

Abstract
The cross‐species transmission of viruses into new host populations, termed virus emergence, is a significant issue in public health, agriculture, wildlife management, and related fields. Virus emergence requires overlap between host populations, alterations in virus genetics to permit infection of new hosts, and adaptation to novel hosts such that between‐host transmission is sustainable, all of which are the purview of the fields of ecology and evolution. A firm understanding of the ecology of viruses and how they evolve is required for understanding how and why viruses emerge. In this paper, I address the evolutionary mechanisms of virus emergence and how they relate to virus ecology. I argue that, while virus acquisition of the ability to infect new hosts is not difficult, limited evolutionary trajectories to sustained virus between‐host transmission and the combined effects of mutational meltdown, bottlenecking, demographic stochasticity, density dependence, and genetic erosion in ecological sinks limit most emergence events to dead‐end spillover infections. Despite the relative rarity of pandemic emerging viruses, the potential of viruses to search evolutionary space and find means to spread epidemically and the consequences of pandemic viruses that do emerge necessitate sustained attention to virus research, surveillance, prophylaxis, and treatment.
Keywords: host range expansion, host shift, virus evolution, virus ecology, emerging infectious diseases
Introduction
Despite their limited genetic repertoires and typically specific host tropisms, viruses have a remarkable ability to infect new, often highly unrelated host types. The incidence and spread of novel viruses in host populations is termed virus emergence. In recent years, the incidence of newly identified viruses in human populations appears to be increasing, but whether this is a consequence of improved detection or more emergence events is a topic of considerable debate.1, 2, 3 Nevertheless, there are some general patterns for virus emergence. Emergence events typically occur when novel host and reservoir populations experience significant changes in range, demographics, aggregation and dispersal behavior, contact rates, environmental and climatic conditions, or vector distributions. In humans, there appear to be several hotspots for emergence, typically regions of high human population activity and density. These emergence hotspots include the Eastern United States, Western Europe, Japan, and Southeastern Australia.3 Sources of emerging viruses tend to be phylogenetically closely related organisms. For example, zoonotic viruses of wildlife are overrepresented among the emerging human viruses.3 In addition, there is a tendency for emerging viruses to have RNA genomes.4 This pattern may be a consequence of the high evolvability of RNA viruses.5 Despite these generalities, efforts to predict potential emerging viruses have not progressed as fast as hoped.
Mechanistically, virus emergence is a three‐step process. In the first step, a virus acquires the ability to infect new host cells. The second step consists of virus adaption to the novel host such that transmission between hosts is facilitated. Finally, to achieve full emergence, the virus gains the ability to spread epidemically through the host population. While the first two steps entail genetic changes in the virus, the third step may require changes on the part of the vector or host populations, such as through increased contact rates, range changes, or other ecological or environmental shifts.6 Virus emergence is, therefore, by definition, an eco‐evolutionary process.7 In this review, I address our current understanding of the evolutionary ecology of virus emergence, highlight new approaches to its study, and assess the future prospects for the management of risks associated with pandemic infectious viruses. Examples selected to illustrate basic concepts tend to come from medically and agriculturally important viruses, primarily because these viruses are the best studied (Table 1). However, it should be noted that virus emergence is a biologically universal phenomenon and occurs across all phyla. A major future challenge is to broaden our perspective of emerging viruses to include bacterial, archaeal, fungal, and invertebrate viruses.
Table 1.
Recent virus emergence events and their ecological circumstances
| Virus | Family | First reported outbreak | Location | Original host | New host | Transmission route | Factors contributing to emergence | References |
|---|---|---|---|---|---|---|---|---|
| Bluetongue virus | Reoviridae | 1998 | Europe | Ruminants | Ruminants, especially sheep | Culicoides midges | Warmer temperatures in Europe | 41, 42 |
| Chikungunya virus | Togaviridae | 2005 | India | Nonhuman primates | Humans | Aedes albopictus | Vector switch | 21, 25 |
| East African cassava mosaic virus (Uganda variant) | Geminiviridae | 1988 | Uganda | Cassava plant | Cassava plant | Whiteflies | Recombination between coinfecting strains | 43, 44 |
| Ebola virus | Filovirdae | 1976 | Congo, Sudan | Fruit bats | Humans | Contact with body fluids | Encroachment into wildlife habitat | 45, 46 |
| Human immunodeficiency virus | Retroviridae | Early 20th century | Cameroon | Chimpanzees, gorillas, sooty mangabeys | Humans | Contact with body fluids | Exposure to primate body fluid | 47, 48 |
| Influenza A virus H1N1 | Orthomyxoviridae | 2009 | Mexico | Swine | Humans | Airborne, fomites | Reassortment of virus segments | 49, 50 |
| Nipah virus | Paramyxoviridae | 1998 | Malaysia | Fruit bats | Swine, humans | Pig consumption of fruit pulp, droplet transmission to humans | Changing agricultural patterns | 51, 52 |
| Parvovirus | Parvoviridae | 1978 | Europe | Cats, raccoons | Canines | Oral contact with feces, fomites | Rapid evolution | 53, 54 |
| SARS coronavirus | Coronaviridae | 2003 | China | Bats | Humans | Handling of contaminated animals | Encroachment of wildlife habitat | 55, 56 |
| Sin Nombre virus | Bunyaviridae | 1993 | Southwestern United States | Mice | Humans | Contact with fomites, feces | El Niño–driven increases in rodent populations | 57, 58 |
| West Nile virus | Flaviviridae | 1999 | New York City | Birds | Humans | Culex mosquitoes | Broad host range, bird migration | 59, 60 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
The study of virus emergence necessarily touches on a broad range of interdependent ecological and evolutionary phenomena. Conceptually, I begin by addressing changes concomitant with emergence in viruses themselves, then expand to consider broader population, ecosystem, and global changes associated with virus emergence. Since gaining the ability to infect a novel host and the initial adaptation to that host is an inherently evolutionary process, I begin this discussion by focusing on virus genetics and evolution. I then link evolutionary changes to ecological processes in the context of host and virus population dynamics.
Virus evolution
Virus quasispecies may facilitate host range expansion
Viruses are among the smallest nucleic acid–based replicating entities and possess characteristics associated with exceptionally fast evolutionary change: small genomes, short generation times, high mutation rates, large population sizes, high levels of genetic diversity, and strong selection pressures.8, 9 Because viruses often lack nucleic acid–proofreading mechanisms, virus mutation rates are commonly on the order of 10–3–10–6 per nucleotide each time the genome is copied.10 Given the small sizes of viral genomes (typically ∼10–30 kb), viruses with high mutation rates can generate genetically different progeny each time the genome is replicated. This error‐prone replication can produce viral quasispecies (i.e., vast populations of closely related genotypes).5, 11 Although sequence spaces are unfathomably large (the sequence space of a 10‐kb virus genome is 410,000), the immense virus populations can contain many alternative variants; thus, viruses can rapidly sample sequence space and locate high fitness combinations of mutations.6
The quasispecies nature of viruses may facilitate virus emergence. The initial gain‐of‐function mutations permitting infection of novel hosts tend to be associated with host entry, namely the binding of the virus to a specific molecule on the host cell's surface. In many cases, changes in cell tropism can be accomplished through a few (or even one) nucleotide substitutions.12, 13, 14, 15, 16, 17 A typical example is provided by the alphaviruses, a group of 29 nonsegmented, positive‐sense, single‐stranded RNA (+ssRNA) viruses mainly vectored by mosquitoes.18, 19 Examples of alphaviruses include Eastern equine encephalitis virus, chikungunya virus (CHIKV), Ross River virus, and Sindbis virus. Despite the striking architectural similarities among the different strains, these viruses can infect an exceptionally broad range of hosts, including mammals, fish, amphibians, reptiles, birds, and insects.18, 19 Emergence events can result in hundreds of thousands of human infections.20, 21 The molecular basis of this broad host range stems from the malleability of the alphavirus host attachment protein, the E2 envelope glycoprotein, and its ability to bind the highly conserved laminin receptor.
Single amino acid substitutions in the E2 envelope glycoprotein projecting from the alphavirus capsid surface have been linked with host range expansion.17 Many of these substitutions were shown to occur in a specific domain of E2, approximately spanning amino acid residues 190–260 (part of domain B; see Fig. 1).22, 23, 24, 25, 26, 27, 28, 29, 30, 31 Mutations in this region are also associated with escape from monoclonal antibody–mediated neutralization.32, 33, 34 Smith et al. used cryoelectron microscopy to show that fragment antigen‐binding (Fab) fragments bind the outermost tip of the E2 glycoprotein of Sindbis and Ross River viruses, roughly corresponding to amino acids 190–260 of the E2 protein.35 A class III PDZ domain binding motif was identified at residues 213–216.36 PDZ domains are protein‐interaction modules that recognize short amino acid motifs at the C‐termini of target proteins.37 It is this region that presumably binds the laminin receptor, and changes in the amino acid sequence constituting this region likely permit binding to different laminin receptor variants. Furthermore, alphavirus host shifts may be facilitated by the indispensability and lack of structural flexibility of the highly conserved laminin receptor.39, 40 The E2 host receptor binding site has likely been shaped by natural selection for flexibility to bind different host laminin receptors, providing alphaviruses with the ability to shift hosts easily.
Figure 1.
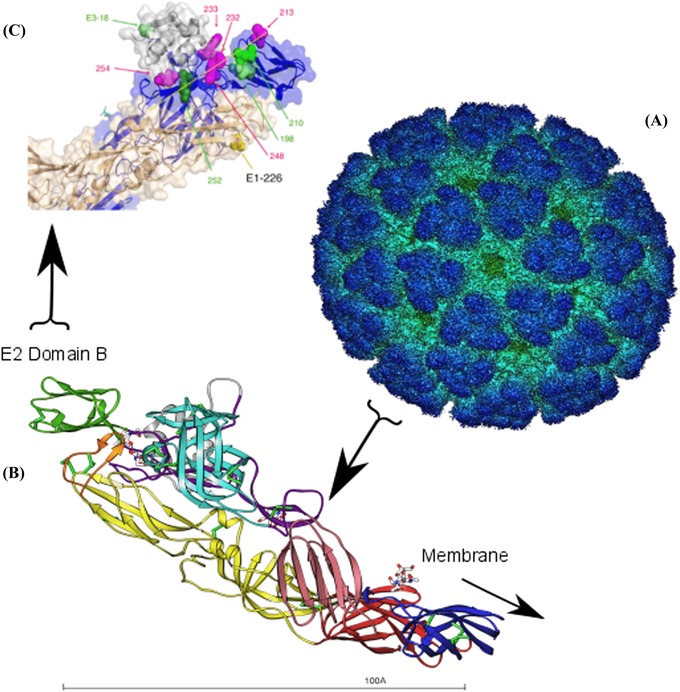
Alphavirus infection of host cells is mediated by two viral glycoproteins, E1 and E2. In this figure, the chikungunya virus cryoelectron structure is used to illustrate the location of E2 domain B, an immunodominant region of E2 where host range–expanding mutations are commonly found. (A) The cryoelectron structure of the entire icosahedral chikungunya virus particle is shown. The E1 and E2 glycoproteins dimerize and form 80 membrane‐embedded trimeric spikes across the virus capsid surface, shown in dark blue. E2 binds the host cell laminin receptor, and E1 induces the fusion of the viral and host cell membranes, allowing the nucleocapsid to enter the cell. Figure downloaded from Protein Data Bank Japan (PDBj). (B) A ribbon diagram shows the E1–E2 heterodimer. Each E1–E2 heterodimer is aligned so that E2 domain B (shown in green) is at the membrane distal end, and the opposite end (E1 domains I and III in red and blue, respectively) is closest to the virus lipid bilayer. Figure courtesy of Marie‐Christine Vaney and Félix Rey, Institute Pasteur; reprinted from Ref. 24. (C) A close‐up of E2 domain B (blue) shows the locations of substitutions (pink and green spheres) that increase chikungunya virus fitness in A. albopictus (except for a T213Q substitution, see Ref. 38). Figure modified from Ref. 38.
Another well‐studied example of changes in binding avidity concomitant to virus host shifts comes from influenza A viruses (IAV). In birds, IAV host attachment hemagglutinins prefer to bind oligosaccharides that terminate with a sialic acid linked to galactose by α2,3‐linkages. By contrast, human IAVs prefer sialic acids with α2,6‐linkages.61 As few as one or two mutations in hemagglutinin can significantly alter binding avidity to favor either human or avian sialic acids.62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73 Several studies have suggested that subtle changes in the electrostatic properties of the virus host receptor domain and increased opportunities for hydrogen bonding with complementary domains on host sialic acids permit host range expansion.61, 62, 63, 64, 65 Crucially, mutations permitting binding to human receptors do not preclude binding to avian receptors, and vice versa, although binding affinities are typically reduced.61, 68, 71, 72, 73 The ability to bind both human and avian receptors allows the possibility that a broad host range virus could be amplified in one host while periodically infecting the other in what is described in ecological theory as a source–sink scenario,74, 75 Source–sink evolution and its relevance to viral ecology will be discussed later in this paper.
Emergence by the numbers
The small number of mutations required to shift hosts has significant implications for virus emergence. Given small viral genomes and large population sizes, the number of potential host range mutants in a sample can be surprisingly large. A 10‐kb genome has a maximum of 3 × 104 potential independent single mutants, which is well below the average population size of viruses. Given that a considerable proportion of these mutants are not biologically feasible owing to lethal mutations,76 two or three rounds of replication of a single virus can generate every possible viable mutation one step away from the parental genotype.
A simple thought experiment illustrates the ease of generating host range mutants in high mutation rate viruses. A single sneeze can expel 20,000 droplets, with each droplet containing 1000 virus particles.77 Assuming that a single nucleotide substitution A→T in a 10‐kb genome provides the ability to infect a novel host, there could be 10,000 virus particles in that sneeze that possess mutations allowing them to infect a novel host. Moreover, while most virus mutations are highly deleterious, even lethal,76 host range–expanding mutations often incur minimal impacts on viral fitness on native hosts, although this is by no means universal.78, 79, 80, 81 Since the effectiveness of purifying selection is proportional to the strength of selection, one consequence is that slightly deleterious mutations may persist for long periods in quasispecies swarms in primary host populations.82, 83 Due to genetic drift and other stochastic processes, their frequencies in host populations could even increase. In our example, 10,000 host range mutants per sneeze could be a significant underestimate of the number of particles able to infect a novel host. It is clear that the first step of virus emergence is not the rate‐limiting step.
Virus emergence through recombination or reassortment
Virus host shifts can also occur via large‐scale genomic rearrangements among related viruses, that is, through the recombination of homologous genetic sequences, the acquisition of nonhomologous sequences (i.e., illegitimate recombination), or the reassortment of virus genome segments (Fig. 2).84, 85 Recombination is relatively common among the DNA viruses.86, 87, 88 While recombination frequencies vary considerably, some DNA viruses have extremely high rates of recombination. In one study of cauliflower mosaic virus, 53.8% of the genomes recovered after a single passage were recombinants.89 In the brome mosaic virus, practically every virus is a recombinant.90 Nonreplicative recombination uses enzyme‐mediated breakage‐repair mechanisms, similar to DNA‐based genomes.91 Thus, the function(s) of recombination in DNA viruses may be similar to the hypothesized functions of recombination in other clonal organisms. That is, recombination in DNA viruses may serve to repair DNA, remove mutations, break down linkage disequilibrium, and maintain genome integrity.92 Moreover, recombination may allow viruses to capture and use host‐encoded genes to manipulate hosts and increase infectivity (Fig. 2).93 For example, the Epstein–Barr herpesvirus likely acquired, via recombination, a homolog of human interleukin‐10, called BRCFI. BRCFI inhibits the synthesis of antiviral cytokines and stimulates the production of B cells in which the virus replicates.94
Figure 2.
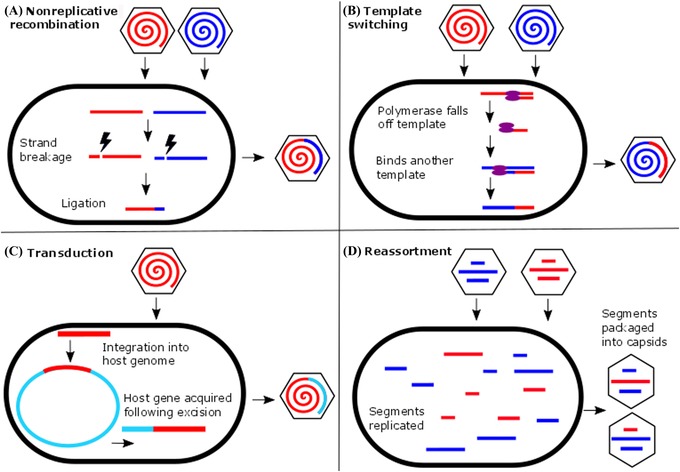
Major mechanisms of virus genetic recombination. (A) In nonreplicative recombination, nucleic acid strand breakage and repair permit the recombination of genetic material from different sources into the same viral genome. Recombination can occur between homologous or nonhomologous sequences and between coinfecting viruses or between virus and foreign nucleic acid strands. (B) In replicative recombination or template switching, a polymerase molecule changes template during the process of replicating a nucleic acid strand. If the templates are derived from different sources, then novel genetic material can be introduced into the virus genome. (C) During the process of virus integration and excision from a host genome, viruses can acquire genetic material from the host. These genes can increase infectivity or aid in host suppression. (D) Reassortment occurs following coinfection of a host cell by multiple segmented viruses. Replicated genome segments are packaged into procapsids irrespective of the parent of origin. In this manner, segments from two or more parents can be packaged into the same procapsid, giving rise to progeny that are genetically different from either parent.
In contrast to DNA virus recombination, RNA virus recombination mainly occurs through a different mechanism, termed copy‐choice recombination, although some RNA virus recombination can occur through nonreplicative mechanisms.95, 96, 97 In copy‐choice recombination, an RNA‐dependent RNA polymerase molecule switches from one template RNA molecule to another during the process of genome replication (Fig. 2). If the two templates contain divergent genetic information, these differences can be recombined into the same RNA molecule. Recombination frequency varies widely among the RNA viruses. This variation in recombination frequency may reflect the diverse nature of RNA genomes. RNA virus genomes may consist of +ssRNA, negative‐sense, single‐stranded RNA (–ssRNA), or double‐stranded RNA (dsRNA). Recombination is rare in –ssRNA viruses,95 but frequent in retroviruses, particularly in HIV, where recombination rates can exceed mutation rates.98 Recombination frequencies in the +ssRNA viruses are mixed. Some groups show high recombination rates (e.g., Picornaviridae), while, in others, recombination is rare or nonexistent (e.g., some Flaviviridae and Leviviridae).84
Some of these differences may derive from mechanistic constraints on recombination. For example, –ssRNA viruses are often bound in ribonucleoprotein complexes, which limits opportunities for hybridization between complementary sequences. Encapsidation of dsRNA viruses in nucleocapsids during genome replication prohibits recombination. However, in other cases, the rarity of recombination may have more to do virus and host life history. For example, recombination has not been observed among the tick‐borne flaviviruses. Ticks usually feed once during each life history stage (as larva, nymph, and adult). Therefore, the likelihood of an infected tick feeding on a host infected by a different virus strain is low. Since recombination requires coinfection of the same host by multiple virus strains, the rarity of tick coinfection may explain the failure to detect recombination among the tick‐borne flaviviruses.99
Virus recombination has been shown to facilitate virus emergence.88, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109 A recombination event may provide a host receptor gene with a new genetic background, providing the recombinant virus with the ability to gain access into a host cell without triggering host active immunity. For example, the spike glycoprotein (host receptor) gene of avian coronavirus infectious bronchitis virus, an upper‐respiratory virus of chickens, was replaced by a spike gene from an unknown source.101 The chimeric virus gained the ability to infect and cause disease in turkeys, but as an enteric, not respiratory, virus. Moreover, this new virus, now called turkey coronavirus, no longer possessed the ability to infect chickens.
A good case study of recombination‐facilitated emergence is provided by the geminiviruses, a large group of vector‐transmitted viruses infecting plants. Geminiviruses are unique in the virus world by virtue of encapsidating their ∼3‐kb single‐stranded DNA genome in two incomplete icosahedra to form a twinned particle.110 Most geminiviruses, with the exception of the bipartitite‐genomed Begomoviruses, are monopartite. These viruses have been implicated in a number of devastating plant diseases, including maize streak, cassava mosaic, cotton leaf curl, and tomato yellow leaf curl diseases.104, 105, 106, 107, 108, 109, 111, 112
Interspecies recombination rates among the geminiviruses are among the highest of all viruses.104, 106, 113 Some of these recombination events have given rise to destructive new variants. For example, in 1988, an extremely virulent form of mosaic virus appeared in Uganda, destroying crops of the staple food cassava and causing food shortages and famine. The new pathogen spread from its source at a rate of 20–25 km/year.114 The Uganda variant (EACMV‐UG2), as it was called, was shown to be a recombinant of African cassava mosaic virus (ACMV) and East African cassava mosaic virus (EACMV), which despite their similar names are not closely related.105, 114 Specifically, the EACMV‐UG2 strain was generated when the EACMV strain acquired a fragment of the ACMV coat protein. Given the role of geminivirus coat proteins in intraplant movement and between‐plant vector transmission, it should not come as a surprise that the protein is linked to an emergence episode.115 Similar recombination‐mediated emergence events may have occurred in other geminiviruses, including EACMV emergence in Cameroon,116 tomato yellow leaf curl emergence in Spain and the Mediterranean basin,108, 117, 118 cotton leaf curl virus in Pakistan,107, 119 and sugarcane yellow leaf virus in Hawaii.120
The frequency of recombination among the geminiviruses may be due to its significant role in virus biology. Geminivirus genomes are replicated using recombination‐dependent mechanisms.121, 122 Recombination may also be a mechanism for DNA mutation repair.123 Despite this, recombination may play a role in the generation of virus genetic diversity and the facilitation of host shifts.104, 106, 112, 117, 124, 125, 126, 127 In fact, there is considerable evidence that recombination in the geminiviruses occurs at specific locations in the genome, termed hotspots.108, 113, 118, 128, 129, 130 Many of these hotspots tend to preserve gene integrity because the break points are found in intergenic regions.131 By contrast, gene regions, such as that encoding the coat protein cp, are recombination coldspots.132 One common recombination hotspot in geminiviruses is located at the coat protein/small intergenic region (cp/SIR) interface.129, 130, 133, 134 It is plausible that the genome architecture of geminiviruses has been selected to expedite the swapping of the coat protein module among different virus strains, thus facilitating host switching.
Reassortment only occurs in segmented or multipartite viruses. When two or more segmented viruses infect the same cell, their replicated segments can be packaged into procapsids regardless of the parent of origin, producing viruses with a mixture of segments acquired from both parents (Fig. 2).100, 135, 136 Alternatively, such as in the multipartite viruses, each genome segment is independently packaged into a separate virus particle, but all particles are required for successful host infection.137 Reassortment is common in some viruses, including the reoviruses (e.g., rotavirus),138 orthomyxoviruses (e.g., influenza virus),139, 140, 141 bunyaviruses (e.g., hantavirus),142, 143, 144 and bromoviruses (e.g., Cucumber mosaic virus).145, 146 It may be that reassortment fulfills a similar role to recombination by generating genetic diversity and facilitating host shifts.
Perhaps the most famous case of reassortment leading to the expansion of virus host range comes from the 2009 IAV H1N1 pandemic. This virus is believed to have originated from a triple‐reassortment event in 1998, where IAV segments from birds, swine, and humans were combined into one H3N2 IAV, which circulated in swine.147, 148, 149 This virus contained nucleoprotein (NP), matrix protein (M), and nonstructural protein (NS) segments from a swine influenza strain; hemagglutinin (H3), neuramidase (N2), and polymerase subunit (PB1) segments from the seasonal human influenza strain; and polymerase subunits (PB2, PA) from an avian influenza strain.147 Subsequently, this virus reassorted with and acquired hemagglutinin (H1) and neuramidase (N1) segments from classical swine influenza.147, 150, 151 In 2009, this H1N1 swine strain reassorted with an avian‐derived H1N1 swine strain from Europe, acquired the M and N1 segments, and subsequently shifted hosts and emerged in human populations (Fig. 3). By the time the pandemic subsided, the World Health Organization reported 18,631 laboratory‐confirmed deaths, but studies suggest the total death toll was 10‐fold higher.152
Figure 3.
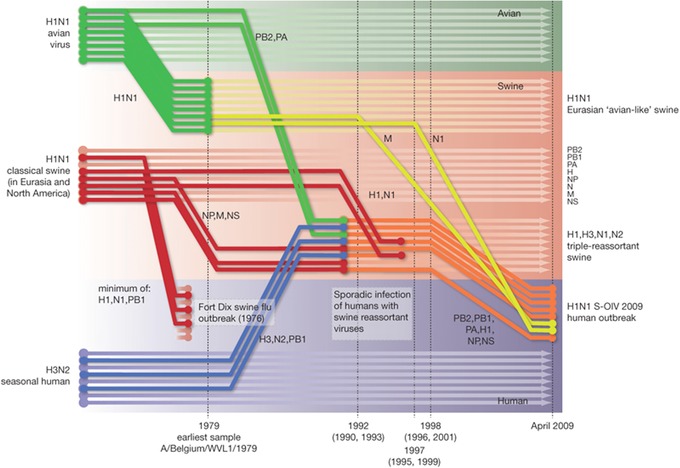
Reconstruction of the sequence of reassortment events leading up to the emergence of pandemic H1N1/09 virus. Shaded boxes represent host species: avian (green), swine (orange), and human (purple). Colored lines represent interspecies‐transmission pathways of influenza genome segments. The eight genomic segments are represented as parallel lines in descending order of size. Dates marked with dashed vertical lines on “elbows” indicate the mean time of divergence of the pandemic H1N1/09 segments from corresponding virus lineages. Reassortment events not involved with the emergence of human disease are omitted. Fort Dix refers to the last major outbreak of swine‐derived influenza H1N1 in humans. The first triple‐reassortant swine viruses were detected in 1998, but, to improve clarity, the origin of this lineage is placed earlier. Figure reprinted from Ref. 147.
Adaptive landscapes, pleiotropy, epistasis, and emergence
A virus can be said to occupy a position on an adaptive landscape corresponding to its host. An adaptive landscape is defined as a network of genotypes connected by mutational paths.153, 154 This network of genotypes is visualized as a three‐dimensional topology of peaks and valleys corresponding to the fitness values of the associated genotypes. The relevance of adaptive landscapes for virus emergence stems from the fact that most emergence events require a virus to significantly increase its fitness on the novel host such that between‐host transmission can be sustained. When a virus emerges in a new host, it transitions to a new adaptive landscape. Unlike the landscape corresponding to its previous host, it is exceedingly unlikely that the emerging virus is preadapted to the new host; thus, the virus would need to ascend a new fitness peak in order to fully emerge in the novel host.
An example of the challenges a virus faces when emerging on a novel host comes from the bacteriophage φ6. One or two mutations allow the bacteriophage φ6 to infect a novel host, Pseudomonas pseudoalcaligenes. However, the phage's absolute fitness on the new host is approximately an order of magnitude lower than its fitness on the original host (Fig. 4).74, 78 Moreover, each mutation incurred up to a 10% reduction in absolute fitness in the original host, Pseudomonas phaseolicola (Fig. 4).78 In order to increase their fitness on P. pseudoalcaligenes to a level comparable to the original host, these φ6 mutants need to navigate a new adaptive landscape of unpredictable topology before going extinct.
Figure 4.
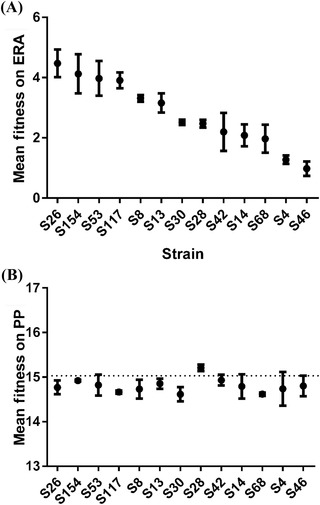
Mean absolute fitness of 13 independent host range–expanding mutations in original (A) and novel hosts (B). Each point is the mean of five replicate measurements of fitness. Bars are ±1 SE. Mutations incur a ∼10% fitness cost on the original host, Pseudomonas phaseolicola, indicative of antagonistic pleiotropy. Mean fitness values are much more variable on the novel host P. pseudoalcaligenes, but are approximately one order of magnitude lower than fitness on the original host. These data are suggestive of significant maladaptation between the bacteriophage and its novel host. Figure adapted from Ref. 78.
When a virus mutates to a genotype that allows infection of a novel host, the exact topology of the adaptive landscape on the new host will depend on the fitness values of genotypes adjacent to this new position. In order to ascend a fitness‐peak host from its current position on the landscape, the mutant virus needs to navigate an adaptive trajectory that passes through these intervening genotypes. Several adaptive landscape characteristics can affect a virus's ability to ascend a new fitness peak, including the genetic distance from the present location to the genotype representing the new fitness peak, any interactions between mutations along this adaptive trajectory, and the ruggedness of the adaptive landscape.
Genetic distance refers to the number of mutations separating any two genotypes on an adaptive landscape. There are at least two factors limiting the likelihood of traversing greater distances in adaptive trajectories. First, since the probabilities of acquiring specific mutations are additive, the greater the genetic distance between two genotypes, the lower the probability that the virus can navigate from one to the other. Assuming no recombination and equal probabilities of mutations across all loci, the probability (P) of acquiring multiple mutations in the same lineage is (u)n, where u is the per generation mutation rate and n is the number of mutations that need to be acquired. Therefore, if a virus with a mutation rate of 10–6 needs to acquire five mutations in order to fully emerge on a novel host, then the probability of acquiring all five mutations in the same lineage is 10–30. By comparison, the probability of a coin coming up heads in 99 consecutive coin flips is ∼3 × 10–30.
This example clearly illustrates that these five mutations would need to be acquired sequentially rather than simultaneously, which brings about a second problem. If mutations are acquired sequentially, many mutational trajectories may not be accessible. While the five mutations may have high fitness together, other intermediate combinations would not necessarily have higher fitness than the starting genotype. In fact, they could have lower fitness than the starting genotype. In this situation, a virus would need to cross a fitness valley or find an alternative path to reach the fitness peak. What this means is that mutations may have to be acquired in a specific order, which further reduces the probability of traversing the intervening genetic distance. Experiments exploring mutational trajectories have seldom been performed, but one example comes from the acquisition of β‐lactamase resistance in Escherichia coli. Five point mutations provide to E. coli a high degree of β‐lactamase resistance. In principle, there are five or 120 possible mutational trajectories. Weinreich et al. showed that 102 of 120 trajectories were inaccessible to natural selection, and many of the remaining were unlikely to occur.155 Thus, it was demonstrated that the acquisition of β‐lactamase resistance in E. coli could only occur via a limited number of mutational paths. The specific order in which mutations need to be acquired is a function of epistasis and pleiotropy.
Pleiotropy refers to situations where mutations in a single gene affect multiple traits. A mutation that increases fitness in one context may simultaneously reduce fitness in a different context, a phenomenon termed antagonistic pleiotropy. An example of antagonistic pleiotropy was introduced earlier in this paper when I described how bacteriophage φ6 mutations permitting infection of a novel host decreased fitness in the original host (see also Fig. 4).78 While the prevalence and magnitude of antagonistic pleiotropy in viruses have been largely unexplored, antagonistic pleiotropy may make some mutational trajectories nonviable because mutations along that trajectory may severely reduce fitness in some contexts. The compactness, multifunctionality, and lack of redundancy characteristic of virus genomes is expected to make antagonistic pleiotropy a frequent consequence of mutation.156 High frequencies of antagonistic pleiotropic effects among viral mutations will limit adaptive evolution in viruses infecting novel hosts.
Epistasis is the phenomenon where the fitness effects of a mutation depend on the genetic background in which it occurs. Sign epistasis refers to a specific class of epistatic interactions where the fitness effect of a mutation is beneficial in some backgrounds but deleterious in others.157 Reciprocal sign epistasis occurs when mutations are individually deleterious, but beneficial when they appear together.158 In the absence of sign epistasis, the adaptive landscape is smooth, fitness effects are additive, and mutations can be incorporated in any order. In the presence of sign epistasis, adaptive trajectories are constrained, and fewer paths to increased fitness are available.157 As reciprocal sign epistasis increases, the adaptive landscape becomes more rugged, and it becomes increasingly difficult to ascend the highest fitness peak without being trapped on local maxima.158 Experimental studies have shown that epistasis is pervasive among viruses.159, 160, 161, 162 The prevalence of epistasis, particularly reciprocal sign epistasis, restricts virus adaptive evolution by limiting accessible evolutionary trajectories and by making it more difficult for viruses to ascend global fitness maxima.
An example of the limiting effects of epistasis comes from CHIKV, which exploits the vector Aedes aegypti to facilitate its own transmission. A single nucleotide substitution (E1‐A226V) allows CHIKV to infect Aedes albopictus, but despite the high abundance of Ae. albopictus in Southeast Asia, CHIKV has never become established in this vector in this region.163 The failure of the Asian CHIKV lineage to exploit Ae. albopictus as a vector can be attributed to a negative epistatic interaction between the E1‐A226V mutation and a threonine residue at position E1‐98.164 Another CHIKV lineage, the East/Central/South African genotype (ECSA), has alanine at position E1‐98, which has no negative epistatic interaction with E1‐226V. In fact, the E1‐T98A substitution generated a nearly 100‐fold increase in infectivity of Ae. albopictus by E1‐226V. Thus, this ECSA lineage was able to acquire the A226V mutation, adapt to Ae. albopictus, and invade the Southeast Asia niche.164 Since Ae. albopictus has a broader range than Ae. aegypti in temperate zones,165 this lineage switch could lead to the expansion of CHIKV into new regions.
Together, epistasis, pleiotropy, and genetic distance may help explain why, despite the ease of acquiring host range–expanding mutations, pandemic emerging viruses are relatively rare. These features of adaptive landscapes may explain why some viruses, such as severe acute respiratory syndrome virus (SARS), fail to emerge in novel host populations. In 2002, an outbreak of SARS in Guangdong, China eventually infected thousands of people in Southeast Asia and North America. However, relatively low fitness of SARS (basic reproductive number, R 0 = 1.6), a high degree of host population heterogeneity in transmission potential (k), and the rapid mobilization of containment procedures led to the eradication of the outbreak by late 2003.166 The implication here is that SARS was unable to effectively navigate the adaptive trajectory required to increase its basic reproductive number and cause a wider outbreak. Rugged adaptive landscapes associated with host switches may make host adaptation too difficult to achieve in the light of the other ecological and environmental constraints on virus reproduction and transmission. The ability of organisms to navigate adaptive trajectories is referred to as evolvability.
Virus evolvability
Evolvability is defined as the capacity of a population to increase its fitness over time in response to changes in the environment.5, 167 Components of evolvability include high mutation rates, high levels of standing genetic variation, large population sizes, facility for genetic rearrangements, modular genomes, and short generation times, all of which are characteristic of viruses.5, 167 Since evolvability can be expressed as an organism's speed in responding to environmental change or, more formally, an organism's nonsynonymous substitution rate,168 viruses are exceptionally evolvable. Virus nucleotide substitution rates estimated through phylogenetic analyses typically range between 10–2 and 10–5 substitutions per site per year,169, 170 although there is considerable variation in substitution rates.171
Viruses and their genomes show many signs of having been selected for increased evolvability, and this may be a consequence of the ecological demands placed on viruses to change rapidly. High mutation rates and rapid adaptation were cited as the primary factors driving the emergence of parvovirus in canines.172 In HIV, the envelope (env) gene fixed adaptive mutations, on average, every 3.3 months or every 25 viral generations, which was the fastest adaptive rate ever recorded.173 This exceptionally fast rate of evolution is testament not only to HIV's evolvability, which facilitates evolutionary change, but also to the strength of selection imposed by the human immune system, which necessitates evolutionary change.
The capacity for rapid evolution makes viruses especially treacherous, as they can quickly escape host immunity, host resistance, and antiviral treatment and can rapidly adapt to increase their fitness on novel hosts. This feature of virus evolution is especially prominent in within‐host HIV evolution, where virus phylogenies show a ladder‐like structure over time. Here, successive waves of virus genotypes expand and contract in response to host‐imposed diversifying selection, particularly in the envelope (env) gene.174 These phylogenies dramatically contrast with between‐host HIV evolution, which show more branched distributions as different lineages survive and differentiate over time.174
High viral mutation rates, however, may be a double‐edged sword. While facilitating the acquisition of beneficial mutations, the high frequency of deleterious mutations may “sandbag” virus populations and slow adaptive evolution.175, 176 The effects of mutation load are more pronounced in small populations or when populations pass through bottlenecks.177, 178 By nature, viruses experience severe bottlenecks during between‐host transmission. Only a tiny fraction of a virus population's within‐host growth is expected to successfully encounter a new host. These bottlenecks can result in a loss of genetic diversity, mutation accumulation, and fitness losses. For example, in an evolutionary study of the potyvirus tobacco etch virus, strong bottlenecks imposed on serially passaged virus populations produced fitness losses of ∼5% per passage.179
Since the number of viruses transmitted between hosts is proportional to its within‐host growth, viruses that grow poorly within a host have a lower probability of being transmitted. This point is especially pertinent for emerging viruses, because they are expected to be poorly adapted to novel hosts.16 Because emerging viruses are often poorly adapted, within‐host replication is reduced, and, correspondingly, the number of viruses released from the host is also reduced. This has two consequences. First, the number of viruses in transmission vehicles (e.g., sneeze droplets, fomites, etc.) is decreased, reducing the chance that they will be picked up by another host. Second, if a new host is encountered, the number of particles entering the host is also decreased, reducing the probability that the virus can establish itself in that host without being eliminated by the immune system, inactivated, or washed out of the organism. In fact, these effects are likely compounded during each transmission event in a vicious cycle, and likely give rise to stuttering transmission chains that eventually go extinct.180, 181 In this sense, the population dynamics of preemerged viruses can be described by the source–sink paradigm of ecology, which will be discussed in greater detail later in this paper.
Emergence and the evolution of virulence
An enduring paradox in evolutionary biology is the harm done to hosts by the parasites that depend on them for survival. The impairment and/or death of a host would appear to be disadvantageous to the parasite that depends on it for growth and transmission. Early theory suggesting that viruses evolve to a benign coexistence with hosts has matured into theory emphasizing the benefits and costs of virulence (trade‐offs) for virus transmission. In brief, every virus has an optimal virulence that maximizes between‐host transmission. Increases or decreases in virulence will evolve insofar as they also increase virus transmission.182 With respect to virus emergence, the relevance of virulence evolution largely stems from the supposed mismatch between virus virulence in a novel host and its optimal virulence in that host. Since an emerging virus has, by definition, not adapted to its host, it is unlikely that its virulence in that host is optimal.
We can speculate that many virus spillover events do not progress to full‐blown emergence because of less‐than‐optimal virus virulence. For example, Ebola virus (EBOV) is remarkable for its extreme virulence in humans. Mortality rates due to EBOV are approximately 50%.183 Given that EBOV is spread through direct contact with infected individuals, EBOV's excessive virulence curtails the duration of infectivity and person‐to‐person contact rates, thus reducing the transmission potential of the virus. This reduction in transmission may be the reason why EBOV outbreaks are, so far, spatially limited to certain regions of Africa. However, EBOV outbreaks have been trending upward in both numbers of individuals infected and area affected. The most recent outbreak in West Africa resulted in 25,000 cases of infection and 10,000 deaths across three nations, which exceeds the totals for all previous EBOV outbreaks combined.184 Reports examining EBOV evolution during the latest outbreak caution that the virus might eventually evolve greater transmission and fully emerge in human populations.185, 186
Excessive virulence is not the only reason why a virus may fail to emerge. An insufficiently virulent virus may suffer the same fate because within‐host growth is not high enough to effect transmission to a new host. In addition, the weakly virulent virus may lose in direct competition for resources with a more virulent genotype. An example of the competitive replacement of a weakly virulent virus by a more virulent genotype comes from the dengue virus serotype 3 (DENV‐3). Before 1989, DENV‐3 was endemic to the Indian subcontinent, but caused mild or no disease in humans.187 After 1989, the virulence of DENV‐3 changed and caused outbreaks of dengue hemorrhagic fever, a severe, often fatal form of the disease. Messer et al. showed that the sudden onset of dengue hemorrhagic fever was caused by the competitive displacement of group A DENV‐3 strain by group B DENV‐3 strain in the Indian subcontinent.187 Subsequent work showed that the group B DENV3 strain infects and disseminates in Ae. aegypti more efficiently than the group A DENV3 strain, suggesting that the invasive strain is transmitted more effectively.188
Virus ecology
Viral population dynamics and source–sink evolution
In a source–sink scenario, a primary host population (the source) contributes virus “migrants” to a secondary host population (the sink), where the virus is poorly adapted.74, 189, 190 By definition, populations existing in sinks have a negative population growth rate; thus, they are expected to go extinct in the absence of migration.75 The influx of migrants can provide genetic diversity and contribute to density‐dependent growth in the sink population, allowing the acquisition and fixation of mutations increasing fitness in the secondary host.75 Eventually, fitness gains are such that the virus is able to escape the sink by achieving positive population growth, even in the absence of migration.189, 190
The difficulties encountered in the evolutionary transition required to turn an ecological sink into a source are probably the strongest constraining factors in virus emergence. In a sink, a virus needs to increase in fitness fast enough to counteract the relentless downward pressure imposed by negative population growth. The synergistic effects of genetic drift and erosion, positive density‐dependent growth, and mutational and migrational meltdown all act to limit the fixation of beneficial alleles.178, 190, 191, 192 Moreover, sink populations are more susceptible to chance fluctuations and demographic stochasticity,193, 194 leading to broken transmission chains and virus extinction. Ultimately, these factors may help explain the discrepancy between the relative ease with which host range–expanding mutations are acquired and the relative lack of pandemically spreading emerging infectious diseases infecting humans.
One means of evading these difficulties is the possibility that viruses can migrate from sink hosts to source hosts for further amplification and spread. To use IAVs as an example, imagine a mutant IAV in waterfowl containing the ability to bind both avian and human sialic acids. Despite being competitively inferior to other IAV genotypes, this mutant does not necessarily go extinct in waterfowl. In fact, it is common for multiple competing isogenic variants of similar relative fitnesses to circulate in an adapting asexual population.195, 196 This mutant IAV may epidemically spread through a waterfowl population with occasional spillover into human populations. The mutant virus may initially be a poor fit in humans but able to replicate in immunocompromised individuals, for example. While human‐to‐human transmission is not observed at this point, the high affinity for avian receptors may lead to human‐to‐waterfowl transmission of IAV genotypes that have increased infective/replicative ability in humans. In this manner, a broad host range IAV is amplified by repeated transmission between humans and waterfowl until sustained transmission between humans is achieved.
Though it is recognized that host populations must overlap in order for virus emergence to take place, virus host alternation has seldom been explored outside of a few laboratory and theoretical studies. However, these studies show that, contrary to the prevailing perception, natural selection can simultaneously improve virus fitness on multiple different hosts.197, 198, 199, 200 In the vernacular of evolutionary ecology, there seem to be few barriers to evolving to be a host generalist, and viruses are not necessarily constrained to specialize on certain hosts.
The broader issue of parasite generalism–specialism has a long and complicated history.201, 202 Commonly, it was believed that adaptation to a specific host necessitated a reduction in fitness in other hosts due to between‐host trade‐offs (i.e., antagonistic pleiotropy). Such between‐host trade‐offs can and do occur,203, 204 but the frequency of observing no trade‐offs has caused the question to be inverted to ask why broad host range viruses are not more common.205, 206 It may be that, while adaptation to multiple hosts is possible, ecological circumstances usually promote virus adaptation to a single host type. In other words, host specialization is caused by the absence of selection in favor of maintaining host generalism. If there are no alternative hosts for a virus in a particular habitat, then the virus will adapt to the currently available host (i.e., specialize). During the process of adaptation to a single host, mutations are fixed that are either beneficial or neutral to the present host but are deleterious to the alternate host. Once host specialization has begun, linkage between loci affecting host preference and performance on that host may make it increasingly difficult to reverse direction and reevolve broad host ranges. By contrast, if multiple hosts are available, then a virus may adapt to both hosts simultaneously in the absence of fitness‐reducing trade‐offs. It is likely that, in most emergence situations, antagonistic pleiotropy is not a factor, and alternating exposure to multiple hosts can lead to increased virus fitness on both hosts.
When host ecology changes, it may result in increased contact between primary and secondary host species, providing opportunities for multihost evolution. In humans, relevant ecological changes include altered local demographics, changing travel or immigration patterns, intensified agricultural practices, changing land‐use patterns and encroachment into wildlife habitat, climate change in human occupied areas, and range expansion of virus vectors or reservoirs.208 The reciprocal transmission of viruses following increased interspecies contact rates may select for virus variants with increased fitness in one or both species. An interesting and open question is how the relative sizes and dynamics of the two host populations affect emergence probabilities.
Environmental change and impacts on virus ecology
Host utilization, host range, virus life history, transmission routes, and kindred subjects are all aspects of virus ecology; that is, interactions between viruses and the biotic and abiotic environment. Environmental heterogeneity across space and time affects the incidence of disease in vectors and reservoir hosts and can lead to emergence events. An illustrative example comes from outbreaks of an arbovirus—bluetongue virus (BTV)—among ruminants in Europe. In its native range, BTV is vectored by Culicoides imicola midges.209 In recent years, a sub‐Saharan strain of the virus expanded northward from Africa, first to Southern and Eastern Europe and then into Northern and Western Europe, leading to widespread mortality among livestock.209 Changes in climate have been implicated as a major driver in the expansion of BTV's range.210 The most compelling evidence supporting this hypothesis is the observation of BTV and C. imicola in regions that have warmed the most since the 1990s, but not in regions that stayed cool during this period.210
The northward shift in BTV's range likely stemmed from the expansion of the African–Asian vector C. imicola northward,211 where it came into contact with indigenous European Culicoides species.209, 212, 213 BTV then switched vectors from C. imicola midges to C. obsoletus and C. pulicaris midges adapted to the cooler, wetter climes of Northern Europe.212, 213 The overlapping Culicoides populations may have allowed BTV to increase its infection competence in the latter vectors.213
Environmental variables of elevation, temperature, and rainfall had the biggest impact on BTV spread because they affected vector abundance and host availability.214, 215 Temperature, in particular, affected the magnitude of BTV's basic reproductive number (R 0), a measure of the transmission potential of a disease.216 R 0 was maximal when temperatures ranged from 20 to 25°C, but declined outside that range.214 The connection likely stems from the fact that Culicoides midges show increased activity, reproduction, and feeding behavior at higher temperatures.209, 217 In addition, milder winter temperatures promote midge overwinter survival and are crucial for the maintenance of BTV presence in any particular region.218
Hantavirus and El Niño
In the spring of 1993, an unknown pathogen infecting humans appeared in the Southwestern United States, resulting in death in more than 40% of cases.219 The causative agent was soon identified as a previously unknown hantavirus (Bunyaviridae), subsequently named Sin Nombre virus (SNV).220 SNV's reservoir host was subsequently identified as the deer mouse Peromyscus maniculatus, a common inhabitant of the deserts and dwellings of the southwest.221 Peromyscus population densities were shown to have increased threefold to 20‐fold between 1992 and 1993. The Peromyscus population surge was linked to the El Niño–Southern Oscillation event of 1992, which resulted in increased temperatures and precipitation in the region during the fall, winter, and spring of 1992–1993.219 The outcome was a trophic cascade. The increased precipitation recharged the thirsty soil and increased primary production. More plants led to increased rodent forage, such as seeds, berries, nuts, and insects. Improved forage augmented rodent survival and reproductive output. Greater rodent densities led to increased contact rates among rodents and pushed them to their range margins, such as human dwellings. The higher rodent densities likely facilitated increased SNV infection rates, triggering spillover infections in consanguineous human populations.219
Rodent populations in the area subsequently crashed, reaching a nadir during the La Niña event (a dry and cold period) in the spring of 1996.219 Human SNV infections diminished as well.219 In 1997–1998, another El Niño episode appeared, and rodent populations resurged.219 However, fewer human SNV infections were reported in 1998 than in 1993.219 Curiously, human cases doubled the following year in 1999.219 While rodent densities in 1999 were lower than in 1998, the density of SNV‐infected rodents in 1999 was an order of magnitude higher than at any other time during this study.219 This pattern was repeated in subsequent El Niño events.222 The precise causes of this time‐lagged infection prevalence are unclear, but it is hypothesized that prevalence of infection in Peromyscus populations is proportional to the population densities reached during the previous reproductive season.223
The greatest SNV risk to humans, therefore, occurs 2–3 years following the increased precipitation event.219 However, strictly speaking, SNV has not yet fully emerged according to the three‐step process established earlier in this paper. Sustained human‐to‐human transmission has not yet been observed.224 In fact, person‐to‐person transmission of hantaviruses has been documented only for the Andes virus.225 Given that SNV is mainly transmitted to humans by exposure to rodent urine and feces, human sanitary habits may reduce the opportunities for human‐to‐human transmission of SNV. While the specific mechanism of Andes virus human‐to‐human transmission has not been established, some evidence suggests that close exposure among sex partners and family members may be responsible.226 It stands to reason that hantaviruses may be optimized for indirect transmission but can gain the ability to be spread by bodily contact, as the Andes virus may have done. If a hantavirus, such as SNV, gains the ability to spread via direct human contact, the prospects for emergence are troublesome. The specific obstacles to hantavirus human‐to‐human transmission urgently need to be determined in order to assess the likelihood that hantaviruses can eventually fully emerge in human populations.
The greater lesson from these studies is that climatic disruption alters species ranges and demographics and can have unforeseen consequences for virus dynamics. It is plausible that other viruses that episodically appear in human populations, (e.g., EBOV) may be driven by as‐yet unknown environmental and ecological changes, but many virus outbreaks are not well documented, especially in underdeveloped countries. Particularly worrisome is the encroachment of tropical climates in typically temperate areas, along with the spread of vectors and reservoirs that are climate limited in their distributions. Animal and human populations in temperate regions are fertile ground for virus emergence and spread.
How can we defend against emerging viruses?
Virus emergence poses tremendous challenges to human society, and ecological and environmental trends suggest that the problem will get worse. Urbanization, globalization, encroachment of wildlife habitat, intensified agricultural practices, and climate‐driven range shifts will tend to increase the incidence and spread of infectious viruses. Exhortations in favor of more stringent epidemiological surveillance, improved predictive modeling, enhanced diagnostic tools, increased vaccination, and better public health infrastructure are common. While these approaches are valuable, their long‐term economic and logistic feasibility as primary means to interdict emerging viruses is questionable. There are simply too many potential emerging viruses, and containment measures are too porous.
More than 219 human pathogenic viruses have been described.227 If similar numbers exist for each mammalian species, we can liberally estimate roughly 320,000 viruses among the ∼5500 known mammals.228 This figure suggests that enormous numbers of viruses are at least superficially similar enough to infect our phylogenetic relatives, and thus are likely suspects for potential emergence events. Add the viruses of other animals, and potential pathogens may number in the millions. Simply identifying these viruses would cost billions.228 Actively tracking their frequencies and evolutionary trajectories is a daunting, perhaps impossible task.
If surveillance of the viruses of economically important animals and plants are included, it is clear from these rough numbers that wholesale virus surveillance to prevent new viruses from entering new host populations will never be feasible. While a more limited triage approach may be possible, the ability of viruses to escape interdiction and spread globally (e.g., 2009 IAV H1N1) does not inspire much confidence that this approach will generally work. Moreover, a surveillance approach does not account for laboratory biocontainment failures or even the possibility of deliberately engineered and released pathogenic viruses. The ease of DNA synthesis, genome sequencing and editing, and cell culture have frightening implications in the hands of state‐sponsored actors or terrorist organizations. Can we rule out that a dedicated research effort with sufficient funds and expertise could produce a highly virulent and transmissible synthetic virus?
One priority should be the development of broad‐spectrum antiviral drugs. While the “one bug–one drug” approach has many successes, it is not well suited to countering new emerging viruses, as it suffers from a “closing the barn door after horse has bolted” problem because of the time required to develop and implement a new treatment regime. Broad‐spectrum antivirals will allow rapid treatment of newly infected individuals before the pathogenic agent is identified and epidemiologically characterized. The highly conserved nature of many virus structures and functions may aid the discovery of broad‐spectrum antiviral agents. Broad‐spectrum drugs targeting shared virus mechanisms, such as membrane fusion,229 capsid assembly,230 receptor binding,231 metal nanoparticles,232 transcription, and translation have already been developed.233 From an evolutionary perspective, antiviral drug resistance will always be an issue, but they can be the knockout punch that interdicts an emerging virus before it goes pandemic.
Another promising approach is to find improved means of training immune systems to broadly recognize and defend against pathogenic viruses. Leblanc et al. developed the VaxCelerate platform to generate self‐assembling vaccines against specific pathogen targets in less than 120 days.234 Other efforts rely on computational and bioinformatic tools to identify highly conserved segments of antigenic genes or proteins and their binding epitopes (e.g., Conservatrix, EpiMatrix).235 Well‐developed platforms can be modified for speedy use against novel viruses.236
Since the majority of emerging viruses are arthropod‐borne, special attention should be paid to developing gene‐driven mechanisms to eliminate the most common vectors of emerging pathogens. The development of CRISPR‐CAS genome editing techniques has permitted the introduction of gene‐drive mechanisms into insect vectors that disrupt reproduction.237 For example, Hammond et al. identified, in the malaria vector Anopheles gambiae, three genes that, when disrupted, confer recessive female sterility.238 CRISPR‐Cas9 gene–drive constructs designed to target and edit each gene were introduced into A. gambiae. The drive was transmitted to offspring at rates ranging from 91.4% to 99.6%.238 The drive successfully induced infertility in homozygous mutant females, while males and heterozygous females showed normal fertility, which is necessary for the drive system to spread within the population (Fig. 5). While it is unlikely that gene drives can result in vector extinction because of the evolution of gene‐drive resistance, such gene drives could suppress vector populations below levels required for sustained virus transmission, breaking epidemics and preventing further virus dissemination.
Figure 5.
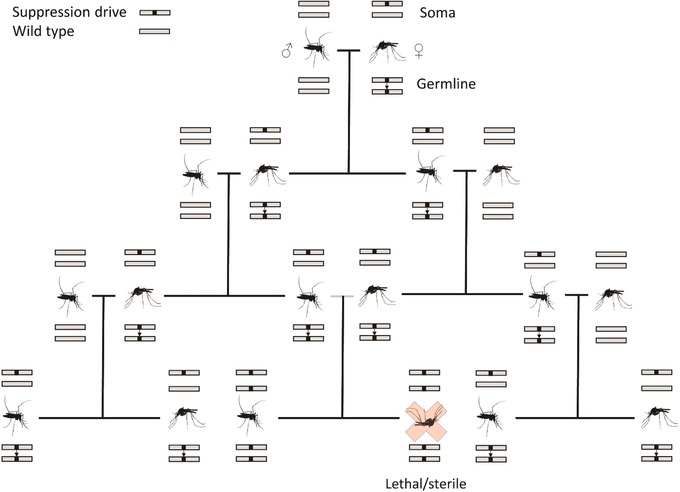
A suppression drive targeting a recessive gene required for viability or fertility will spread rapidly from heterozygotes with the drive, but would create an increasing number of sterile or unviable homozygotes, eventually resulting in a population crash. Figure modified from Ref. 237.
Finally, education should factor into any plan to combat emerging infectious viruses. Dispensing knowledge on the importance of avoiding risky behaviors (e.g., contact with animals), eliminating vectors, encouraging the use of antimicrobial surfaces (e.g., copper), preventing virus transmission (e.g., limiting travel, using face masks), controlling the spread of invasive animals and plants, sustaining wildlife habitat, and ameliorating climate change may go a long way to prevent the introduction of dangerous viruses into human populations. Ultimately, this knowledge must come from scientists themselves and is spread through their engagement with the public.
Conflicts of interest
The author declares no conflicts of interest.
References
- 1. Rosenberg, R. 2015. Detecting the emergence of novel, zoonotic viruses pathogenic to humans. Cell. Mol. Life Sci. 72: 1115–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rosenberg, R. , Johansson M.A., Powers A.M. & Miller B.R.. 2013. Search strategy has influenced the discovery rate of human viruses. Proc. Nat. Acad. Sci. USA 110: 13961–13964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jones, K.E. , Patel N.G., Levy M.A., et al 2008. Global trends in emerging infectious diseases. Nature 451: 990–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cleaveland, S. , Laurenson M.K., & Taylor L.H.. 2001. Diseases of humans and their domestic mammals: pathogen characteristics, host range and the risk of emergence. Phil. Trans. Roy. Soc. Lond. B ‐ Biol. Sci. 356: 991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lauring, A.S. , Frydman J. & Andino R.. 2013. The role of mutational robustness in RNA virus evolution. Nat. Rev. Microbiol. 11: 327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Elena, S.F. , Bedhomme S., Carrasco P., et al 2011. The evolutionary genetics of emerging plant RNA viruses. Mol. Plant Microbe Interact. 24: 287–93. [DOI] [PubMed] [Google Scholar]
- 7. Luo, S.S. & Koelle K.. 2013. Navigating the devious course of evolution: the importance of mechanistic models for identifying eco‐evolutionary dynamics in nature. Am. Nat. 181: S58–S75. [DOI] [PubMed] [Google Scholar]
- 8. Elena, S.F. & Sanjuan R.. 2007. Virus evolution: Insights from an experimental approach. Annu. Rev. Ecol. Evol. Syst. 38: 27–52. [Google Scholar]
- 9. Holmes, E.C. 2009. The evolutionary genetics of emerging viruses. Annu. Rev. Ecol. Evol. Syst. 40: 353–72. [Google Scholar]
- 10. Duffy, S. , Shackelton L.A., & Holmes E.C.. 2008. Rates of evolutionary change in viruses: patterns and determinants. Nat. Rev. Genet. 9: 267–76. [DOI] [PubMed] [Google Scholar]
- 11. Andino, R. & Domingo E.. 2015. Viral quasispecies. Virology 479: 46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Konstantoulas, C.J. , Lamp B., Rumenapf T.H. & Indik S.. 2015. Single amino acid substitution (G42E) in the receptor binding domain of mouse mammary tumour virus envelope protein facilitates infection of non‐murine cells in a transferrin receptor 1‐independent manner. Retrovirology 12: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baranowski E., Ruiz‐Jarabo C.M., Pariente N., et al 2003. Evolution of cell recognition by viruses: a source of biological novelty with medical implications. Adv. Virus Res. 62: 19–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boyd, M.T. , Simpson G.R., Cann A.J., et al 1993. A single amino acid substitution in the V1 loop of human immunodeficiency virus type 1 gp120 alters cellular tropism. J. Virol. 67: 3649–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Qi X., Gao H., Gao Y., et al 2009. Naturally occurring mutations at residues 253 and 284 in VP2 contribute to the cell tropism and virulence of very virulent infectious bursal disease virus. Antiviral Res. 84: 225–33. [DOI] [PubMed] [Google Scholar]
- 16. Vahlenkamp, T.W. , Verschoor E.J., Schuurman N.N., et al 1997. A single amino acid substitution in the transmembrane envelope glycoprotein of feline immunodeficiency virus alters cellular tropism. J. Virol. 71: 7132–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brault, A.C. , Powers A.M., Ortiz D., et al 2004. Venezuelan equine encephalitis emergence: enhanced vector infection from a single amino acid substitution in the envelope glycoprotein. Proc. Natl. Acad. Sci. USA. 101: 11344–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Strauss, J.H. & Strauss E.G.. 1994. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 58: 491–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weaver, S.C. , Winegar R., Manger I.D. & Forrester N.L.. 2012. Alphaviruses: population genetics and determinants of emergence. Antiviral Res. 94: 242–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weaver, S.C. , Ferro C., Barrera R., et al 2004. Venezuelan equine encephalitis. Annu. Rev. Entomol. 49: 141–74. [DOI] [PubMed] [Google Scholar]
- 21. Powers, A.M. & Logue C.H.. 2007. Changing patterns of chikungunya virus: re‐emergence of a zoonotic arbovirus. J. Gen. Virol. 88: 2363–77. [DOI] [PubMed] [Google Scholar]
- 22. Weger‐Lucarelli, J. , Aliota M.T., Wlodarchak N., et al 2015. Dissecting the role of E2 protein domains in alphavirus pathogenicity. J. Virol. 90: 2418–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Anishchenko, M. , Bowen R.A., Paessler S., et al 2006. Venezuelan encephalitis emergence mediated by a phylogenetically predicted viral mutation. Proc. Natl. Acad. Sci. USA. 103: 4994–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Voss, J.E. , Vaney M.C., Duquerroy S., et al 2010. Glycoprotein organization of chikungunya virus particles revealed by x‐ray crystallography. Nature. 468: 709–12. [DOI] [PubMed] [Google Scholar]
- 25. Tsetsarkin, K.A. , McGee C.E., Volk S.M., et al 2009. Epistatic roles of E2 glycoprotein mutations in adaption of Chikungunya virus to Aedes albopictus and Ae. aegypti mosquitoes. PLoS ONE. 4:e6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Myles, K.M. , Pierro D.J. & Olson K.E.. 2003. Deletions in the putative cell receptor‐binding domain of Sindbis virus strain MRE16 E2 glycoprotein reduce midgut infectivity in Aedes aegypti . J. Virol. 77: 8872–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kerr, P.J. , Weir R.C. & Dalgarno L.. 1993. Ross River virus variants selected during passage in chick embryo fibroblasts: serological, genetic, and biological changes. Virology. 193: 446–9. [DOI] [PubMed] [Google Scholar]
- 28. Heil, M.L. , Albee A., Strauss J.H. & Kuhn R.J.. 2001. An amino acid substitution in the coding region of the E2 glycoprotein adapts Ross River virus to utilize heparan sulfate as an attachment moiety. J. Virol. 75: 6303–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Karlsen, M. , Andersen L., Blindheim S.H., et al 2015. A naturally occurring substitution in the E2 protein of Salmonid alphavirus subtype 3 changes viral fitness. Virus Res. 196: 79–86. [DOI] [PubMed] [Google Scholar]
- 30. Arias‐Goeta, C. , Moutailler S., Mousson L., et al 2014. Chikungunya virus adaptation to a mosquito vector correlates with only few point mutations in the viral envelope glycoprotein. Infect. Genet. Evol. 24: 116–26. [DOI] [PubMed] [Google Scholar]
- 31. Mossel, E.C. , Ledermann J.P., Phillips A.T., et al2013. Molecular determinants of mouse neurovirulence and mosquito infection for Western equine encephalitis virus. PLoS ONE. 8:e60427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Strauss, E.G. , Stec D.S., Schmaljohn A.L. & Strauss J.H.. 1991. Identification of antigenically important domains in the glycoproteins of Sindbis virus by analysis of antibody escape variants. J. Virol. 65: 4654–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang, K.S. & Strauss J.H.. 1991. Use of a lambda gt11 expression library to localize a neutralizing antibody‐binding site in glycoprotein E2 of Sindbis virus. J. Virol. 65: 7037–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Porta, J. , Jose J., & Roehrig J.T.. 2014. Locking and blocking the viral landscape of an alphavirus with neutralizing antibodies. J. Virol. 88: 9616–9623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Smith, T.J. , Cheng R.H., Olson N.H., et al 1995. Putative receptor binding sites on alphaviruses as visualized by cryoelectron microscopy. Proc. Natl. Acad. Sci. USA. 92: 10648–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Asnet, M.J. , Paramasivan R., Tyagi B.K., et al 2013. Identification of structural motifs in the E2 glycoprotein of Chikungunya involved in virus‐host interaction. J. Biomol. Struct. Dyn. 31: 1077–85. [DOI] [PubMed] [Google Scholar]
- 37. Lee, H.‐J.& ; Zheng J.J.. 2010. PDZ domains and their binding partners: structure, specificity, and modification. Cell Comm. Sig. 8: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tsetsarkin, K.A. , Chen R., Yun R., et al 2014. Multi‐peaked adaptive landscape for chikungunya virus evolution predicts continued fitness optimization in Aedes albopictus mosquitoes. Nat. Commun. 5: 4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang, K.S. , Kuhn R.J., Strauss E.G., et al 1992. High‐affinity laminin receptor is a receptor for Sindbis virus in mammalian cells. J. Virol. 66: 4992–5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Beck, K. , Hunter I. & Engel J.. 1990. Structure and function of laminin: anatomy of a multidomain glycoprotein. FASEB J. 4: 148–160. [DOI] [PubMed] [Google Scholar]
- 41. Wilson, A.J. & Mellor P.S.. 2009. Bluetongue in Europe: past, present and future. Phil. Trans. R. Soc. Lond. B: Biol. Sci. 364: 2669–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Purse, B.V. , Mellor P.S., Rogers D.J., et al 2005. Climate change and the recent emergence of bluetongue in Europe. Nat. Rev. Microbiol. 3: 171–81. [DOI] [PubMed] [Google Scholar]
- 43. Zhou, X.P. , Liu Y.L., Calvert L., et al 1997. Evidence that DNA‐A of a geminivirus associated with severe cassava mosaic disease in Uganda has arisen by interspecific recombination. J. Gen. Virol. 78: 2101–11. [DOI] [PubMed] [Google Scholar]
- 44. Pita, J.S. , Fondong V.N., Sangare A., et al 2001. Recombination, pseudorecombination and synergism of geminiviruses are determinant keys to the epidemic of severe cassava mosaic disease in Uganda. J. Gen. Virol. 82: 655–65. [DOI] [PubMed] [Google Scholar]
- 45. Bowen, E.T. , Lloyd G., Harris W.J., et al 1977. Viral haemorrhagic fever in southern Sudan and northern Zaire. Preliminary studies on the aetiological agent. Lancet. 1: 571–3. [DOI] [PubMed] [Google Scholar]
- 46. Pattyn, S. , van der Groen G., Jacob W., et al 1977. Isolation of Marburg‐like virus from a case of haemorrhagic fever in Zaire. Lancet. 1: 573–4. [DOI] [PubMed] [Google Scholar]
- 47. Worobey, M. , Gemmel M., Teuwen D.E., et al 2008. Direct evidence of extensive diversity of HIV‐1 in Kinshasa by 1960. Nature. 455: 661–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sharp, P.M. & Hahn B.H.. 2011. Origins of HIV and the AIDS pandemic. Cold Spring Harbor Persp. Med. 1: a006841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smith, G.J.D. , Vijaykrishna D., Bahl J., et al 2009. Origins and evolutionary genomics of the 2009 swine‐origin H1N1 influenza A epidemic. Nature. 459: 1122–1125. [DOI] [PubMed] [Google Scholar]
- 50. Dawood, F.S. , Jain S., Finelli L., et al 2009. Emergence of a novel swine‐origin influenza A (H1N1) virus in humans novel swine. New Engl. J. Med. 360: 2605–15. [DOI] [PubMed] [Google Scholar]
- 51. Lo Presti, A. , Cella E., Giovanetti M., et al 2016. Origin and evolution of Nipah virus. J. Med. Virol. 88: 380–8. [DOI] [PubMed] [Google Scholar]
- 52. Pulliam, J.R.C. , Epstein J.H., Dushoff J., et al 2012. Agricultural intensification, priming for persistence and the emergence of Nipah virus: a lethal bat‐borne zoonosis. J. Roy. Soc. Interface. 9: 89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hoelzer, K. , Shackelton L.A., Parrish C.R. & Holmes E.C.. 2008. Phylogenetic analysis reveals the emergence, evolution and dispersal of carnivore parvoviruses. J. Gen. Virol. 89: 2280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hueffer, K. & Parrish C.R.. 2003. Parvovirus host range, cell tropism and evolution. Curr. Opin. Microbiol. 6: 392–8. [DOI] [PubMed] [Google Scholar]
- 55. Guan, Y. , Zheng B.J., He Y.Q., et al 2003. Isolation and characterization of viruses related to the SARS coronavirus from animals in Southern China. Science. 302: 276–8. [DOI] [PubMed] [Google Scholar]
- 56. Stadler, K. , Masignani V., Eickmann M., et al 2003. SARS–beginning to understand a new virus. Nat. Rev. Microbiol. 1: 209–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yates, T.L. , Mills J.N., Parmenter C.A., et al 2002. The ecology and evolutionary history of an emergent disease: Hantavirus pulmonary syndrome. Bioscience. 52: 989–98. [Google Scholar]
- 58. Calisher, C.H. , Sweeney W., Mills J.N. & Beaty B.J.. 1999. Natural history of Sin Nombre virus in western Colorado. Emerg. Infect. Dis. 5: 126–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hayes, E.B. & Gubler D.J.. 2006. West Nile virus: epidemiology and clinical features of an emerging epidemic in the United States. Annu. Rev. Med. 57: 181–194. [DOI] [PubMed] [Google Scholar]
- 60. Kramer, L.D. , Styer L.M. & Ebel G.D.. 2008. A global perspective on the epidemiology of West Nile virus. Annu. Rev. Entomol. 53: 61–81. [DOI] [PubMed] [Google Scholar]
- 61. de Graaf, M. & Fouchier R.A.. 2014. Role of receptor binding specificity in influenza A virus transmission and pathogenesis. EMBO J. 33: 823–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tharakaraman, K. , Raman R., Viswanathan K., et al 2013. Structural determinants for naturally evolving H5N1 hemagglutinin to switch its receptor specificity. Cell. 153: 1475–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lin, Y.P. , Xiong X., Wharton S.A., et al 2012. Evolution of the receptor binding properties of the influenza A(H3N2) hemagglutinin. Proc. Natl. Acad. Sci. USA. 109: 21474–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gamblin, S.J. , Haire L.F., Russell R.J., et al 2004. The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science. 303: 1838–42. [DOI] [PubMed] [Google Scholar]
- 65. Liu, J. , Stevens D.J., Haire L.F., et al 2009. Structures of receptor complexes formed by hemagglutinins from the Asian Influenza pandemic of 1957. Proc. Natl. Acad. Sci. USA. 106: 17175–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Glaser, L. , Stevens J., Zamarin D., et al 2005. A single amino acid substitution in 1918 influenza virus hemagglutinin changes receptor binding specificity. J. Virol. 79: 11533–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Matrosovich, M. , Tuzikov A., Bovin N., et al 2000. Early alterations of the receptor‐binding properties of H1, H2, and H3 avian influenza virus hemagglutinins after their introduction into mammals. J. Virol. 74: 8502–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Auewarakul, P. , Suptawiwat O., Kongchanagul A., et al 2007. An avian influenza H5N1 virus that binds to a human‐type receptor. J. Virol. 81: 9950–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gambaryan, A. , Tuzikov A., Pazynina G., et al 2006. Evolution of the receptor binding phenotype of influenza A (H5) viruses. Virology. 344: 432–8. [DOI] [PubMed] [Google Scholar]
- 70. Yamada, S. , Suzuki Y., Suzuki T., et al 2006. Haemagglutinin mutations responsible for the binding of H5N1 influenza A viruses to human‐type receptors. Nature. 444: 378–82. [DOI] [PubMed] [Google Scholar]
- 71. Zhang, W. , Shi Y., Qi J., et al 2013. Molecular basis of the receptor binding specificity switch of the hemagglutinins from both the 1918 and 2009 pandemic influenza a viruses by a D225G substitution. J. Virol. 87: 5949–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pappas, C. , Viswanathan K., Chandrasekaran A., et al 2010. Receptor specificity and transmission of H2N2 subtype viruses isolated from the pandemic of 1957. PLoS ONE. 5: e11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chutinimitkul, S. , Herfst S., Steel J., et al 2010. Virulence‐associated Ssubstitution D222G in the hemagglutinin of 2009 pandemic influenza A(H1N1) virus affects receptor binding. J. Virol. 84: 11802–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dennehy, J.J. , Friedenberg N.A., Holt R.D. & Turner P.E.. 2006. Viral ecology and the maintenance of novel host use. Am. Nat. 167: 429–39. [DOI] [PubMed] [Google Scholar]
- 75. Holt, R.D. 1996. Adaptive evolution in source‐sink environments: direct and indirect effects of density‐dependence on niche evolution. Oikos. 75: 182–92. [Google Scholar]
- 76. Sanjuán, R. , Moya A. & Elena S.F.. 2004. The distribution of fitness effects caused by single‐nucleotide substitutions in an RNA virus. Proc. Natl. Acad. Sci. USA. 101: 8396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Milton, D.K. , Fabian M.P., Cowling B.J., et al 2013. Influenza virus aerosols in human exhaled breath: particle size, culturability, and effect of surgical masks. PLoS Pathog. 9: e1003205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ford, B.E. , Sun B., Carpino J., et al 2014. Frequency and fitness consequences of bacteriophage phi 6 host range mutations. PLoS ONE. 9: e113078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Duffy, S. , Turner P.E. & Burch C.L.. 2006. Pleiotropic costs of niche expansion in the RNA bacteriophage Phi 6. Genetics. 172: 751–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ferris, M.T. , Joyce P. & Burch C.L.. 2007. High frequency of mutations that expand the host range of an RNA virus. Genetics. 176: 1013–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Li, W. , Zhang C., Sui J., et al 2005. Receptor and viral determinants of SARS‐coronavirus adaptation to human ACE2. EMBO J. 24: 1634–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tully, D.C. & Fares M.A.. 2009. Shifts in the selection‐drift balance drive the evolution and epidemiology of foot‐and‐mouth disease virus. J. Virol. 83: 781–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hughes, A.L. 2005. Evidence for abundant slightly deleterious polymorphisms in bacterial populations. Genetics. 169: 533–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lai, M.M.C. 1992. RNA recombination in animal and plant viruses. Microbiol. Rev. 56: 61–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Pérez‐Losada, M. , Arenas M., Galán J.C., et al 2015. Recombination in viruses: mechanisms, methods of study, and evolutionary consequences. Infect. Genet. Evol. 30: 296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Robinson, C.M. , Seto D., Jones M.S., et al 2011. Molecular evolution of human species D adenoviruses. Infect. Genet. Evol. 11: 1208–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Thiry, E. , Meurens F., Muylkens B., et al 2005. Recombination in alphaherpesviruses. Rev. Med. Virol. 15: 89–103. [DOI] [PubMed] [Google Scholar]
- 88. Martin, D.P. , Biagini P., Lefeuvre P., et al 2011. Recombination in eukaryotic single stranded DNA viruses. Viruses. 3: 1699–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Froissart, R. , Roze D., Uzest M., et al 2005. Recombination every day: Abundant recombination in a virus during a single multi‐cellular host infection. PLoS Biol. 3: 289–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Urbanowicz, A. , Formanowicz A.M., Blazewicz J., et al 2005. Homologous crossovers among molecules of brome mosaic bromovirus RNA1 or RNA2 segments in vivo. J. Virol. 79: 5732–5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Posada, D. , Crandall K.A. & Holmes E.C.. 2002. Recombination in evolutionary genomics. Annu. Rev. Genet. 36: 75–97. [DOI] [PubMed] [Google Scholar]
- 92. Vos, M. 2009. Why do bacteria engage in homologous recombination? Trends Microbiol. 17: 226–232. [DOI] [PubMed] [Google Scholar]
- 93. Chaston, T.B. & Lidbury B.A.. 2001. Genetic ‘budget’ of viruses and the cost to the infected host: A theory on the relationship between the genetic capacity of viruses, immune evasion, persistence and disease. Immunol. Cell Biol. 79: 62–66. [DOI] [PubMed] [Google Scholar]
- 94. Hsu, D.H. , Malefyt R.D., Fiorentino D.F., et al 1990. Expression of interleukin‐10 activity by Epstein‐Barr virus protein BCRF1. Science 250: 830–832. [DOI] [PubMed] [Google Scholar]
- 95. Han, G.‐Z. & Worobey M.. 2011Homologous recombination in negative sense RNA viruses. Viruses. 3: 1358–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Simon‐Loriere, E. & Holmes E.C.. 2011. Why do RNA viruses recombine? Nat. Rev. Microbiol. 9: 617–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Stedman, K.M. 2015. Deep recombination: RNA and ssDNA genes in DNA virus and host genomes. Annu. Rev. Virol. 2: 203–217. [DOI] [PubMed] [Google Scholar]
- 98. Jetzt, A.E. , Yu H., Klarmann G.J., et al 2000. High rate of recombination throughout the human immunodeficiency virus type 1 genome. J. Virol. 74: 1234–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Twiddy, S.S. & Holmes E.C.. 2003. The extent of homologous recombination in members of the genus flavivirus. J. Gen. Virol. 84: 429–440. [DOI] [PubMed] [Google Scholar]
- 100. Mehle, A. , Dugan V.G., Taubenberger J.K. & Doudna J.A.. 2012. Reassortment and mutation of the avian influenza virus polymerase PA subunit overcome species barriers. J. Virol. 86: 1750–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Jackwood, M.W. , Boynton T.O., Hilt D.A., et al 2010. Emergence of a group 3 coronavirus through recombination. Virology. 398: 98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Jegouic, S. , Joffret M.L., Blanchard C., et al 2009. Recombination between polioviruses and co‐circulating Coxsackie A viruses: role in the emergence of pathogenic vaccine‐derived polioviruses. PLoS Pathog. 5: e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Allison, A.B. , Stallknecht D.E. & Holmes E.C.. 2015. Evolutionary genetics and vector adaptation of recombinant viruses of the Western equine encephalitis antigenic complex provides new insights into alphavirus diversity and host switching. Virology. 474: 154–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Padidam, M. , Sawyer S. & Fauquet C.M.. 1999. Possible emergence of new geminiviruses by frequent recombination. Virology. 265: 218–25. [DOI] [PubMed] [Google Scholar]
- 105. Zhou, X.P. , Liu Y.L., Calvert L., et al 1997. Evidence that DNA‐A of a geminivirus associated with severe cassava mosaic disease in Uganda has arisen by interspecific recombination. J. Gen. Virol. 78: 2101–11. [DOI] [PubMed] [Google Scholar]
- 106. Varsani, A. , Shepherd D.N., Monjane A.L., et al 2008. Recombination, decreased host specificity and increased mobility may have driven the emergence of maize streak virus as an agricultural pathogen. J. Gen. Virol. 89: 2063–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Sanz, A.I. , Fraile A., Garcia‐Arenal F., et al 2000. Multiple infection, recombination and genome relationships among begomovirus isolates found in cotton and other plants in Pakistan. J. Gen. Virol. 81: 1839–49. [DOI] [PubMed] [Google Scholar]
- 108. Monci, F. , Sanchez‐Campos S, Navas‐Castillo J & Moriones E.. 2002. A natural recombinant between the geminiviruses tomato yellow leaf curl Sardinia virus and tomato yellow leaf curl virus exhibits a novel pathogenic phenotype and is becoming prevalent in Spanish populations. Virology. 303: 317–26. [DOI] [PubMed] [Google Scholar]
- 109. Varma, A. & Malathi V.G.. 2003. Emerging geminivirus problems: A serious threat to crop production. Ann. Appl. Biol. 142: 145–64. [Google Scholar]
- 110. Krupovic, M. , Ravantti J.J. & Bamford D.H.. 2009. Geminiviruses: a tale of a plasmid becoming a virus. BMC Evol. Biol. 9: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Nawaz‐ul‐Rehman, M.S. & Fauquet C.M.. 2009. Evolution of geminiviruses and their satellites. FEBS Letters. 583: 1825–32. [DOI] [PubMed] [Google Scholar]
- 112. Mansoor, S. , Briddon R.W., Zafar Y. & Stanley J.. 2003. Geminivirus disease complexes: an emerging threat. Trends Plant Sci. 8: 128–34. [DOI] [PubMed] [Google Scholar]
- 113. Prasanna H.C. & Rai M.. 2007. Detection and frequency of recombination in tomato‐infecting begomoviruses of South and Southeast Asia. Virol. J. 4: 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Pita, J.S. , Fondong V.N., Sangare A., et al 2001. Recombination, pseudorecombination and synergism of geminiviruses are determinant keys to the epidemic of severe cassava mosaic disease in Uganda. J. Gen. Virol. 82: 655–65. [DOI] [PubMed] [Google Scholar]
- 115. Callaway, A. , Giesman‐Cookmeyer D., Gillock E.T., et al 2001. The multifunctional capsid proteins of plant RNA viruses. Annu. Rev. Phytopathol. 39: 419–60. [DOI] [PubMed] [Google Scholar]
- 116. Fondong, V.N. , Pita J.S., Rey M.E., et al 2000. Evidence of synergism between African cassava mosaic virus and a new double‐recombinant geminivirus infecting cassava in Cameroon. J. Gen. Virol. 81: 287–97. [DOI] [PubMed] [Google Scholar]
- 117. Garcia‐Andres, S. , Accotto G.P., Navas‐Castillo J. & Moriones E.. 2007. Founder effect, plant host, and recombination shape the emergent population of begomoviruses that cause the tomato yellow leaf curl disease in the Mediterranean basin. Virology. 359: 302–12. [DOI] [PubMed] [Google Scholar]
- 118. Garcia‐Andres, S. , Tomas DM., Sanchez‐Campos S., et al 2007. Frequent occurrence of recombinants in mixed infections of tomato yellow leaf curl disease‐associated begomoviruses. Virology. 365: 210–9. [DOI] [PubMed] [Google Scholar]
- 119. Saleem, H. , Nahid N., Shakir S., et al 2016. Diversity, mutation and recombination analysis of cotton leaf curl geminiviruses. PLoS ONE. 11: e0151161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Moonan, F. , Molina J. & Mirkov T.E.. 2000. Sugarcane yellow leaf virus: An emerging virus that has evolved by recombination between luteoviral and poleroviral ancestors. Virology. 269: 156–71. [DOI] [PubMed] [Google Scholar]
- 121. Preiss W. & Jeske H.. Multitasking in replication is common among geminiviruses. 2003. J. Virol. 77: 2972–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Jeske, H. , Lutgemeier M. & Preiss W.. 2001. DNA forms indicate rolling circle and recombination‐dependent replication of Abutilon mosaic virus. EMBO J. 20: 6158–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Weller, S.K. & Sawitzke J.A.. 2014. Recombination promoted by DNA viruses: Phage λ to herpes simplex virus. Annu. Rev. Microbiol. 68: 237–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Seal, S.E. , Jeger M.J. & Van den Bosch F.. 2006. Begomovirus evolution and disease management. Adv. Virus Res. 67: 297–316. [DOI] [PubMed] [Google Scholar]
- 125. Legg, J.P. & Fauquet C.M.. 2004. Cassava mosaic geminiviruses in Africa. Plant Mol. Biol. 56: 585–99. [DOI] [PubMed] [Google Scholar]
- 126. Lima, A.T. , Sobrinho R.R., Gonzalez‐Aguilera J., et al 2013. Synonymous site variation due to recombination explains higher genetic variability in begomovirus populations infecting non‐cultivated hosts. J. Gen. Virol. 94: 418–431. [DOI] [PubMed] [Google Scholar]
- 127. Rocha, C.S. , Castillo‐Urquiza G.P., Lima A.T., et al 2013. Brazilian begomovirus populations are highly recombinant, rapidly evolving, and segregated based on geographical location. J. Virol. 87: 5784–5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Martin, D.P. , Lefeuvre P., Varsani A., et al 2011. Complex recombination patterns arising during geminivirus coinfections preserve and demarcate biologically important intra‐genome interaction networks. PLoS Pathog. 7: e1002203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Lefeuvre, P. , Lett J.‐M., Reynaud B. & Martin D.P.. 2007. Avoidance of protein fold disruption in natural virus recombinants. PLoS Pathog. 3(11):e181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Lefeuvre, P. , Martin D.P., Hoareau M., et al 2007. Begomovirus ‘melting pot’ in the south‐west Indian Ocean islands: molecular diversity and evolution through recombination. J. Gen. Virol. 88: 3458–3468. [DOI] [PubMed] [Google Scholar]
- 131. Lefeuvre, P. , Lett J.M., Varsani A. & Martin D.P.. 2009. Widely conserved recombination patterns among single‐stranded DNA viruses. J. Virol. 83: 2697–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Varsani, A. , Shepherd D.N., Monjane A.L., et al 2008. Recombination, decreased host specificity and increased mobility may have driven the emergence of maize streak virus as an agricultural pathogen. J. Gen. Virol. 89: 2063–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Monjane A.L., van der Walt E., Varsani A., et al 2011. Recombination hotspots and host susceptibility modulate the adaptive value of recombination during maize streak virus evolution. BMC Evol. Biol. 11: 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Fauquet, C.M. , Sawyer S., Idris A.M. & Brown J.K.. 2005. Sequence analysis and classification of apparent recombinant begomoviruses infecting tomato in the nile and mediterranean basins. Phytopathology. 95: 549–555. [DOI] [PubMed] [Google Scholar]
- 135. Dugan, V.G. , Chen R., Spiro D.J., et al 2008. The evolutionary genetics and emergence of avian influenza viruses in wild birds. PLoS Pathog. 4: e1000076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Olsen, C.W. 2002. The emergence of novel swine influenza viruses in North America. Virus Res. 85: 199–210. [DOI] [PubMed] [Google Scholar]
- 137. Ladner, J.T. , Wiley M.R., Beitzel B., et al 2016. A multicomponent animal virus isolated from mosquitoes. Cell Host & Microbe. 20: 357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Iturriza‐Gomara, M. , Kang G., & Gray J.. 2004. Rotavirus genotyping: keeping up with an evolving population of human rotaviruses. J. Clin. Virol. 31: 259–265 [DOI] [PubMed] [Google Scholar]
- 139. Webster, R.G. , Bean W.J., Gorman O.T., Chambers T.M. & Kawaoka Y.. 1992. Evolution and ecology of influenza A viruses. Microbiol. Rev. 56: 152–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Zhou, N.N. , Senne D.A., Landgraf J.S., et al 1999. Genetic reassortment of avian, swine, and human influenza A viruses in American pigs. J. Virol. 73: 8851–8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Vijaykrishna, D. , Poon L.L.M., Zhu H.C., et al 2010. Reassortment of pandemic H1N1/2009 influenza A virus in swine. Science. 328: 1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Rodriguez, L.L. , Owens J.H., Peters C.J. & Nichol S.T.. 1998. Genetic reassortment among viruses causing hantavirus pulmonary syndrome. Virology. 242: 99–106. [DOI] [PubMed] [Google Scholar]
- 143. Sall, A.A. , Zanotto P.M.D., Sene O.K., et al 1999. Genetic reassortment of Rift Valley fever virus in nature. J. Virology. 73: 8196–8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Beaty, B.J. , Sundin D.R., Chandler L.J. & Bishop D.H.L.. 1985. Evolution of bunyaviruses by genome reassortment in dually infected mosquitos (Aedes triseriatus). Science. 230: 548–550. [DOI] [PubMed] [Google Scholar]
- 145. Codoner, F.M. & Elena S.F.. 2008. The promiscuous evolutionary history of the family Bromoviridae . J. Gen. Virol. 89: 1739–1747. [DOI] [PubMed] [Google Scholar]
- 146. Roossinck, M.J. 2002. Evolutionary history of cucumber mosaic virus deduced by phylogenetic analyses. J. Virol. 76: 3382–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Smith, G.J.D. , Vijaykrishna D., Bahl J., et al 2009. Origins and evolutionary genomics of the 2009 swine‐origin H1N1 influenza A epidemic. Nature. 459: 1122–1125. [DOI] [PubMed] [Google Scholar]
- 148. Brown, I.H. , Harris P.A., McCauley J.W. & Alexander D.J.. 1998. Multiple genetic reassortment of avian and human influenza A viruses in European pigs, resulting in the emergence of an H1N2 virus of novel genotype. J. Gen. Virol. 79: 2947–55. [DOI] [PubMed] [Google Scholar]
- 149. Webby, R.J. , Swenson S.L., Krauss S.L., et al 2000. Evolution of swine H3N2 influenza viruses in the United States. J. Virol. 74: 8243–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Newman, A.P. , Reisdorf E., Beinemann J., et al 2008. Human case of swine influenza A (H1N1) triple reassortant virus infection, Wisconsin. Emerg. Infect. Dis. 14: 1470–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Shinde, V. , Bridges C.B., Uyeki T.M., et al 2009. Triple‐reassortant swine influenza A (H1) in humans in the United States, 2005–2009. N. Engl. J. Med. 360: 2616–25. [DOI] [PubMed] [Google Scholar]
- 152. Simonsen, L. , Spreeuwenberg P, Lustig R, et al 2013. Global mortality estimates for the 2009 influenza pandemic from the GLaMOR project: a modeling study. PLoS Med. 10: e1001558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Wright, S. 1932. The role of mutation, inbreeding, crossbreeding and selection in evolution. Proc. 6th Int. Congress Genet. 1, 356–366. [Google Scholar]
- 154. Steinberg, B. & Ostermeier M.. 2016. Environmental changes bridge evolutionary valleys. Sci. Adv. 2: e1500921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Weinreich, D.M. , Delaney N.F., Depristo M.A. & Hartl D.L.. 2006. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science. 312: 111–4. [DOI] [PubMed] [Google Scholar]
- 156. Elena S.F., Fraile A. & Garcia‐Arenal F.. 2014. Evolution and emergence of plant viruses. Adv. Virus Res. 88: 161–91. [DOI] [PubMed] [Google Scholar]
- 157. Weinreich, D.M. , Watson RA & Chao L.. 2005. Perspective: Sign epistasis and genetic constraint on evolutionary trajectories. Evolution. 59: 1165–74. [PubMed] [Google Scholar]
- 158. Poelwijk, F.J. , Tanase‐Nicola S., Kiviet D.J. & Tans S.J.. 2011. Reciprocal sign epistasis is a necessary condition for multi‐peaked fitness landscapes. J. Theor. Biol. 272: 141–4. [DOI] [PubMed] [Google Scholar]
- 159. Sanjuán, R. , Moya A. & Elena S.F.. 2004. The contribution of epistasis to the architecture of fitness in an RNA virus. Proc. Nat. Acad. Sci. USA. 101: 15376–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Bernet, G.P. & Elena S.F.. 2015. Distribution of mutational fitness effects and of epistasis in the 5′ untranslated region of a plant RNA virus. BMC Evol. Biol. 15: 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Lalić, J. & Elena S.F.. 2013. Epistasis between mutations is host‐dependent for an RNA virus. Biol. Lett. 9: 20120396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Burch, C.L. & Chao L.. 1999. Evolution by small steps and rugged landscapes in the RNA virus phi 6. Genetics. 151: 921–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Coffey, L.L. , Forrester N., Tsetsarkin K., et al 2013. Factors shaping the adaptive landscape for arboviruses: implications for the emergence of disease. Future Microbiol. 8; 155–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Tsetsarkin, K.A. , Chen R., Leal G., et al 2011. Chikungunya virus emergence is constrained in Asia by lineage‐specific adaptive landscape. Proc. Nat. Acad. Sci. USA. 108: 7872–7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Kraemer, M.U.G. , Sinka M.E., Duda K.A., et al 2015. The global distribution of the arbovirus vectors Aedes aegypti and Ae . albopictus. eLife 4: e08347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Hartfield, M. & Alizon S.. 2013. Introducing the outbreak threshold in epidemiology. PLoS Pathog. 9: e1003277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Kirschner, M. & Gerhart J.. 1998. Evolvability. Proc. Nat. Acad. Sci. USA. 95: 8420–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Li, W.H. , Wu C.I. & Luo C.C.. 1985. A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol. Biol. Evol. 2: 150–74. [DOI] [PubMed] [Google Scholar]
- 169. Jenkins, G.M. , Rambaut A., Pybus O.G. & Holmes E.C.. 2002. Rates of molecular evolution in RNA viruses: a quantitative phylogenetic analysis. J. Mol. Evol. 54: 156–65. [DOI] [PubMed] [Google Scholar]
- 170. Hanada, K. , Suzuki Y. & Gojobori T.. 2004. A large variation in the rates of synonymous substitution for RNA viruses and its relationship to a diversity of viral infection and transmission modes. Mol. Biol. Evol. 21: 1074–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Duffy, S. , Shackelton L.A., Holmes E.C.. 2008. Rates of evolutionary change in viruses: patterns and determinants. Nat. Rev. Genet. 9: 267–76. [DOI] [PubMed] [Google Scholar]
- 172. Shackelton, L.A. , Parrish C.R., Truyen U. & Holmes E.C.. 2005. High rate of viral evolution associated with the emergence of carnivore parvovirus. Proc. Nat. Acad. Sci. USA. 102: 379–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Williamson, S. 2003. Adaptation in the env gene of HIV‐1 and evolutionary theories of disease progression. Mol. Biol. Evol. 20: 1318–25. [DOI] [PubMed] [Google Scholar]
- 174. Lemey, P. , Rambaut A. & Pybus O.G.. 2006. HIV evolutionary dynamics within and among hosts. AIDS Rev. 8: 125–40. [PubMed] [Google Scholar]
- 175. Koelle, K. & Rasmussen D.A.. 2015. The effects of a deleterious mutation load on patterns of influenza A/H3N2's antigenic evolution in humans. Elife. 4: e07361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Pybus, O.G. , Rambaut A., Belshaw R., et al 2007. Phylogenetic evidence for deleterious mutation load in RNA viruses and its contribution to viral evolution. Mol. Biol. Evol. 24: 845–52. [DOI] [PubMed] [Google Scholar]
- 177. Domingo, E. , Sheldon J. & Perales C.. 2012. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 76: 159–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Lynch, M. , Conery J. & Burger R.. 1995. Mutation accumulation and the extinction of small populations. Am. Nat. 146: 489–518. [Google Scholar]
- 179. de la Iglesia, F. & Elena S.F.. 2007. Fitness declines in Tobacco etch virus upon serial bottleneck transfers. J. Virol. 81: 4941–4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Dennehy, J.J. 2009. Bacteriophages as model organisms for virus emergence research. Trends Microbiol. 17: 450–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Blumberg, S. & Lloyd‐Smith J.O.. 2013. Inference of R(0) and transmission heterogeneity from the size distribution of stuttering chains. PLoS Comput. Biol. 9: e1002993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Ewald, P.W. 1983. Host‐parasite relations, vectors, and the evolution of disease severity. Annu. Rev. Ecol. Syst. 14: 465–485. [Google Scholar]
- 183. Guo, Z. , Xiao D., Li D., et al 2016. Predicting and evaluating the epidemic trend of Ebola virus disease in the 2014–2015 outbreak and the effects of intervention measures. PLoS ONE 11: e0152438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Liu, W.B. , Li Z.X., Du Y. & Cao G.W.. 2015. Ebola virus disease: from epidemiology to prophylaxis. Mil. Med. Res. 2: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Gire, S.K. , Goba A., Andersen K.G., et al 2014. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345: 1369–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Hoenen, T. , Safronetz D., Groseth A., et al 2015. Mutation rate and genotype variation of Ebola virus from Mali case sequences. Science 348:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Messer, W.B. , Gubler D.J., Harris E., et al 2003. Emergence and global spread of a dengue serotype 3, subtype III virus. Emerg. Infect. Dis. 9: 800–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Hanley, K.A. , Nelson J.T., Schirtzinger E.E., et al 2008. Superior infectivity for mosquito vectors contributes to competitive displacement among strains of dengue virus. BMC Ecology 8: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Ching, J. , Musheyev S., Chowdhury D., et al 2013. Migration enhances adaptation in bacteriophage populations evolving in ecological sinks. Evolution. 67: 10–7. [DOI] [PubMed] [Google Scholar]
- 190. Dennehy, J.J. , Friedenberg N.A., McBride R.C., et al 2010. Experimental evidence that source genetic variation drives pathogen emergence. Proc. R. Soc. B‐Biol. Sci. 277: 3113–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Vantreuren, R. , Bijlsma R., Vandelden W. & Ouborg N.J.. 1991. The significance of genetic erosion in the process of extinction 1. Genetic differentiation in Salvia pratensis and Scabiosa columbaria in relation to population size. Heredity. 66: 181–9. [Google Scholar]
- 192. Lande, R. 1988. Genetics and demography in biological conservation. Science. 241: 1455–60. [DOI] [PubMed] [Google Scholar]
- 193. Kawecki, T.J. 2008. Adaptation to marginal habitats. Annu. Rev. Ecol. Evol. Syst. 39: 321–42. [Google Scholar]
- 194. Wang, G.M. , Edge W.D. & Wolff J.O.. 2001. Demographic uncertainty in ecological risk assessments. Ecol. Model. 136: 95–102. [Google Scholar]
- 195. Strelkowa, N. & Lassig M.. 2012. Clonal interference in the evolution of influenza. Genetics. 192: 671–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Miralles, R. , Gerrish P.J., Moya A. & Elena S.F.. 1999. Clonal interference and the evolution of RNA viruses. Science. 285: 1745–7. [DOI] [PubMed] [Google Scholar]
- 197. Elena, S.F. 2002. Restrictions to RNA virus adaptation: an experimental approach. Antonie Van Leeuwenhoek. 81: 135–42. [DOI] [PubMed] [Google Scholar]
- 198. Elena, S.F. , Agudelo‐Romero P. & Lalic J.. 2009. The evolution of viruses in multi‐host fitness landscapes. Open Virol. J. 3: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Gompert, Z. , Jahner J.P., Scholl C.F., et al 2015. The evolution of novel host use is unlikely to be constrained by trade‐offs or a lack of genetic variation. Mol. Ecol. 24: 2777–93. [DOI] [PubMed] [Google Scholar]
- 200. Smith‐Tsurkan, S.D. , Wilke C.O. & Novella I.S.. 2010. Incongruent fitness landscapes, not tradeoffs, dominate the adaptation of vesicular stomatitis virus to novel host types. J. Gen. Virol. 91: 1484–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Futuyma, D.J. & Moreno G.. 1988. The evolution of ecological specialization. Annu. Rev. Ecol. Syst. 19: 207–33. [Google Scholar]
- 202. Kassen, R. 2002. The experimental evolution of specialists, generalists, and the maintenance of diversity. J. Evol. Biol. 15: 173–90. [Google Scholar]
- 203. Coffey, L.L. , Vasilakis N., Brault A.C., et al 2008. Arbovirus evolution in vivo is constrained by host alternation. Proc. Nat. Acad. Sci. USA. 105: 6970–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Garcia‐Arenal, F. & Fraile A.. 2013Trade‐offs in host range evolution of plant viruses. Plant Pathol. 62: 2–9. [Google Scholar]
- 205. Bedhomme, S. , Hillung J. & Elena S.F.. 2015. Emerging viruses: why they are not jacks of all trades? Curr. Opin. Virol. 10: 1–6. [DOI] [PubMed] [Google Scholar]
- 206. Fry, J.D. 1996. The evolution of host specialization: are trade‐offs overrated? Am. Nat. 148: S84–S107. [Google Scholar]
- 207. Remold, S. 2012. Understanding specialism when the jack of all trades can be the master of all. Proc. R. Soc. B: Biol. Sci. 279: 4861–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Woolhouse, M. & Gaunt E..2007. Ecological origins of novel human pathogens. Crit. Rev. Microbiol. 33: 231–42. [DOI] [PubMed] [Google Scholar]
- 209. Wilson, A.J. & Mellor P.S.. 2009. Bluetongue in Europe: past, present and future. Phil. Trans. R. Soc. Lond. B: Biol. Sci. 364: 2669–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Purse, B.V. , Mellor P.S., Rogers D.J., et al 2005. Climate change and the recent emergence of bluetongue in Europe. Nat. Rev. Microbiol. 3: 171–81. [DOI] [PubMed] [Google Scholar]
- 211. Acevedo, P. , Ruiz‐Fons F., Estrada R., et al 2010. A broad assessment of factors determining Culicoides imicola abundance: modelling the present and forecasting its future in climate change scenarios. PLoS ONE. 5: e14236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Carpenter, S. , Wilson A. & Mellor P.S.. 2009. Culicoides and the emergence of bluetongue virus in northern Europe. Trends Microbiol. 17: 172–8. [DOI] [PubMed] [Google Scholar]
- 213. Purse, B.V. , McCormick B.J.J., Mellor P.S., et al 2007. Incriminating bluetongue virus vectors with climate envelope models. J. Appl. Ecol. 44: 1231–42. [Google Scholar]
- 214. Pioz, M. , Guis H., Crespin L., Gay E., et al 2012. Why did bluetongue spread the way it did? Environmental factors influencing the velocity of bluetongue virus serotype 8 epizootic wave in France. PLoS ONE. 7: e43360.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Zuliani, A. , Massolo A., Lysyk T., et al 2015. Modelling the northward expansion of Culicoides sonorensis (Diptera: Ceratopogonidae) under future climate scenarios. PLoS ONE. 10: e0130294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Heffernan, J.M. , Smith R.J. & Wahl L.M.. 2005. Perspectives on the basic reproductive ratio. J. Roy. Soc. Interface. 2: 281–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Mullens, B.A. , Gerry A.C., Lysyk T.J. & Schmidtmann E.T.. 2004. Environmental effects on vector competence and virogenesis of bluetongue virus in Culicoides: interpreting laboratory data in a field context. Vet. Ital. 40: 160–6. [PubMed] [Google Scholar]
- 218. Jimenez‐Clavero, M.A . 2012. Animal viral diseases and global change: bluetongue and West Nile fever as paradigms. Front. Genet. 3: 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Yates, T.L. , Mills J.N., Parmenter C.A., et al 2002. The ecology and evolutionary history of an emergent disease: Hantavirus pulmonary syndrome. Bioscience. 52: 989–98. [Google Scholar]
- 220. Nichol, S.T. , Spiropoulou C.F., Morzunov S., et al 1993. Genetic identification of a hantavirus associated with an outbreak of acute respiratory illness. Science. 262: 914–7. [DOI] [PubMed] [Google Scholar]
- 221. Childs, J.E. , Ksiazek T.G., Spiropoulou C.F., et al 1994. Serologic and genetic identification of Peromyscus maniculatus as the primary rodent reservoir for a new hantavirus in the southwestern United States. J. Infect. Dis. 169: 1271–80. [DOI] [PubMed] [Google Scholar]
- 222. Dearing, M.D. & Dizney L.. 2010. Ecology of hantavirus in a changing world. Ann. N. Y. Acad. Sci. 1195: 99–112. [DOI] [PubMed] [Google Scholar]
- 223. Calisher, C.H. , Sweeney W., Mills J.N. & Beaty B.J.. 1999. Natural history of Sin Nombre virus in western Colorado. Emerg. Infect. Dis. 5: 126–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Holmes, E.C. & Zhang Y.Z.. 2015. The evolution and emergence of hantaviruses. Curr. Opin. Virol. 10: 27–33. [DOI] [PubMed] [Google Scholar]
- 225. Kruger, D.H. , Figueiredo L.T.M., Song J.‐W. & Klempa B.. 2015. Hantaviruses—Globally emerging pathogens, J. Clin. Virol. 64: 128–136. [DOI] [PubMed] [Google Scholar]
- 226. Martinez, V.P. , Bellomo C., San Juan J., et al 2005. Person‐to‐person transmission of Andes virus. Emerg. Infect. Dis. 11: 1848–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Woolhouse, M. , Scott F., Hudson Z., et al 2012. Human viruses: discovery and emergence. Phil. Trans. Roy. Soc. B: Biol. Sci. 367: 2864–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Anthony, S.J. , Epstein J.H., Murray K.A., et al 2013. A strategy to estimate unknown viral diversity in mammals. mBio 4: e00598–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Vigant, F.N.C. Santos & Lee B.. 2015. Broad‐spectrum antivirals against viral fusion. Nat. Rev. Microbiol. 13: 426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. ElSawy, K.M. , Twarock R., Verma C.S. & Caves L.S.D.. 2012. Peptide inhibitors of viral assembly: a novel route to broad‐spectrum antivirals. J. Chem. Inf. Model. 52: 770–776. [DOI] [PubMed] [Google Scholar]
- 231. Ichiyama, K. , Yang C., Chandrasekaran L., et al 2016. Cooperative orthogonal macromolecular assemblies with broad spectrum antiviral activity, high selectivity, and resistance mitigation. Macromolecules 49: 2618–2629. [Google Scholar]
- 232. Raia, M. , Deshmukha S.D., Inglea A.P., et al 2016. Metal nanoparticles: The protective nanoshield against virus infection. Crit. Rev. Microbiol. 42: 46–56. [DOI] [PubMed] [Google Scholar]
- 233. Debing, Y. , Neyts J. & Delang L.. 2015. The future of antivirals: broad‐spectrum inhibitors. Curr. Opin. Inf. Dis. 28: 596–602. [DOI] [PubMed] [Google Scholar]
- 234. Leblanc, P.L. Moise , Luza C., Chantaralawan K., et al 2014. VaxCelerate II: rapid development of a self‐assembling vaccine for Lassa fever. Hum. Vaccine Immunother. 10: 3022–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. De Groot A.S. & Martin W.. 2009. Reducing risk, improving outcomes: Bioengineering less immunogenic protein therapeutics. Clin. Immunol. 131: 189–201. [DOI] [PubMed] [Google Scholar]
- 236. Garcia‐Sastre, A. & Mena I.. 2013. Novel vaccine strategies against emerging viruses. Curr. Opin. Virol. 3: 210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Champer, J. , Buchman A. & Akbari O.S.. 2016. Cheating evolution: engineering gene drives to manipulate the fate of wild populations. Nat. Rev. Genet. 17: 146–159. [DOI] [PubMed] [Google Scholar]
- 238. Hammond, A. , Galizi R., Kyrou K., et al 2016. A CRISPR‐Cas9 gene drive system targeting female reproduction in the malaria mosquito vector Anopheles gambiae . Nat. Biotech. 34: 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]