ISQAVHAAHAEINEAGR) peptide (Microchemical Core Facility, Keck School of Medicine, USC, Los Angeles, CA) was emulsified at 3 mg/ml with an equal amount of incomplete Freund's adjuvant (IFA; Sigma‐Aldrich, St. Louis, MO) supplemented with 5 mg/ml Mycobacterium tuberculosis, strain H37Ra (Difco, Detroit, MI). Mice were immunized subcutaneously with 200 μl distributed over two sites on the back. Axillary and inguinal lymph nodes were removed on day 10 postimmunization. Single‐cell suspensions were incubated at 8 × 105 cells/well in RPMI medium supplemented with 10% FCS, 2 mM L‐glutamine, 25 μg/ml genatamicin, nonessential amino acids, sodium pyruvate, and 5 × 10–5 M 2‐mercaptoethanol for 72 hr at 37°C, and 1 μCi/well [3H]thymidine (ICN Radiochemicals, Irvine, CA) was added for the final 16–20 hr. [3H]thymidine incorporation was measured by liquid scintillation spectroscopy.
RESULTS
CNS Restricted Transgene mRNA Expression
Transgene mRNA expression was confined to the brain and spinal cord of adult mice with no transgene mRNA detected in the CNS of control mice. Consistent with the restricted expression of the hGFAP promoter (Brenner et al., 1994), no transgene mRNA was detected in spleen, liver, thymus, or muscle (Fig. 2). CNS and peripheral organs of control wt mice were negative for transgene mRNA (data not shown). Although both lines exhibited identical patterns of transgene mRNA expression, based on the increased level of CNS transgene mRNA detected in mice derived from founder 3 (approximately fourfold higher), further experiments focused on mice derived from the high‐mRNA‐expressing founder‐derived line. At the sixth back‐cross, heterozygous transgene‐positive mice were intercrossed to produce homozygous mice. No phenotype was apparent during generation of the heterozygotic or homozygotic tg mice, which developed and bred normally. To ensure that transgene expression had no effect on astrocyte development, morphology, or distribution, astrocytes in homozygous tg and wt mice were compared. No differences in amount, distribution, or morphology of astrocytes were detected in either brain (Fig. 3) or spinal cord (data not shown) in comparing tg and wt mice. These data suggest that expression of the GFAPγR1Δ transgene mRNA is restricted to the CNS and does not result in any clinically apparent phenotype or adversely affect astrocyte development.
Figure 2.
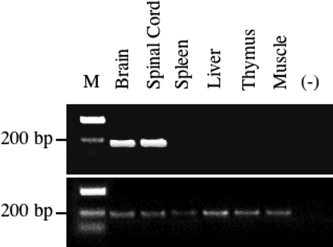
CNS restricted transgene mRNA expression. Transgene mRNA in organs derived from homozygous GFAPγR1Δ tg mice at the seventh back‐cross. Total mRNA was extracted, reverse transcribed, and amplified by PCR using primer pairs specific for either the transgene (top) or HPRT (bottom). M, molecular weight marker. –, Negative control containing buffer components only.
Figure 3.
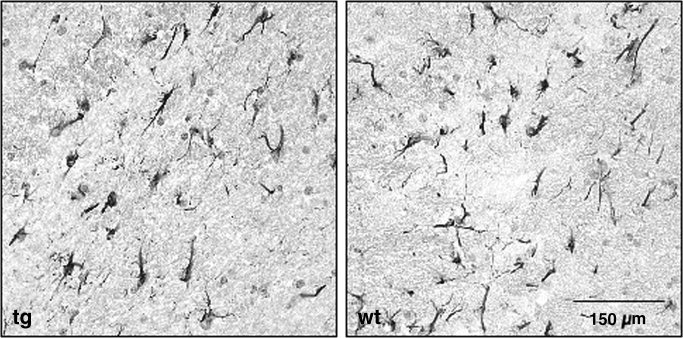
Astrocyte distribution in GFAPγR1Δ mice. GFAP+ astrocytes in brains from GFAPγR1Δ tg (left) and control C57BL/6 wt (right) mice. Sections of temporal lobe white matter counterstained with hematoxylin.
IFN‐γR1 Expression
High‐affinity IFN‐γ binding to its receptor requires expression of an IFN‐γR1/R2 dimer (Bach et al., 1997). Although somatic cells express IFN‐γR, only minimal IFN‐γR1 expression was detected on astrocytes in the CNS of wt mice by immunofluorescence (Fig. 4A), which is consistent with the previously described detection of IFN‐γR1 on CNS‐resident cells by flow cytometry (Gonzalez et al., 2005). In contrast, IFN‐γR1 expression was increased within the CNS of GFAPγR1Δ tg mice (Fig. 4A), which is consistent with the transgene mRNA expression (Fig. 2). Minimal expression of IFN‐γR2 was detected on both wt and tg astrocytes (data not shown). IFN‐γR1 and GFAP colocalization was also detected in the CNS of the GFAPγR1Δ tg mice, supporting astrocyte‐specific transgene expression. To confirm specific expression, the c‐myc tag encoded within the transgene was also examined. No c‐myc expression was detected in wt CNS tissues (Fig. 4B). By contrast, c‐myc and GFAP colocalized in the CNS of the tg mice (Fig. 4B), consistent with increased IFN‐γR1 expression by astrocytes. These data indicate that expression of the transgene driven by the GFAP promoter results in increased IFN‐γR1 expression by astrocytes.
Figure 4.
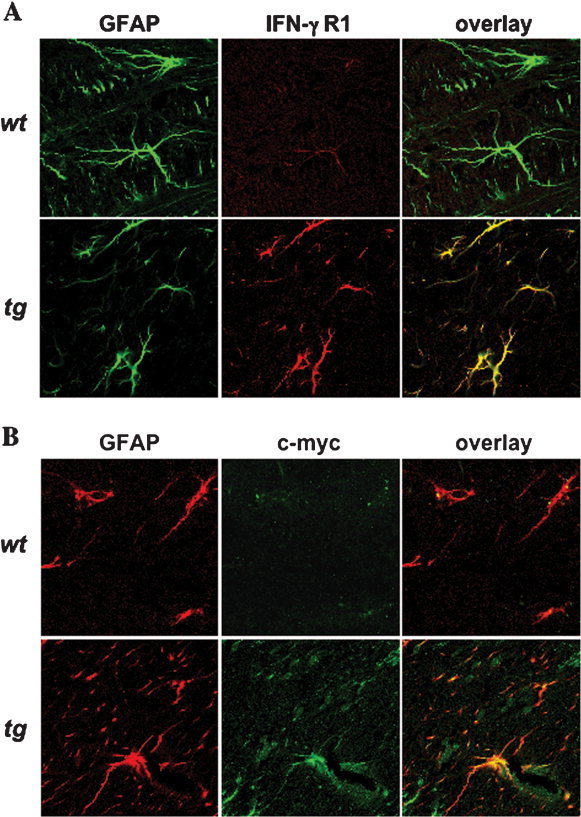
Expression of the tg IFN‐γR1 by astrocytes. IFN‐γR1 (A), c‐myc (B), and astrocyte‐specific expression analyzed by confocal microscopy. Control wt (top) and tg (bottom) mice were examined for IFN‐γR1 expression (A, red) and GFAP (green). Expression of the transgene was confirmed by expression of c‐myc (B, green) and GFAP (red). No evidence for ectopic expression was detected in other CNS cell types in tg or wt mice.
Impaired Astrocyte Response to IFN‐γ
Astrocytes do not constitutively express MHC class II; however, expression is induced by IFN‐γ (Fierz et al., 1985; Benveniste, 1998; Dong and Benveniste, 2001). To demonstrate functional transgene expression, astrocytes in mixed glial cultures derived from tg and wt mice were incubated in the presence or absence of IFN‐γ, and class II expression was analyzed by flow cytometry after 48 hr of induction. Cultures containing 30–40% CD45+ microglia were examined for IFN‐γ‐induced class II expression to provide an internal control population. No class II expression was detected on untreated CD45− astrocytes (Fig. 5A) or microglia (data not shown) from wt or tg mice. By contrast, IFN‐γ induced class II expression on both astrocytes and microglia derived from wt mice. MHC class II induction on microglia obtained from the CNS of wt and GFAPγR1Δ tg mice was similar with respect both to the percentage of microglia expressing class II and to the level of expression based on mean fluorescent intensity (data not shown). IFN‐γ induction of class II on microglia peaked within the first 36 hr of stimulation and then declined at 48 hr, resulting in expression on approximately 20% of microglial cells at 48 hr postinduction (data not shown). In contrast to induction of class II on >60% of astrocytes derived from wt mice, IFN‐γ induced class II expression on <20% of astrocytes derived from the GFAPγR1Δ tg mice (Fig. 5A). A dose‐response analysis showed increased class II expression by astrocytes derived from wt mice with increasing IFN‐γ (Fig. 5B). By contrast, even at the highest IFN‐γ concentration tested, astrocytes derived from GFAPγR1Δ tg mice exhibited markedly diminished class II up‐regulation (Fig. 5B). These data demonstrate that, whereas IFN‐γ signaling in microglia is not affected, IFN‐γ responsiveness is selectively impaired in astrocytes derived from the CNS of the GFAPγR1Δ tg mice.
Figure 5.
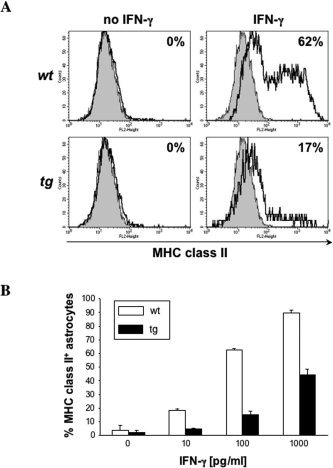
Impaired IFN‐γ signaling in astrocytes from the GFAPγR1Δ mice. IFN‐γ‐dependent MHC class II expression analyzed by flow cytometry (A). Mixed glial cell cultures derived from wt (top) and tg (bottom) mice incubated in the absence (left) or presence (right) of 100 pg/ml IFN‐γ for 48 hr. Histograms are gated on CD45− astrocytes. The gray area represents isotype control and the bold line MHC class II‐specific staining. Numbers indicate percentage of MHC class II+ cells within the astroglial cell population. B: Percentage of class II‐positive astrocytes after 48 hr incubation at various IFN‐γ concentrations. Data represent average results of two separate experiments. Error bars represent standard deviation.
Impaired Astrocyte IFN‐γ Signaling Does Not Alter Virus Replication
GFAPγR1Δ tg and wt mice were immunized with OVA to confirm that transgene expression did not alter T cell‐mediated immunity. No differences in T cell proliferation (Fig. 6A) or antigen‐induced IFN‐γ secretion (data not shown) were detected in comparing tg and wt mice. Infection with a neurotropic coronavirus was used to examine potential effects of diminished astrocyte IFN‐γ signaling. JHMV infects ependymal cells, astrocytes, microglia, macrophages, and oligodendroglia (Wang et al., 1992; Sun et al., 1995; Bergmann et al., 2004). Infectious JHMV peaks in the CNS at approximately day 6 postinfection (p.i.) and then declines to undetectable levels by approximately 14 days p.i. (Wang et al., 1992; Lin et al., 1997; Parra et al., 1999). Prominent astrocytic infection occurs rapidly, and replication in astrocytes is controlled via a perforin‐dependent mechanism mediated by CD8+ T cells (Lin et al., 1997; Bergmann et al., 2004). No differences in clinical disease were noted in comparing JHMV‐infected wt and GFAPγR1Δ tg mice to day 14 p.i. (data not shown). To determine whether reduced IFN‐γ signaling by astrocytes affected virus replication, the levels of infectious virus in the CNS of the tg and wt mice were compared at 7 days p.i. A slight, but not significant, increase in the infectious virus within the CNS of the GFAPγR1Δ was detected compared with wt mice (Fig. 6A). Similarly, no difference in bone marrow‐derived CD45hi CNS inflammatory cells was detected in comparing GFAPγR1Δ tg and wt mice (Fig. 6A), which is consistent with an unaltered immune response to a peripheral antigen (Fig. 6A). To ensure that the relative composition of CNS‐infiltrating cells was not altered, the percentage of NK cells, CD4+ T cells, CD8+ T cells, and macrophages within the inflammatory population was determined via flow cytometry. Consistent with the similar levels of both infectious virus and overall inflammation, similar NK cell, CD4+ T cell, CD8+ T cell, and macrophage infiltrates were detected within the CNS of the GFAPγR1Δ tg and wt mice (Fig. 6A). Finally, to ensure that transgene expression did not alter virus tropism, the distribution of both virus‐infected cells and inflammatory cells was examined. No difference in either the extent or the distribution of either the inflammatory response (Fig. 6B) or the virus‐infected astrocytes was detected in comparing GFAPγR1Δ tg and wt mice (Fig. 7). Similarly, no differences in inflammation or demyelination were detected in comparing the two groups at day 14 p.i. (data not shown).
Figure 6.
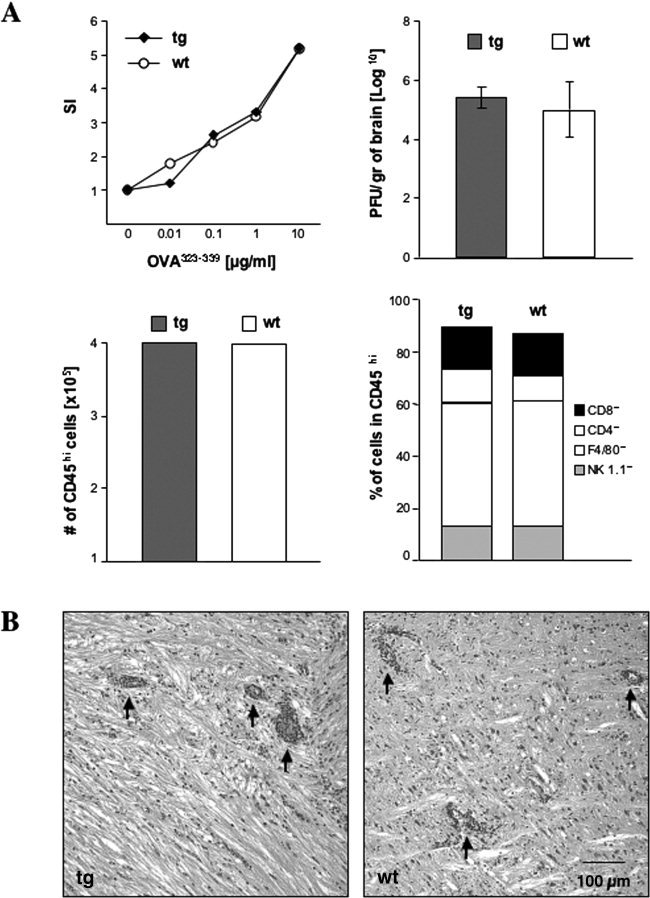
Immune responses in wt and tg mice. A: Antigen‐specific T cell proliferation from OVA323–339‐immunized GFAPγR1Δ tg (diamonds) and C57BL/6 wt (circles) mice. SI, stimulation index. CNS virus titers (average of three individual mice) at day 7 p.i. in GFAPγR1Δ tg (solid bars) and wt (open bars) mice. Error bars represent standard deviation. CNS infiltrating CD45hi cell numbers in GFAPγR1Δ tg (solid bars) and wt (open bars) mice at day 7 p.i. Composition of CNS‐infiltrating cells at day 7 p.i. in GFAPγR1Δ tg and wt mice. T cell proliferation and CNS‐infiltrating cells were analyzed in pooled cell populations from three individual mice. All data shown are representative of three separate experiments. B: CNS inflammation in GFAPγR1Δ tg (left) and wt (right) mice at day 7 p.i. Sections of midupper brainstem stained with hematoxylin and eosin. Arrows indicate perivascular inflammation.
Figure 7.
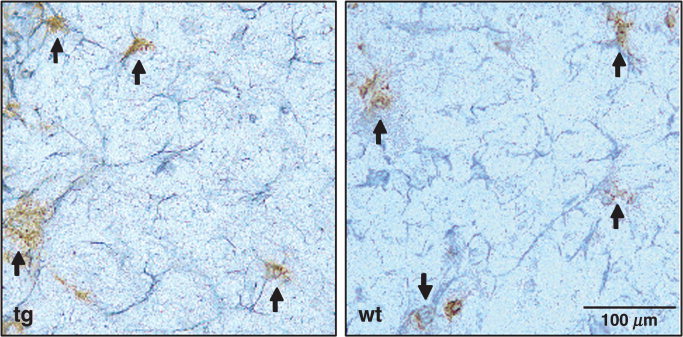
JHMV infection of astrocytes. JHMV antigen in posterior cerebral deep subcortical white matter of GFAPγR1Δ tg (left) and wt (right) mice at day 7 p.i. Immunoperoxidase double staining using astrocyte‐specific anti‐GFAP (gray) and JHMV‐specific J.3.3 (brown) with hematoxylin counterstain. Arrows indicate viral antigen‐positive astrocytes.
DISCUSSION
Mice deficient in the expression of specific immune modulators have facilitated an understanding of the contribution(s) of these modulators to the pathogenesis of inflammatory diseases, including CNS virus infection (Doherty, 1993; Lin et al., 1997; Parra et al., 1999). Analysis of IFN‐γ‐deficient mice confirmed the critical role of IFN‐γ in regulation of a variety of CNS autoimmune or viral infections, and also highlighted its complexity. For example, although EAE is mediated by IFN‐γ‐secreting Th1 CD4+ T cells, inhibition or absence of IFN‐γ increases disease severity (Billiau et al., 1988; Willenborg et al., 1996). Additionally, during CNS infection with both JHMV and Theiler's murine encephalomyelitis virus, viral clearance is IFN‐γ dependent (Parra et al., 1999; Rodriguez et al., 2003; Bergmann et al., 2004), yet IFN‐γ contributes to virus‐induced demyelination (Pewe and Perlman, 2002; Rodriguez et al., 1995). Despite its apparent dual roles as effector and inhibitor in these CNS pathologies, IFN‐γ is clearly critical in controlling virus replication in neurons and oligodendroglia (Parra et al., 1999; Griffin, 2003). Determining the role of IFN‐γ signaling in specific cell types in vivo is hampered both by the nearly ubiquitous expression of the IFN‐γR and by the number of IFN‐γ‐inducible genes and their downstream products (Benveniste, 1998; Chesler and Reiss, 2002; Schroder et al., 2004), which can be differently regulated in adjacent cells as well as the target cell of interest.
Astrocytes, critical to normal CNS function, may play a major role in regulating immune responses within the CNS. Astrocytes express MHC molecules, required for APC activity, upon stimulation with IFN‐γ (Fierz et al., 1985). In addition, IFN‐γ up‐regulates costimulatory molecules required for T cell activation (Nikcevich et al., 1997; Dong and Benveniste, 2001; De Keyser et al., 2003), supporting the concept that antigen presentation by astrocytes might have a pivotal role in CNS immunopathology. Activated astrocytes secrete a variety of proinflammatory cytokines and chemokines, including IL‐6, TNF‐α, and CXCL10 (Aschner, 1998; Benveniste, 1998; Lane et al., 1998; Dong and Benveniste, 2001), elevated during brain injury, CNS inflammation and degenerative diseases. However, astrocytes also secrete immune mediators with potential antiinflammatory effects, which inhibit microglial activation, phagocytosis, and cytokine secretion (Aloisi et al., 1997; DeWitt et al., 1998). Astrocytes also suppress antigen‐specific T cell proliferation via induction of the inhibitory CTLA‐4 molecule and regulatory T cells (Gimsa et al., 2004; Trajkovic et al., 2004). Thus, depending on context, these data suggest that IFN‐γ signaling in astrocytes may result in either pro‐ or antiinflammatory outcomes.
To isolate the responses of astrocytes, a dominant‐negative IFN‐γR1 was expressed to diminish IFN‐γ signaling events by using a promoter regulated similarly to the endogenous GFAP gene (Brenner et al., 1994). Expression of foreign proteins under control of the GFAP promoter can induce astrocyte impairment structurally and functionally and thus adversely impact the CNS. For example, inflammation, demyelination, and neurodegeneration have all been observed after astrocyte‐specific transgenic expression of inflammatory cytokines (Campbell, 1998). Increased secretion of TGF‐β by astrocytes resulted in hydrocephalus, whereas expression of insulin‐like growth factor binding protein‐6 reduced brain and body weight and decreased the number of astrocytes (Galbreath et al., 1995; Bienvenu et al., 2004). Expression of human GFAP caused severe encephalopathy associated with altered astrocyte morphology, including the presence of inclusion bodies (Messing et al., 1998). Thus, although a variety of abnormalities is associated with expression of some transgenes in astrocytes, the GFAPγR1Δ tg mice developed and bred normally and did not exhibit clinical symptoms or abnormalities in astrocyte frequency, distribution, or morphology. Transgene‐specific expression by astrocytes was confirmed by immunofluorescence. Although only minimal IFN‐γR1 expression was detected in wt mice, IFN‐γR1 expression by astrocytes was more readily detected in tg mice. However, expression of the accessory IFN‐γR2 remained low on both wt and tg astrocytes, with no simultaneous increase in IFN‐γR1ΔIC/IFN‐γR1 and IFN‐γR2 surface expression as previously noted in mice with targeted expression of the IFN‐γR1 transgene in oligodendroglia (Gonzalez et al., 2005). These data are consistent with the concept that IFN‐γR1 and IFN‐γR2 are not preassembled (Bach et al., 1996). Although no increase in IFN‐γR2 was detected, the diminished response to IFN‐γ provides evidence for a functional transgene with impaired IFN‐γ signaling. The response to IFN‐γ was substantially reduced in astrocytes derived from the tg mice; however, the response was not completely abolished. Although this suggests that astrocytic responses in these tg mice may be diminished but not completely eliminated, increased transgene expression from the GFAP promoter, for example, during inflammation (Eng and Ghirnikar, 1994), may increase differential responsiveness.
Mice with global defects in IFN‐γ signaling exhibit impaired immune responses (Dalton et al., 1993; Huang et al., 1993). By contrast, impaired IFN‐γ signaling exclusively in astrocytes did not affect peripheral immune responses or the activation or CNS recruitment of inflammatory cells, despite the notion that chemokines secreted by astrocytes influence recruitment of inflammatory cells into the CNS during JHMV infection (Lane et al., 1998). CXCL10 plays an essential role in the antiviral defense following MHV infection of the CNS (Liu et al., 2000). However, CXCL10 can also be induced in a variety of cells, including astrocytes, upon stimulation with type I interferons (Liu et al., 2000). Consistently with the data suggesting that virus replication is controlled in astrocytes via perforin‐mediated cytotoxicity and that MHC class I is regulated by both type I interferons and IFN‐γ (Lin et al., 1997; Parra et al., 1999; Bergmann et al., 2004), no difference in virus replication or cell types infected was detected in comparing JHMV pathogenesis in GFAPγR1Δ tg and wt mice. These data are consistent with induction of MHC class I, required for control of JHMV infection by virus‐specific CD8+ T cells, via type I interferon signaling (Atta et al., 1995), a pathway unimpaired in these tg mice. These data suggest that, in contrast to the IFN‐γ‐dependent MHC class II expression on astrocytes and microglia (Fierz et al., 1985; Bergmann et al., 2003), MHC class I expression, at least at the minimal levels sufficient for CD8+ T cell recognition, is independent of IFN‐γ signaling.
In summary, impaired IFN‐γ signaling in astrocytes does not affect peripheral immunity, viral tropism within the CNS, or recruitment of inflammatory cells into the CNS. Therefore, tg mice, expressing a dominant‐negative IFN‐γR1 specifically on astrocytes will provide a valuable novel tool for examining the impact of IFN‐γ signaling on regulatory events mediated by astrocytes, including IFN‐γ‐dependent interactions with other CNS resident cells, during CNS infection, autoimmunity, and neurodegenerative diseases.
Acknowledgements
The excellent technical assistance of Wenqiang Wei with immunohistochemistry is gratefully acknowledged. We also thank Ni Feng and Emmanuel V. Dimacali for technical assistance and Eva A. Borucka and Maria R. Ramirez for maintaining the transgenic animal colony.
REFERENCES
- Aloisi F, Penna G, Cerase J, Menendez Iglesias B, Adorini L. 1997. IL‐12 production by central nervous system microglia is inhibited by astrocytes. J Immunol 159: 1604–1612. [PubMed] [Google Scholar]
- Aschner M. 1998. Astrocytes as mediators of immune and inflammatory responses in the CNS. Neurotoxicology 19: 269–281. [PubMed] [Google Scholar]
- Atta MS, Irving WL, Powell RJ, Todd I. 1995. Enhanced expression of MHC class I molecules on cultured human thyroid follicular cells infected with reovirus through induction of type 1 interferons. Clin Exp Immunol 101: 121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach EA, Tanner JW, Marsters S, Ashkenazi A, Aguet M, Shaw AS, Schreiber RD. 1996. Ligand‐induced assembly and activation of the gamma interferon receptor in intact cells. Mol Cell Biol 16: 3214–3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach EA, Aguet M, Schreiber RD. 1997. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol 15: 563–591. [DOI] [PubMed] [Google Scholar]
- Benveniste EN. 1998. Cytokine actions in the central nervous system. Cytokine Growth Factor Rev 9: 259–275. [DOI] [PubMed] [Google Scholar]
- Bergmann CC, Parra B, Hinton DR, Chandran R, Morrison M, Stohlman SA. 2003. Perforin‐mediated effector function within the central nervous system requires IFN‐gamma‐mediated MHC up‐regulation. J Immunol 170: 3204–3213. [DOI] [PubMed] [Google Scholar]
- Bergmann CC, Parra B, Hinton DR, Ramakrishna C, Dowdell KC, Stohlman SA. 2004. Perforin and gamma interferon‐mediated control of coronavirus central nervous system infection by CD8 T cells in the absence of CD4 T cells. J Virol 78: 1739–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard F, Brenner M, Nakatani Y, Chao R, Purohit HJ, Freese E. 1991. Multiple interacting sites regulate astrocyte‐specific transcription of the human gene for glial fibrillary acidic protein. J Biol Chem 266: 18877–18883. [PubMed] [Google Scholar]
- Bienvenu G, Seurin D, Grellier P, Froment P, Baudrimont M, Monget P, Le Bouc Y, Babajko S. 2004. Insulin‐like growth factor binding protein‐6 transgenic mice: postnatal growth, brain development, and reproduction abnormalities. Endocrinology 145: 2412–2420. [DOI] [PubMed] [Google Scholar]
- Billiau A, Heremans H, Vandekerckhove F, Dijkmans R, Sobis H, Meulepas E, Carton H. 1988. Enhancement of experimental allergic encephalomyelitis in mice by antibodies against IFN‐gamma. J Immunol 140: 1506–1510. [PubMed] [Google Scholar]
- Brenner M, Kisseberth WC, Su Y, Besnard F, Messing A. 1994. GFAP promoter directs astrocyte‐specific expression in transgenic mice. J Neurosci 14: 1030–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IL. 1998. Structural and functional impact of the transgenic expression of cytokines in the CNS. Ann N Y Acad Sci 840: 83–96. [DOI] [PubMed] [Google Scholar]
- Chesler DA, Reiss CS. 2002. The role of IFN‐gamma in immune responses to viral infections of the central nervous system. Cytokine Growth Factor Rev 13: 441–454. [DOI] [PubMed] [Google Scholar]
- Dalton DK, Pitts‐Meek S, Keshav S, Figari IS, Bradley A, Stewart TA. 1993. Multiple defects of immune cell function in mice with disrupted interferon‐gamma genes. Science 259: 1739–1742. [DOI] [PubMed] [Google Scholar]
- De Keyser J, Zeinstra E, Frohman E. 2003. Are astrocytes central players in the pathophysiology of multiple sclerosis? Arch Neurol 60: 132–136. [DOI] [PubMed] [Google Scholar]
- Dettke M, Scheidt P, Prange H, Kirchner H. 1997. Correlation between interferon production and clinical disease activity in patients with multiple sclerosis. J Clin Immunol 17: 293–300. [DOI] [PubMed] [Google Scholar]
- DeWitt DA, Perry G, Cohen M, Doller C, Silver J. 1998. Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer's disease. Exp Neurol 149: 329–340. [DOI] [PubMed] [Google Scholar]
- Dighe AS, Campbell D, Hsieh CS, Clarke S, Greaves DR, Gordon S, Murphy KM, Schreiber RD. 1995. Tissue‐specific targeting of cytokine unresponsiveness in transgenic mice. Immunity 3: 657–666. [DOI] [PubMed] [Google Scholar]
- Doherty PC. 1993. Virus infections in mice with targeted gene disruptions. Curr Opin Immunol 5: 479–483. [DOI] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. 2001. Immune function of astrocytes. Glia 36: 180–190. [DOI] [PubMed] [Google Scholar]
- Eng LF, Ghirnikar RS. 1994. GFAP and astrogliosis. Brain Pathol 4: 229–237. [DOI] [PubMed] [Google Scholar]
- Fierz W, Endler B, Reske K, Wekerle H, Fontana A. 1985. Astrocytes as antigen‐presenting cells. I. Induction of Ia antigen expression on astrocytes by T cells via immune interferon and its effect on antigen presentation. J Immunol 134: 3785–3793. [PubMed] [Google Scholar]
- Fleming JO, Trousdale MD, el‐Zaatari FA, Stohlman SA, Weiner LP. 1986. Pathogenicity of antigenic variants of murine coronavirus JHM selected with monoclonal antibodies. J Virol 58: 869–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frucht DM, Fukao T, Bogdan C, Schindler H, O'Shea JJ, Koyasu S. 2001. IFN‐gamma production by antigen‐presenting cells: mechanisms emerge. Trends Immunol 22: 556–560. [DOI] [PubMed] [Google Scholar]
- Galbreath E, Kim SJ, Park K, Brenner M, Messing A. 1995. Overexpression of TGF‐beta 1 in the central nervous system of transgenic mice results in hydrocephalus. J Neuropathol Exp Neurol 54: 339–349. [DOI] [PubMed] [Google Scholar]
- Gimsa U, Oren A, Pandiyan P, Teichmann D, Bechmann I, Nitsch R, Brunner‐Weinzierl MC. 2004. Astrocytes protect the CNS: antigen‐specific T helper cell responses are inhibited by astrocyte‐induced upregulation of CTLA‐4 (CD152). J Mol Med 82: 364–372. [DOI] [PubMed] [Google Scholar]
- Gonzalez JM, Bergmann CC, Fuss B, Hinton DR, Kangas C, Macklin WB, Stohlman SA. 2005. Expression of a dominant negative IFN‐gammareceptor on mouse oligodendrocytes. Glia 51: 22–34. [DOI] [PubMed] [Google Scholar]
- Griffin DE. 2003. Immune responses to RNA‐virus infections of the CNS. Nat Rev Immunol 3: 493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart MN, Fabry Z. 1995. CNS antigen presentation. Trends Neurosci 18: 475–481. [DOI] [PubMed] [Google Scholar]
- Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, Vilcek J, Zinkernagel RM, Aguet M. 1993. Immune response in mice that lack the interferon‐gamma receptor. Science 259: 1742–1745. [DOI] [PubMed] [Google Scholar]
- Lane TE, Asensio VC, Yu N, Paoletti AD, Campbell IL, Buchmeier MJ. 1998. Dynamic regulation of alpha‐ and beta‐chemokine expression in the central nervous system during mouse hepatitis virus‐induced demyelinating disease. J Immunol 160: 970–978. [PubMed] [Google Scholar]
- Li Y, Fu L, Gonzales DM, Lavi E. 2004. Coronavirus neurovirulence correlates with the ability of the virus to induce proinflammatory cytokine signals from astrocytes and microglia. J Virol 78: 3398–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Stohlman SA, Hinton DR. 1997. Mouse hepatitis virus is cleared from the central nervous systems of mice lacking perforin‐mediated cytolysis. J Virol 71: 383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MT, Chen BP, Oertel P, Buchmeier MJ, Armstrong D, Hamilton TA, Lane TE. 2000. The T cell chemoattractant IFN‐inducible protein 10 is essential in host defense against viral‐induced neurologic disease. J Immunol 165: 2327–2330. [DOI] [PubMed] [Google Scholar]
- Marten NW, Stohlman SA, Bergmann CC. 2001. MHV infection of the CNS: mechanisms of immune‐mediated control. Viral Immunol 14: 1–18. [DOI] [PubMed] [Google Scholar]
- Messing A, Head MW, Galles K, Galbreath EJ, Goldman JE, Brenner M. 1998. Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am J Pathol 152: 391–398. [PMC free article] [PubMed] [Google Scholar]
- Minagar A, Shapshak P, Fujimura R, Ownby R, Heyes M, Eisdorfer C. 2002. The role of macrophage/microglia and astrocytes in the pathogenesis of three neurologic disorders: HIV‐associated dementia, Alzheimer disease, and multiple sclerosis. J Neurol Sci 202: 13–23. [DOI] [PubMed] [Google Scholar]
- Nikcevich KM, Gordon KB, Tan L, Hurst SD, Kroepfl JF, Gardinier M, Barrett TA, Miller SD. 1997. IFN‐gamma‐activated primary murine astrocytes express B7 costimulatory molecules and prime naive antigen‐specific T cells. J Immunol 158: 614–621. [PubMed] [Google Scholar]
- Olsson T. 1992. Cytokines in neuroinflammatory disease: role of myelin autoreactive T cell production of interferon‐gamma. J Neuroimmunol 40: 211–218. [DOI] [PubMed] [Google Scholar]
- Panitch HS, Hirsch RL, Haley AS, Johnson KP. 1987. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet 1: 893–895. [DOI] [PubMed] [Google Scholar]
- Parra B, Hinton DR, Marten NW, Bergmann CC, Lin MT, Yang CS, Stohlman SA. 1999. IFN‐gamma is required for viral clearance from central nervous system oligodendroglia. J Immunol 162: 1641–1647. [PubMed] [Google Scholar]
- Pewe L, Perlman S. 2002. Cutting edge: CD8 T cell‐mediated demyelination is IFN‐gamma dependent in mice infected with a neurotropic coronavirus. J Immunol 168: 1547–1551. [DOI] [PubMed] [Google Scholar]
- Popko B, Corbin JG, Baerwald KD, Dupree J, Garcia AM. 1997. The effects of interferon‐gamma on the central nervous system. Mol Neurobiol 14: 19–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M, Pavelko K, Coffman RL. 1995. Gamma interferon is critical for resistance to Theiler's virus‐induced demyelination. J Virol 69: 7286–7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M, Zoecklein LJ, Howe CL, Pavelko KD, Gamez JD, Nakane S, Papke LM. 2003. Gamma interferon is critical for neuronal viral clearance and protection in a susceptible mouse strain following early intracranial Theiler's murine encephalomyelitis virus infection. J Virol 77: 12252–12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin LL, Staddon JM. 1999. The cell biology of the blood–brain barrier. Annu Rev Neurosci 22: 11–28. [DOI] [PubMed] [Google Scholar]
- Schroder K, Hertzog PJ, Ravasi T, Hume DA. 2004. Interferon‐gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75: 163–189. [DOI] [PubMed] [Google Scholar]
- Sedgwick JD, Schwender S, Imrich H, Dorries R, Butcher GW, ter Meulen V. 1991. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci U S A 88: 7438–7442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll G, Jander S, Schroeter M. 1998. Inflammation and glial responses in ischemic brain lesions. Prog Neurobiol 56: 149–171. [DOI] [PubMed] [Google Scholar]
- Sun N, Grzybicki D, Castro RF, Murphy S, Perlman S. 1995. Activation of astrocytes in the spinal cord of mice chronically infected with a neurotropic coronavirus. Virology 213: 482–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teismann P, Schulz JB. 2004. Cellular pathology of Parkinson's disease: astrocytes, microglia and inflammation. Cell Tissue Res 318: 149–161. [DOI] [PubMed] [Google Scholar]
- Trajkovic V, Vuckovic O, Stosic‐Grujicic S, Miljkovic D, Popadic D, Markovic M, Bumbasirevic V, Backovic A, Cvetkovic I, Harhaji L, Ramic Z, Mostarica Stojkovic M. 2004. Astrocyte‐induced regulatory T cells mitigate CNS autoimmunity. Glia 47: 168–179. [DOI] [PubMed] [Google Scholar]
- Tyler KL, Gonzales‐Scarano F. 1996. Viral diseases of the central nervous system. In: Nathanson N, editor. Viral pathogenesis. Philadelphia: Lippincott‐Raven. p 837–853. [Google Scholar]
- Wang FI, Hinton DR, Gilmore W, Trousdale MD, Fleming JO. 1992. Sequential infection of glial cells by the murine hepatitis virus JHM strain (MHV‐4) leads to a characteristic distribution of demyelination. Lab Invest 66: 744–754. [PubMed] [Google Scholar]
- Weidinger G, Henning G, ter Meulen V, Niewiesk S. 2001. Inhibition of major histocompatibility complex class II‐dependent antigen presentation by neutralization of gamma interferon leads to breakdown of resistance against measles virus‐induced encephalitis. J Virol 75: 3059–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. 1996. IFN‐gamma plays a critical down‐regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein‐induced autoimmune encephalomyelitis. J Immunol 157: 3223–3227. [PubMed] [Google Scholar]
- Williamson JS, Stohlman SA. 1990. Effective clearance of mouse hepatitis virus from the central nervous system requires both CD4+ and CD8+ T cells. J Virol 64: 4589–4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EH, Hunter CA. 2004. The role of astrocytes in the immunopathogenesis of toxoplasmic encephalitis. Int J Parasitol 34: 543–548. [DOI] [PubMed] [Google Scholar]
- Zhang X, Hinton DR, Cua DJ, Stohlman SA, Lai MM. 1997. Expression of interferon‐gamma by a coronavirus defective‐interfering RNA vector and its effect on viral replication, spread, and pathogenicity. Virology 233: 327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]