

Abstract
Ste24 enzymes, a family of eukaryotic integral membrane proteins, are zinc metalloproteases (ZMPs) originally characterized as “CAAX proteases” targeting prenylated substrates, including a‐factor mating pheromone in yeast and prelamin A in humans. Recently, Ste24 was shown to also cleave nonprenylated substrates. Reduced activity of the human ortholog, HsSte24, is linked to multiple disease states (laminopathies), including progerias and lipid disorders. Ste24 possesses a unique “α‐barrel” structure consisting of seven transmembrane (TM) α‐helices encircling a large intramembranous cavity (~14 000 Å3). The catalytic zinc, coordinated via a HExxH…E/H motif characteristic of gluzincin ZMPs, is positioned at one of the cavity's bases. The interrelationship between Ste24 as a gluzincin, a long‐studied class of soluble ZMPs, and as a novel cavity‐containing integral membrane protein protease has been minimally explored to date. Informed by homology to well‐characterized soluble, gluzincin ZMPs, we develop a model of Ste24 that provides a conceptual framework for this enzyme family, suitable for development and interpretation of structure/function studies. The model consists of an interfacial, zinc‐containing “ZMP Core” module surrounded by a “ZMP Accessory” module, both capped by a TM helical “α‐barrel” module of as yet unknown function. Multiple sequence alignment of 58 Ste24 orthologs revealed 38 absolutely conserved residues, apportioned unequally among the ZMP Core (18), ZMP Accessory (13), and α‐barrel (7) modules. This Tripartite Architecture representation of Ste24 provides a unified image of this enzyme family.
Keywords: computational biology, endopeptidase, lipodystrophy, membrane proteins, metalloprotease, progeria, zinc
1. INTRODUCTION
Proteases, abundant enzymes across all forms of life, conduct a remarkably broad range of biological functions.1 Approximately 2% of genome‐encoded mammalian proteins are proteases.2 Due to their abundance and copious biological roles, protease dysfunction can lead to deleterious health consequences.1 Proteases are targeted to virtually all cellular compartments from cytosolic/luminal space to the membranes of organelles and the cell exterior. Because of their “gatekeeping” status, peripheral and integral membrane protein proteases play further specialized and vital roles in metabolic and signaling pathways.3, 4, 5
Ste24 enzymes, a family of integral membrane proteins, function as zinc metalloproteases (ZMPs) and are found in all eukaryotic organisms.6 The first Ste24 family member, Ste24p (where “p” is an abbreviation for protein), was discovered and characterized in the yeast Saccharomyces cerevisiae, where it is involved in posttranslational modification of the peptide‐derived a‐factor mating pheromone.7, 8, 9 Nascent a‐factor processing involves three proteolytic cleavages, two of which Ste24p is competent to perform.6 One Ste24p cleavage occurs between a farnesylated Cys and the sequence Val‐Ile‐Ala, which form a C‐terminal “CAAX box” motif (C is Cys; A is typically an aliphatic amino acid; and X is one of several amino acids less restricted in characteristics).10 Ste24p is capable of, though not required for, carrying out the CAAX box cleavage,9 and uniquely required for one of the two other nonprenylated site cleavages.8 The ZMP protease Axlp, or its homolog Ste23p, is required for ultimately completing a‐factor maturation via cleavage of the remaining site.11
The human HsSte24 enzyme, also known as FACE‐1 and ZMPSTE24, is localized to the endoplasmic reticulum and the inner nuclear membrane.6, 12 The earliest functional characterization of HsSte24 demonstrated its role in the processing of prelamin A to mature intermediate filament protein lamin A.13, 14 Lamins provide mechanical stability to the nuclear envelope and function as scaffolds for DNA repair and replication complexes.15, 16 Prelamin A is a CAAX box‐containing substrate that undergoes two proteolytic cleavages, at the CAAX box and an N‐terminal site.17 Both cleavages can be performed by HsSte24, with a unique requirement for HsSte24 cleavage of the nonprenylated site (analogous to Ste24p).17, 18, 19, 20 The Ras Converting CAAX Endopeptidase 1 (Rce1), an unrelated cysteine protease, can also perform CAAX box cleavage of both a‐factor7 and prelamin A.17
Deficiencies in HsSte24 activity, whether by mutation18, 21, 22 or inhibition,23, 24 restrict prelamin A maturation and lead to disease states known as laminopathies, which range from progerias (premature aging syndrome) to lipodystrophies.25, 26 Inhibition of HsSte24 has particular clinical relevance, as AIDS patients receiving viral protease inhibitors as part of their drug regimen develop lipodystrophies because of off‐target interaction of HIV protease inhibitor drugs with HsSte24.24, 27 The cellular role for Ste24 family enzymes has expanded recently, beyond its role in prenylated protein processing. Both fungal (yeast) and human Ste24 have been shown to clear clogged translocons.28, 29 Importantly, recent in vitro studies demonstrate that Ste24 can act as a generalized, integral membrane protein protease upon diverse substrates, without requiring the presence of a farnesyl moiety for substrate recognition and cleavage.30 Recently, a nonenzymatic role for HsSte24p as a broad spectrum viral restriction factor has also been reported.31, 32 Interferon‐inducible Transmembrane Proteins (IFITMs), produced by the (mammalian) host innate immune response, have been reported to reduce infectivity of more than 20 viruses,33, 34 including Dengue, Ebola, HIV, Hepatitis C, MERS‐ and SARS‐Coronaviruses, West Nile, and Zika. Protein interaction studies using the IFITM3 protein as “bait” discovered HsSte24 as a downstream effector.31, 32 Strikingly, overexpression of HsSte24 in IfitmDel cells (all ifitm genes deleted) yielded the same reduced viral infection phenotype as seen in IFITM‐expressing cells, suggesting that HsSte24 is the antiviral “causative agent.”31 Also, catalytically inactive HsSte24 in these experiments produced the same results, indicating that proteolytic function is not required.
Yeast30, 33 and human23, 30, 34, 35, 36, 37 Ste24 orthologs have been the most rigorously characterized Ste24 family members both functionally and structurally. Additionally, functional complementation between human and yeast Ste24 orthologs has been established in vivo where HsSte24 rescues defects in a‐factor biogenesis associated with Ste24p knockout yeast (ste24Δ).9 Crystal structures of yeast (Saccharomyces mikatae; SmSte24)33 and human Ste24 (HsSte24) reveal these two Ste24 structures to be highly similar (RMSD of Cα atoms ~1.7 Å).23, 35, 37 The structure of Ste24, as exemplified by SmSte24 (PDB: 4IL3), possesses a striking seven‐transmembrane (TM) helical “α‐barrel” structure encapsulating a voluminous reaction cavity (~14 000 Å3; Figure 1A). The zinc‐coordinated active site is localized at one “end” of the approximately cylindrical reaction cavity (Figure 1A,B). Ste24 is associated with the gluzincin subclan of Zincin ZMPs (MEROPS38 designation MA[E]), which use a glutamate, Glu390 in SmSte24, or histidine residue as the third ligand coordinating the zinc atom (ie, HExxH…E/H) (Figure 1B,C).39, 40, 41 Despite the status of the Ste24 family as a gluzincin, structural homology to and sequence conservation with gluzincins (or particular subsets of gluzincin families) has been scantly investigated. Such an analysis is especially important for identifying conserved structural elements and catalytically essential residues, in addition to the HExxH…E/H gluzincin motif, which form the complete Ste24 active‐site. Furthermore, the description of Ste24 as a gluzincin describes only a portion of this large, multidomain, membrane‐inserted enzyme.
Figure 1.
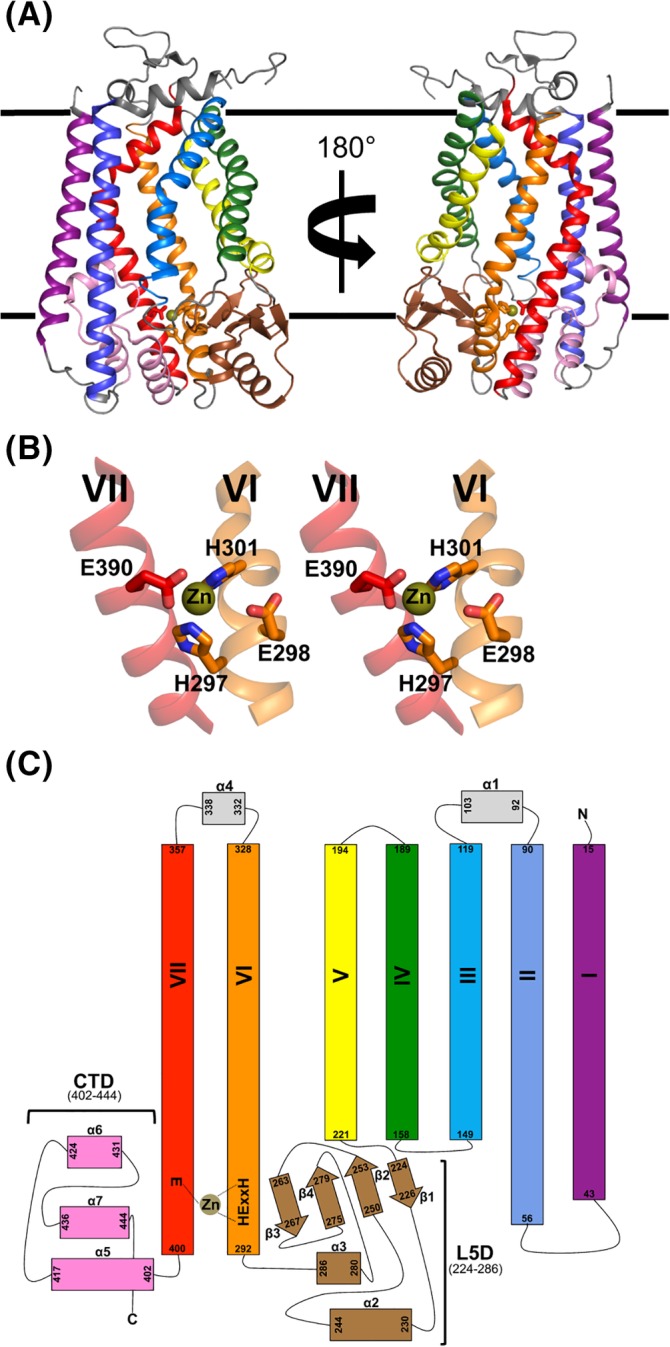
Structural summary of the Ste24 family. A, Ribbon representation of the structure of Ste24 from the fungus (yeast) S. mikatae (PDB 4IL3).33 A total of seven transmembrane (TM) α‐helices span the membrane (indicated by parallel black lines). The zinc ion (shown as a small gold sphere) is positioned at the “lower” membrane interface and ligated by residues from two of the TM helices. B, Stereo view of the Ste24 gluzincin structural motif. The image is generated from the left‐hand orientation of the Ste24 molecule as seen in panel A. Ste24 possesses the gluzincin 297HExxH301…E390 consensus sequence motif located at the base of TM helices VI and VII. Residues His297, His301, and Glu390 coordinate the zinc ion, with Glu298 acting as the general base during turnover.37, 40 C, Topology diagram of Ste24. The color‐coding of the ribbon diagram in panels A and B is maintained. TM α‐helices are labeled I‐VII. Two, large membrane interfacial regions flank the coordinated zinc ion: the mixed α‐helix‐β‐sheet Loop 5 Domain (L5D) located between helix V and helix VI, and the α‐helical C‐terminal Domain (CTD)
In this study, we develop a conceptual framework for Ste24 that encompasses its identities as both a gluzincin and a complex, cavity‐containing integral membrane protein protease. Rather than a structural model, we develop a representational architecture informed by both functional and structural homology data. Through a combination of bioinformatics and structural comparison to homologous, soluble gluzincins, we have developed a Tripartite Architecture representation of the Ste24 enzyme family. We believe this model provides a better framework for understanding Ste24 mechanism, and will be conducive to better rationalize future functional characterization of Ste24 enzymes.
2. MATERIALS AND METHODS
2.1. Identification of soluble gluzincin ZMPs homologs
The Dali server42 was used to identify structurally homologous ZMP structures. A query model of Ste24 was developed containing elements only from the Loop 5 Domain (L5D), C‐terminal Domain (CTD), and the portions of TM α‐helices VI and VII containing the 297HExxH301…E390 gluzincin motif. For analysis and comparison, structures of homologous gluzincin ZMPs were aligned with the SmSte24 structure (PDB 4IL3) by use of the Pymol program SUPER.43 The four conserved gluzincin residues, HExxH…E, of the orthologs were used to seed the structural alignment algorithm.
2.2. Generation of the Ste24 enzyme multiple sequence alignment
Homologous sequences to S mikatae Ste24 (SmSte24) were identified through iterative searching using the blastp suite available on the NCBI web server (https://blast.ncbi.nlm.nih.gov/Blast.cgi).44, 45 In total, 58 homologous sequences were identified from fungi, plant, animal, and protist kingdoms. The program Clustal Omega (EMBL)46 was used to construct a multiple sequence alignment (MSA) from these identified sequences, which identified 38 absolutely conserved residues. Sequence identity and similarity scores between each sequence and SmSte24 are reported in Table S1, and were calculated using the EMBOSS Needle global sequence alignment tool available from EMBL‐EBI (http://www.ebi.ac.uk/Tools/psa/emboss_needle/).47, 48 The MSA was graphically represented using ESPript 3.0 (http://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi).49
3. RESULTS
3.1. The Ste24 active‐site pocket: homology to soluble gluzincins
Inspection of the zinc‐coordinating residues of Ste24 reveals a characteristic HExxH motif of the Zincin family of ZMPs (Figure 1B).40 Zincins are further subdivided by the identity of the third amino acid ligand to the zinc (ie, beyond the two histidine residues of the HExxH motif).39, 40, 50, 51 In gluzincins, a glutamate residue most often provides the third ligand; therefore, all gluzincins share the consensus motif “HExxH…E/H.”40 Invariantly, all residues of the gluzincin motif originate from a neighboring pair of α‐helices.39, 40 Among Ste24 enzymes, the base of the two TM α‐helices VI and VII contribute these four residues (Figure 1B). SmSte24 residues His297, His301, and Glu390 are the zinc ligands and Glu298 acts as a catalytic base.33, 37 Gluzincin ZMPs also possess a region of β‐sheet orthogonal to these α‐helices,50 where a β‐strand contributes catalytically essential residues.39, 50 In the case of Ste24 enzymes, the L5D contributes this β‐sheet (Figure 1A,C). Finally, gluzincins contain a region of α‐helix opposite the β‐sheet contributing further catalytically essential residues.39, 50 In the case of Ste24 enzymes, a CTD composed of a series of loop‐connected α‐helices located between TM α‐helices I and VII is positioned in such a fashion (Figure 1C).
Currently, 29 distinguishable gluzincin members have been assigned in the MEROPS database,38 with Ste24 designated as MA(E)/M48A. Therefore, a structure‐based search of the PDB database using the DALI server42 was used to identify those gluzincin families most homologous to Ste24. Currently available structures of Ste24 comprise fungal (SmSte24)33 and human (HsSte24)23, 35, 37 orthologs. While SmSte24 and HsSte24 structures are highly similar, Ste24 mammalian orthologs contain a variable length insert between the α3‐helix of the L5D and TM α‐helix VI (37 residues in HsSte24) (Figure S1); this insert is disordered in all HsSte24 crystal structures. Therefore, the SmSte24 fungal ortholog structure (PDB 4IL3) was used as a representative query for structural ZMP homologs.
Use of the entire SmSte24 monomer (Figure 1A) as a search model for structurally homologous soluble gluzincins failed because the majority of the DALI‐identified structures recognized the TM α‐helices of SmSte24 as opposed to the region immediately surrounding the gluzincin motif. Therefore, a truncated SmSte24 “gluzincin‐only” search model, including the L5D, the CTD, and portions of TM α‐helices VI (residues 292‐301) and VII (residues 390‐408) was constructed and input to the DALI server (http://ekhidna2.biocenter.helsinki.fi/dali/). The query identified soluble homologs belonging to three gluzincin families: thermolysin (M4), neprilysin (M13), and an uncharacterized ZMP from Geobacter sulfurreducens (M48) (Figure 2A), whose active sites all possess homology with Ste24 (Figure 2B). Structural homology of Ste24 to M13 gluzincins has not been previously reported, while homology to M4 and M48 gluzincins has previously been established.33, 37 (The ZMP from G sulfurreducens is in the M48B subfamily, MA(E)/M48B, and is the only full‐length structure of a non‐M48A subfamily member.)
Figure 2.
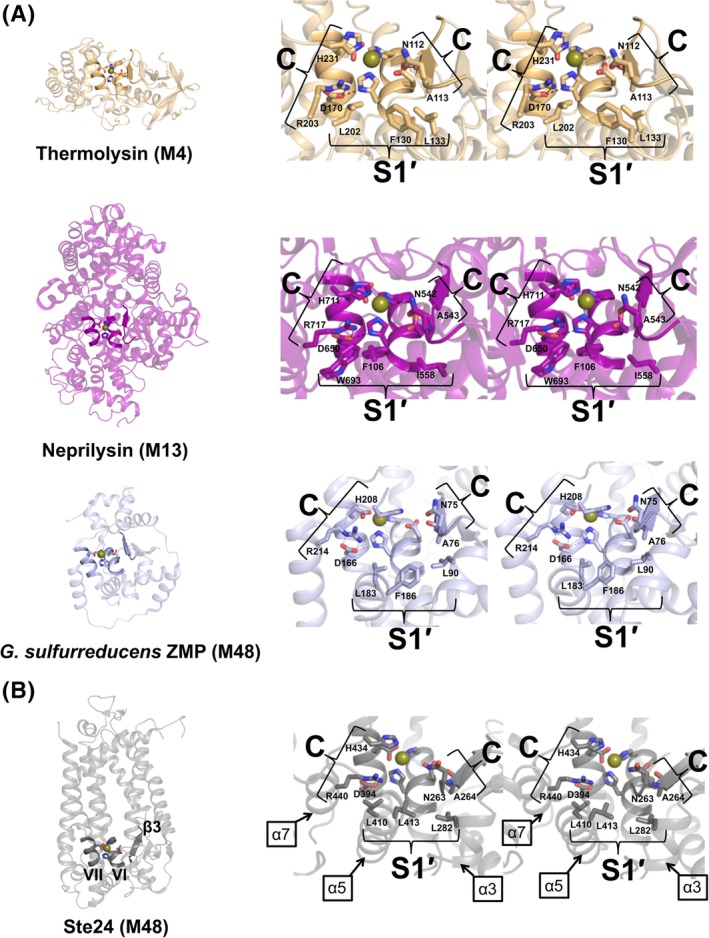
Identification of essential residues for Ste24 protease activity outside the HExxH…E/H gluzincin motif. A, Gluzincin ZMPs homologous to Ste24. The structures are sorted from least similar (top) to most similar (bottom) based on the Dali similarity or Z score between the Ste24 search model and the identified structures. Inspection of output from the Dali server42 revealed thermolysin (PDB 1LNF, Dali Z score 3.6), neprilysin (PDB 1DMT, Dali Z score 4.9), and the ZMP from G sulfurreducens (PDB 3C37, Dali Z score 6.3) as soluble, gluzincin structures most homologous to Ste24. The functional annotations for the selected gluzincins thermolysin (M4), neprilysin (M13), and the G sulfurreducens ZMP (M48) are based on MEROPS38 database assignments. Conserved α‐helical (containing the HExxE…E motif residues shown as sticks) and β‐strand elements are highlighted and represent a canonical gluzincin structure motif. Note the neprilysin and G sulfurreducens ZMP structures carry an extra domain situated above this motif. Closer inspection of each individual core region identifies residues (shown as sticks) essential for optimal substrate cleavage (indicated by C) and specificity for the side chain of the amino acid immediately C‐terminal to the scissile bond (indicated by S1′) where the prime (′) designation indicates enzyme specificity sites for amino acid side‐chain(s) C‐terminal to the scissile bond.65 ZMP residues participating in peptide bond cleavage (ie, catalytic) are absolutely conserved and positioned among the three gluzincins. ZMP residues involved in substrate recognition (ie, side‐chain specificity) are chemically conserved and positioned orthogonally to the catalytic ZMP residues forming a sandwich‐like motif. (B) Putative catalytic and specificity sites within Ste24. Ste24 TM α‐helices V, VI and the β3‐strand of the L5D form the equivalent canonical gluzincin structure motif found in the homologous gluzincin structures. As in the neprilysin and G sulfurreducens ZMP structures, Ste24 possesses an extra domain, in this case a transmembrane (TM) heptahelical “α‐barrel,” situated above the gluzincin motif. Investigation of this region in Ste24 implicates additional residues of the α3 element of the L5D and the α6/α7‐helices of the CTD as important for catalytic and specificity roles. Note that the four catalytic residues Asn263, Ala264, His434, and Arg440 (SmSte24 numbering) are absolutely conserved and identically positioned among all four gluzincins
Protease active‐site residues can be categorized into two interdependent functional classes: (a) residues interacting with peptide backbone atoms and/or promoting cleavage of the scissile bond (ie, catalytic “C” residues) or (b) residues responsible for recognizing specific peptide side‐chain identities or chemical properties (ie, specificity “S” residues) with those C‐terminal to the substrate scissile bond designated with the prime (′) symbol.52 Alignment of the active‐sites of the three soluble gluzincin homologs (Figure 2A, right‐hand panels) to SmSte24 (Figure 2B; right‐hand panel) reveals an arrangement of opposing C and S residues that is typical for a protease active site.52 The dependence of Ste24 activity on identified C residues (Asn263/Ala264 on the β3‐strand of L5D, and His434/Arg440 on the α7‐helix of CTD) has been previously documented (Figure 2B).33, 37 The S residues identified for Ste24 (Leu282 on α3‐helix of L5D and Leu410/Leu413 of α5‐helix of CTD) are predicted to form the S1′ surface that interacts with the substrate residue side chain immediately C‐terminal to the scissile bond (ie, the P1′ residue).53, 54 For all four structural homologs, S1′ forms a hydrophobic pocket (Figure 2). This observation is consistent with the nonpolar or hydrophobic character of the non‐CAAX box P1′ cleavage sites in a‐factor and prelamin A, for which Ste24 is essential.14, 55 in vitro cleavage of nonprenylated biologically derived peptides also occurs at nonpolar or hydrophobic sites.30
3.2. The tripartite architecture of Ste24
The position of the SmSte24 molecule relative to the membrane bilayer was determined via the Orientation of Membrane Protein server.56 The structural elements of the active site are external to or at the membrane interface (Figure 1A). Positioning of the Ste24 active site at the membrane surface, vs. within the bilayer interior, is consistent with identification of soluble, gluzincin ZMPs as Ste24 homologs. In contrast, the active sites of intramembrane ZMPs, such as Site‐2 Protease, are located within the most hydrophobic portion of the membrane interior.57 Comparison of Ste24 enzymes to homologous, soluble gluzincins indicates that residues essential for catalysis arise from TM α‐helices VI and VII, the L5D (β3‐strand and α3‐helix), and the CTD (α5‐ and α7‐helices) (Figure 2B). For the purposes of this study, we refer to this grouping as the “ZMP Core” module (Figure 3A). The designation of a “ZMP Core” module, shared by Ste24 with soluble ZMPs, is further supported by our recent structure of HsSte24 complexed with the classical ZMP inhibitor phosphoramidon (Figure 3B).35 Phosphoramidon is a competitive inhibitor and transition‐state analog of several soluble ZMPs, particularly the gluzincins thermolysin (M4)58 and neprilysin (M13).59 Phosphoramidon also inhibits HsSte24 proteolysis and its binding mode, localized solely within the “ZMP Core” module, is highly conserved among these three families (M4, M13, and M48) of ZMPs.35
Figure 3.
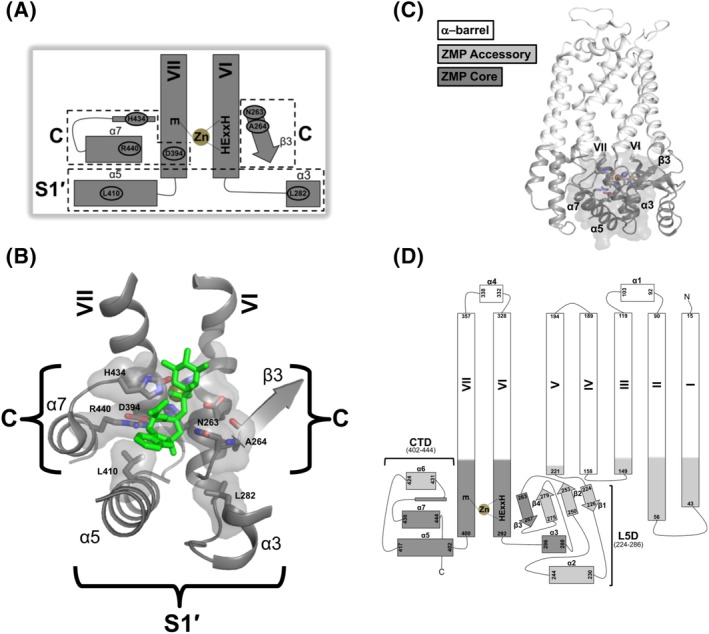
The Tripartite Architecture of Ste24. A, The “ZMP Core” module of Ste24. Functional and structural analyses of homologous, soluble gluzincin orthologs (see Figure 2A) reveal the complete ZMP Core module of Ste24 to be comprised of elements from the CTD (α5‐helix, α7‐helix, and α7‐loop), TM α‐helices (VI and VII), and L5D (β3‐strand and α3‐helix) (see Figure 2B). For clarity, the loop element leading to α7 within CTD is represented by a thin rectangle but does not possess α‐helical secondary structure. Catalytic and substrate specificity regions are indicated by C and S1′, respectively; SmSte24 residues which are absolutely conserved in Ste24 and within these regions are explicitly labeled and bracketed. B, Superposition of the unbound SmSte24 structure with our recently determined structure of HsSte24 complexed with the ZMP inhibitor phosphoramidon (PDB 6BH8).35 For clarity, only the phosphoramidon molecule (shown as green sticks) of the HsSte24:phosphoramidon structure is displayed. The three‐dimensional image of the ZMP Core module is oriented and labeled to match the schematic cartoon presented in panel (a). Residues contributing to the catalytic (C) and specificity (S1′) functions are explicitly labeled, shown as sticks, and their respective surfaces shown. The phosphoramidon inhibitor is bound and coordinated by residues solely within the ZMP Core module of Ste24, in a binding pose analogous to that observed in its complexes with soluble gluzincins.35 C, The Ste24 Tripartite Architecture. The “ZMP Core” module (dark gray ribbon and van der Waals surface) is shown in the three‐dimensional context of the entire Ste24 structure. Individual helix and strand elements constituting the ZMP core are labeled. Analogous to soluble gluzincins, the ZMP Core module is embedded between accessory structural elements (gray ribbon). Such “ZMP Accessory” modules are highly variable in size, scope, and functional role even among highly homologous gluzincins. The ZMP core region is “capped” by a large, membrane‐spanning region, the “α‐barrel” module, whose contribution to Ste24 catalytic activity is currently unknown (light gray ribbon). D, Ste24 topology diagram mapped with the Tripartite Architecture. The color‐coding scheme is maintained from panel C and labeling scheme from Figure 1C
Among soluble gluzincins, the ZMP Core module is only a portion of the entire structure (Figure 2A, left‐hand panel). Accessory structural elements are responsible for positioning the ZMP Core module. We refer to this grouping of elements as the “ZMP Accessory” module. In the case of Ste24 enzymes, the termini of TM α‐helices I–V and the remaining elements from the L5D and CTD buttress the ZMP Core module and form the ZMP Accessory module (Figure 1A,C). Additionally, the ZMP Core module may be further “capped” to restrict substrate access to the ZMP Core. The ZMP Core module of Ste24 is capped by the large, intramembranous cavity formed by TM α‐helices I–VII (Figure 2B), though the role for this cavity in Ste24 catalysis is currently unknown. In neprilysin (M13) and G sulfurreducens ZMP (M48), their ZMP Core modules are capped by extended (soluble) protein domains (Figure 2A). The ZMP Core module of thermolysin (M4) does not possess this structural “hat” (Figure 2A).
Based upon these observations, we propose a modular Tripartite Architecture for the Ste24 family of integral membrane protein ZMPs consisting of: (a) a largely cytoplasmic active‐site “ZMP Core” module, (b) a mixed soluble/membrane‐interfacial “ZMP Accessory” module (in close apposition to the ZMP Core module), and (c) a TM heptahelical “α‐barrel” module (of currently undetermined/unknown function) adjacent to the ZMP Core and ZMP Accessory modules (Figure 3C,D).
3.3. The Ste24 family MSA
Fifty‐eight eukaryotic sequences homologous to SmSte24 were identified by querying the NCBI nonredundant protein sequence database. Homologs were identified in multicellular and unicellular organisms (Table S1). The pair‐wise identity between SmSte24 and each ortholog was greater than 30%. A multiple sequence alignment (MSA) revealed 38 absolutely conserved residues (8% of the SmSte24 sequence), including the HExxH…E/H gluzincin motif (Figure S1). This set includes three progeria‐associated mutations of HsSte24 that map to SmSte24 (Pro246, Asn263, and Leu437).18, 21, 60
The ZMP Core, ZMP Accessory, and α‐barrel modules of SmSte24 contain 79, 135 and 247 residues, respectively (Figure 3D). The percentages of absolutely conserved residues in each module scale with our current level of understanding of the functions of these domains: 23%, 10% and 3%, respectively. Inspection of SmSte24 and HsSte24 structures alongside those of soluble, homologous gluzincins allows assignment of putative roles for these 38 absolutely conserved amino acid residues (Table 1), which are mapped onto the Tripartite Architecture model in topological and three‐dimensional representations (Figure 4 and Figure S2, respectively). Based on predicted catalytic (C) or substrate (S) binding roles in the ZMP Core domain, or structural roles of the ZMP Accessory domain, we have identified 17, absolutely conserved Ste24 residues that share conservation with all (or a subset) of the identified gluzincin homologs. None of these positions are located in the α‐barrel module (Figure 4 and Figure S2). The presence of the majority (31/38, ~82%) of absolutely conserved residues being in the Core ZMP and Accessory ZMP modules, and the ability of most of these (17/31, ~55%) to be rationalized by comparison to soluble gluzincins, is consistent with and supports our Tripartite Model modular assignments based on structural homology.
Table 1.
Tripartite Architecture module location and functional roles for absolutely conserved SmSte24 residues
| Module | Residue | Attribution |
|---|---|---|
| ZMP Core | His297 | Zinc Ligand |
| ZMP Core | His301 | Zinc Ligand |
| ZMP Core | Glu390 | Zinc Ligand |
| ZMP Core | Glu298 | Active site Base |
| ZMP Core | Asn263 | Catalytic |
| ZMP Core | Ala264 | Catalytic |
| ZMP Core | His434 | Catalytic |
| ZMP Core | Arg440 | Catalytic |
| ZMP Core | Leu282 | S1′ pocket |
| ZMP Core | Leu410 | S1′ pocket |
| ZMP Core | Asp394 | Second Sphere Ligand |
| ZMP Core | His306 | Second Sphere Ligand |
| ZMP Core | Gly300 | Structural: Core Only |
| ZMP Core | Pro435 | Structural: Core Only |
| ZMP Core | Asp280 | Structural: Accessory Interface |
| ZMP Core | Thr281 | Structural: Accessory Interface |
| ZMP Core | Arg387 | Structural: Accessory Interface |
| ZMP Core | Leu437 | Structural: Core & Accessory Interface |
| ZMP Accessory | Arg36 | Structural: Accessory Only |
| ZMP Accessory | Gln37 | Structural: Accessory Only |
| ZMP Accessory | Tyr65 | Structural: Accessory Only |
| ZMP Accessory | Gly152 | Structural: Accessory Only |
| ZMP Accessory | Asn154 | Structural: Accessory Only |
| ZMP Accessory | Asp424 | Structural: Accessory Only |
| ZMP Accessory | Phe145 | Membrane Interface |
| ZMP Accessory | Phe221 | Membrane Interface |
| ZMP Accessory | Pro47 | Structural: Core Interface |
| ZMP Accessory | Pro246 | Structural: Core Interface |
| ZMP Accessory | Ser256 | Structural: Core Interface |
| ZMP Accessory | Gly268 | Structural: Accessory & Core Interface |
| ZMP Accessory | Val277 | Structural: Accessory & Core Interface |
| ZMP Accessory | Pro219 | Structural: α‐barrel Only |
| α‐barrel | Pro138 | Structural: α‐barrel Only |
| α‐barrel | Pro373 | Structural: α‐barrel Only |
| α‐barrel | Tyr142 | Membrane Interfacing |
| α‐barrel | Asp164 | Structural: Accessory/α‐barrel Interface |
| α‐barrel | Trp97 | Structural: α‐barrel & Membrane Interface |
| α‐barrel | Phe340 | Structural: α‐barrel & Membrane Interface |
Note: A total of 38 absolutely conserved residues are assigned to their respective module (ZMP Core, ZMP Accessory, or α‐barrel) within the Tripartite Architecture representation. Those positions, whose functionality can be assigned via comparisons to structural and/or functional characterization of themolysin (M4), neprilysin (M13), and the G sulfurreducens ZMP (M48), or a subset of the three, are indicated with bolded font. Based on their uniqueness among Ste24 family enzymes, as opposed to those positions with overlap among well‐characterized, soluble ZMPs, several positions are simply labeled as “Structural” or “Membrane Interfacial” based solely on inspection of available Ste24 family crystal structures. In several instances, a residue participates in interfacial interactions between two specific modules: the ZMP Core and Accessory or ZMP Accessory and α‐barrel modules. None of the 38 residues form structural interactions between the ZMP Core and α‐barrel modules.
Figure 4.
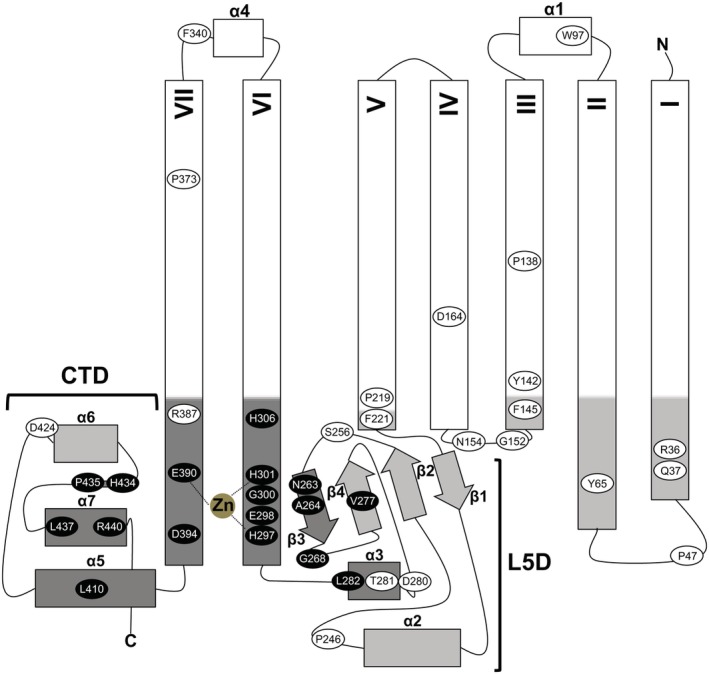
Absolutely conserved SmSte24 residues mapped to the Tripartitie Architecture of Ste24. The topology diagram “backbone” is gray scale shaded as in Figure 3D. Functional roles of 17 of the 38 residues can be assigned via comparisons to structural and/or functional characterization of themolysin (M4), neprilysin (M13), and the G sulfurreducens ZMP (M48) and are displayed as black oval with white font (also see Table 1). The remaining 21 residues, whose structural and functional homology to gluzincins is currently lacking or are unique to the Step24 family, are displayed as white ovals with black font
4. DISCUSSION
4.1. Bioinformatic and structural analyses yield a functionally insightful modular Tripartite Architecture of the Ste24 enzyme family
A structure‐based search was used to identify gluzincin ZMPs homologous to the Ste24 family. The search identified thermolysin (M4), neprilysin (M13), and the G sulfurreducens ZMP (M48). These results enabled us to develop a modular Tripartite Architecture model for Ste24 (Figure 3). We posit that one essential role of the ZMP Accessory and α‐barrel modules is to ensure that the ZMP Core is properly formed and situated to proteolyze substrates. These modules differentiate the Ste24 family from its soluble gluzincin counterparts, as the zinc‐centered active‐site sits at the membrane interface and requires molecular interactions from the extra‐membrane buttress (ie, ZMP Accessory) and membrane‐spanning, α‐barrel “hat” (ie, α‐barrel module) (Figures 1 and 3). As expected, structural homology to soluble gluzincins is scant in the ZMP Accessory module and completely absent in the α‐barrel module (Figure 4 and Table 1).
4.2. The ZMP Core module S1′ specificity pocket
Among M4 and M13 gluzincins, the S1′ specificity residues form a pocket that is the chief determinant of substrate specificity.61, 62, 63 In thermolysin (M4) and neprilysin (M13) X‐ray crystal structures, S1′ specificity pockets are composed of aromatic and hydrophobic residues favoring substrate P1′ side chains that possess complimentary hydrophobic character. Our analysis reveals an analogous hydrophobic pocket in structures of G sulfurreducens ZMP (M48) and SmSte24 (Figure 2). In Ste24, this pocket is composed of Leu282 and Leu410, both absolutely conserved, and Leu413, conserved in 57 of 58 orthologs (Figures 2 and 3, Table 1). Consistent with this observation, SmSte24 has been shown to have preference for cleaving nonprenylated substrates at the hydrophobic P1′ position.30
4.3. TM α‐helices VI & VII and the α6/α7 CTD loop contribute additional residues to the ZMP Core
The ZMP Core module contains 18 absolutely conserved residues of which 15 can be readily rationalized by direct comparison to all three gluzincin structural homologs of Ste24: thermolysin (M4), neprilysin (M13), and the G sulfurreducens ZMP (M48) (Figure 2 and Table 1).
Residues Gly300, His306, Arg387, Pro435, and Leu437 derive from four separate elements within the ZMP Core module. Residues His306 and Arg387 lie above the zinc center and derive from TM α‐helices VI (His306) and VII (Arg387), whereas Gly300, located in TM α‐helix VI, is positioned between the zinc ligands His297 and His301 (Figure 4). The Pro435 residue is in a short loop between helices α6 and α7, and is positioned approximately across from the zinc center in proximity to catalytic residue His434 (Figures 2B and 3B). Leu437 is situated at the N‐terminus of the α7‐helix, preceding the catalytic residue Arg440. Gly300, His306, Pro435, and Leu437 all facilitate the positioning of residues crucial for proteolysis (Table 1). Pro435 and Leu437 share structural analogy to neprilysin (M13) and G sulfurreducens ZMP (M48), whereas His306 shares analogy only with the G sulfurreducens ZMP (M48).
Gly300, with no structural analogies in thermolysin (M4), neprilysin (M13), or G sulfurreducens ZMP (M48), is in the zincin HExxH motif, preceding the C‐terminal histidine residue (Figure 4). The identity of the residues located between the catalytic glutamate base and histidine zinc ligand can be highly variable among gluzincins with the only restriction being that neither position is occupied by an amino acid with a charged side chain.40, 51 Absolute conservation of this glycine is not unique to the Ste24 family, however, as the M2 gluzincin family, which includes angiotensin‐converting enzyme (ACE), does contain glycine at the equivalent position.64
The His306 imidazole forms a second‐sphere hydrogen bond with the His301 imidazole, which itself is a direct ligand to the zinc. Among gluzincins, the identity of this second sphere ligand to the C‐terminal, HExxH histidine is often variable, but the presence of a second sphere ligand is maintained. This variation is in contrast to the other absolutely conserved second sphere ligand, an aspartate, identical in all four proteases and forming absolutely conserved hydrogen bond networks; in SmSte24, Asp394 interacts with the zinc ligand His297 and the catalytic residue Arg440 (Figure 2 and Table 1). Arg387 is the only MSA‐identified residue of TM α‐helix VII that has an interfacial structural role (Table 1) and, among the four proteases, is unique to Ste24. The catalytic His434 residue is located in the α6/α7‐loop. The Pro435 residue breaks the α7‐helix at its N‐terminus, and is the final residue in a loop connecting the α6‐ and α7‐helices (Figure 4). A similar proline‐containing structural motif is observed in neprilysin (M13) and G sulfurreducens ZMP (M48). The α7‐helix contains the catalytic Arg440 of Ste24.
The Leu437 side chain is part of a hydrophobic, leucine‐zipper motif between ZMP Core module helices α5 and α7 of the CTD. The use of a hydrophobic, leucine‐zipper like motif is also conserved in neprilysin (M13) and G sulfurreducens ZMP (M48) structures. Ste24 and neprilysin share an additional conserved feature in this region. In Ste24, a loop element between TM α‐helices I and II contributes further hydrophobic residues near the α5/α7 interface, with the absolutely conserved Pro47 residing within this loop; in neprilysin (M13), a perpendicular α‐helical element contributes residues fulfilling the same role.
4.4. Residues in the ZMP Core and ZMP Accessory modules coordinate the L5D β3‐strand and surrounding turns
A β‐strand positioned on a side of the zinc‐centered active‐site is a common structural feature among ZMPs and donates catalytically essential residues (both side‐chain and main‐chain moieties) for proteolysis39, 50 (Figure 2). In Ste24, the L5D β3‐strand of the ZMP Core module fulfills this role, and is “catalytically aligned” by the ZMP Core module (residues of the α3‐helix of the L5D, Figure 3C,D) and by the ZMP Accessory module (residues of the β3/4‐turn and β4‐strand of the L5D β‐sheet, TM α‐helix III/IV loop, and the TM α‐helix II (Figure 3D).
The β2/β3‐turn of the L5D is a nexus of coordinated molecular interfaces from multiple structural elements; interacting absolutely conserved residues include Asp280 and Thr281 (α3‐helix of the L5D) of the ZMP Core module, and Tyr65 (TM α‐helix II), Gly152 and Asn154 (TM α‐helix III/IV loop) of the ZMP Accessory module (Figure 4). Residues Asp280 and Thr281, in the L5D α3‐helix of the ZMP Core module, form hydrogen bonds with side‐chain and main‐chain residues of the β2/β3‐turn, respectively. Asp280 forms a hydrogen bond with the Ser256 hydroxyl‐bearing side chain. His261, conserved in 56 of 58 orthologs (Figure S1), forms two hydrogen bonds with absolutely conserved Ste24 residues: via its backbone carbonyl to Thr281 (ZMP Core Module) and via its sidechain to Tyr65 (ZMP Accessory module). (The remaining two orthologs have a glutamate at this position, suggesting that a hydrogen bond at this location may be conserved in the Ste24 family [Figure S1]). The TM α‐helix III/IV loop contacts the β2/β3‐turn, forming a short β‐sheet with hydrogen bonds occurring between the two elements. Residues Gly152 and Asn154 are located in the loop and turn elements, and appear to facilitate their positioning. The Gly152 residue sits at the terminus of the TM α‐helix III, while the Asn154 side‐chain amide forms a hydrogen bond with the carbonyl of the TM α‐helix IV terminus residue Thr157, conserved in 56 of 58 orthologs (Figure S1).
The β3/β4‐turn and β4‐strand of the L5D present the other significant interface to the ZMP Core β3‐strand. Among MSA‐identified residues, Gly268 preempts the β3/β4‐turn, and the Val277 side chain forms the base of a hydrophobic pocket localized approximately at the center of the β4‐strand and surrounded by aromatic and hydrophobic residues from the neighboring β3‐ and β2‐strands of the L5D β‐sheet. Furthermore, the presence of an analogous glycine and valine‐centered hydrophobic interface is also present on equivalent elements of thermolysin (M4), neprilysin (M13), and the G sulfurreducens ZMP (M48) structures, suggesting a common structural motif among these closely related gluzincins.
4.5. ZMP Accessory module residues from TM α‐helix I and the CTD α6‐helix complete a unique Ste24 family structural motif
A significant portion of the Ste24 enzyme family ZMP Core module is formed by the CTD, including the antiparallel α5‐ and α7‐helices and the α6/α7‐loop (Figures 3 and 4). The α6‐helix of the CTD, associated with the ZMP Accessory module, is positioned at the membrane interface, approximately perpendicular to the α5‐ and α7‐helices of the ZMP Core module. Residues of the ZMP Core and Accessory modules form structural interactions with the N‐ and C‐termini of this α‐helix, respectively. The C‐terminal end of the α6‐helix forms a salt‐bridge with Arg387, in a portion of TM α‐helix VII within the ZMP Core module. The N‐terminus, via Asp424, forms a second salt‐bridge with Arg36 of the neighboring TM α‐helix I of the ZMP Accessory module. The Gln37 residue of the TM α‐helix I is optimally positioned to form hydrogen bond interactions with the peptide backbones of residues forming the α5/α6‐loop. The structures of SmSte24 and HsSte24 contain large gaps (“fenestrations”) between TM α‐helices, and these fenestrations were posited to function as entry (TM α‐helices V/VI)33, 37 and exit (TM α‐helices I/VII)33 portals to the active site, as it is encircled by TM α‐helices and located within the membrane interior (Figure 1A).
4.6. The Ste24 α‐barrel module
A TM heptahelical “α‐barrel” module, absent in soluble gluzincins and unique to the Ste24 enzyme family, contacts the ZMP Core and ZMP Accessory modules (Figures 3C and 4). The α‐barrel module of Ste24, composed of amphipathic α‐helices, forms a voluminous cavity (~14 000 Å3 in SmSte2433) within the membrane interior. The vast majority of TM α‐helices in integral membrane proteins are hydrophobic; therefore, use of the SmSte24 α‐barrel module in a Dali search yielded both soluble and membrane proteins with structural similarity. The highest‐scoring result (Z score of 17.3, with the next highest of Z score less than six) was HsSte24, reinforcing the complete structural novelty of the α‐barrel module. Of the top 100 “hits,” approximately two thirds were to soluble protein complexes, reflecting the amphipathic nature of the helices in the α‐barrel search model. Absolutely conserved Ste24 residues Trp97, Pro138, Tyr142, Asp164, Pro219, Phe340, and Pro373 reside in the α‐barrel module (Figure 4). Of these seven residues, Pro138, Asp164, and Pro373 are located within the membrane. Trp97 and Phe340 are predicted to be located at the membrane interface opposite to that of the ZMP Core and Accessory modules, while Tyr142 and Pro219 share the same membrane boundary as the ZMP Core and Accessory modules (Table 1).
Residues Tyr142 and Asp164 participate in significant membrane‐interfacial interactions with the ZMP Accessory module. Tyr142 and Phe145, both located within TM α‐helix III, are positioned at the membrane interface between the α‐barrel and ZMP Accessory modules, respectively (Table 1). The Asp164 residue forms an interfacial salt bridge with an Arg258 located at the β2/β3‐turn of the L5D in the ZMP Accessory module. The three absolutely conserved prolines of the α‐barrel module are all located within TM α‐helices. Pro138 introduces a small kink in TM α‐helix III; its location approximately one turn away from the membrane interfacial residue Tyr142 may mitigate its effect upon proteolytic function. In contrast, Pro373 introduces a large kink and realignment (~90° with respect to the membrane normal) of TM α‐helix VII, which contains the zinc ligand Glu390 and second‐sphere ligand Asp394. The site of this kink is ~30 Å from the zinc‐center. The Pro219 residue is positioned near the terminus of TM α‐helix V and is a half turn away from the ZMP Accessory module residue Phe221, which is predicted to rest at the membrane. Residues Trp97 and Phe340 are furthest removed from the zinc center, and are positioned on the opposing membrane leaflet. Trp97 is located in the α1‐helix and rests at the center of an interfacial, hydrophobic pocket formed from residues of TM α‐helices III and VII, and Phe340 of helix α4. Although functions of absolutely conserved residues “distal” to the Ste24 active site are currently unknown, recent studies implicating HsSte24 as a “translocon unclogger” indicate the presence of functional “hot spots” far from the active site.28, 29 Roles for absolutely conserved residues of the α‐barrel module in essential “nonenzymatic” functions may also exist; these could include residues essential for biogenesis or protein stability. Also, residues conserved more narrowly, within animals rather than also in fungi, plants and protists (Table S1), may provide insight into the ability of HsSte24 to function (nonenzymatically) as a broad spectrum viral restriction factor.31, 32
5. CONCLUSION
A detailed bioinformatic and structural analysis of the Ste24 family of ZMPs, in the context of well‐characterized soluble gluzincin orthologs, enabled development of the Tripartite Architecture representation of this integral membrane protein ZMP. The ZMP Core and Accessory modules are capped by a membrane‐spanning α‐barrel module, so named because of our current lack of understanding of its functional role. In light of the ever‐growing literature of Ste24 family orthologs and emergent clinically relevant roles of HsSte24,28, 29, 31 the Tripartite Architecture will aid in the interpretation of disparate functional data. Furthermore, our proposed Tripartite Architecture can act as a platform to design experiments to better understand both the functional overlap and individual uniqueness of Ste24 family enzymes among different family orthologs (eg, fungal/mammalian/plant/protist). Additionally, the Tripartite Architecture will be of utility in the overall context of ZMPs, and may also be applicable to other integral membrane protein enzymes whose active sites reside at a membrane interface.
Supporting information
Supplementary Figure S1 Multiple sequence alignment (MSA) of 58 orthologous Ste24 sequences. Sequence origins range across fungi, animal, plant, and protist. A red box highlights absolutely conserved residues. Black arrows indicate the residues compromising the conserved gluzincin “HExxH…E” motif. The locations of the progeria inducing mutations in the human ortholog of Ste24, HsSte24, are indicated by an asterisk (*)
Supplementary Figure S2 Absolutely conserved SmSte24 residues mapped onto the three‐dimensional SmSte24 structure (PDB 4IL3).33 Gray‐scaled shading and surface coloring for the Tripartite Architecture is identical to that of Figure 3C. The coloring scheme for the 38 Ste24 conserved residues is identical to that of Figure 4. For clarity, the gluzincin 297HExxH301…E390 motif residues surrounding the zinc atom (gold sphere) are shown as sticks. The locations of the remaining point mutant positions are indicated as spheres centered at the position of their alpha carbon
Supplementary Table S1 Summary of identified Ste24 family sequences used in this study. Sequences are grouped by kingdom (fungi, animal, plant, and protist), and ordered within each kingdom from highest to lowest percent identity as compared to the wild‐type SmSte24 query sequence. All identified sequences in this table are utilized in the multiple sequence alignment (MSA) presented in Figure S1
ACKNOWLEDGMENTS
We thank Dr. Walter Schmidt (University of Georgia) for his comments on this manuscript prior to submission. This research was supported by the National Institutes of Health (Grants R01GM108612 and R56AI141627 to M.C.W.).
Goblirsch BR, Pryor EE Jr, Wiener MC. The tripartite architecture of the eukaryotic integral membrane protein zinc metalloprotease Ste24. Proteins. 2020;88:604–615. 10.1002/prot.25841
Present addressEdward E. Pryor Jr, Anatrace Products, LLC, Maumee, OH 43537.
Peer Review The peer review history for this article is available at https://publons.com/publon/10.1002/prot.25841.
Funding information National Institutes of Health, Grant/Award Number: R01GM108612; National Institutes of Health, Grant/Award Number: R56AI141627
REFERENCES
- 1. Lopez‐Otin C, Bond JS. Proteases: multifunctional enzymes in life and disease. J Biol Chem. 2008;283(45):30433‐30437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ordonez GR, Puente XS, Quesada V, Lopez‐Otin C. Proteolytic systems: constructing degradomes. Methods Mol Biol. 2009;539:33‐47. [DOI] [PubMed] [Google Scholar]
- 3. Avci D, Lemberg MK. Clipping or extracting: two ways to membrane protein degradation. Trends Cell Biol. 2015;25(10):611‐622. [DOI] [PubMed] [Google Scholar]
- 4. Ossovskaya VS, Bunnett NW. Protease‐activated receptors: contribution to physiology and disease. Physiol Rev. 2004;84(2):579‐621. [DOI] [PubMed] [Google Scholar]
- 5. Verhelst SHL. Intramembrane proteases as drug targets. FEBS J. 2017;284(10):1489‐1502. [DOI] [PubMed] [Google Scholar]
- 6. Michaelis S, Barrowman J. Biogenesis of the Saccharomyces cerevisiae pheromone a‐factor, from yeast mating to human disease. Microbiol Mol Biol Rev. 2012;76(3):626‐651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boyartchuk VL, Ashby MN, Rine J. Modulation of Ras and a‐factor function by carboxyl‐terminal proteolysis. Science. 1997;275(5307):1796‐1800. [DOI] [PubMed] [Google Scholar]
- 8. Fujimura‐Kamada K, Nouvet FJ, Michaelis S. A novel membrane‐associated metalloprotease, Ste24p, is required for the first step of NH2‐terminal processing of the yeast a‐factor precursor. J Cell Biol. 1997;136(2):271‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tam A, Nouvet FJ, Fujimura‐Kamada K, Slunt H, Sisodia SS, Michaelis S. Dual roles for Ste24p in yeast a‐factor maturation: NH2‐terminal proteolysis and COOH‐terminal CAAX processing. J Cell Biol. 1998;142(3):635‐649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Trueblood CE, Boyartchuk VL, Picologlou EA, Rozema D, Poulter CD, Rine J. The CaaX proteases, Afc1p and Rce1p, have overlapping but distinct substrate specificities. Mol Cell Biol. 2000;20(12):4381‐4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Adames N, Blundell K, Ashby MN, Boone C. Role of yeast insulin‐degrading enzyme homologs in propheromone processing and bud site selection. Science. 1995;270(5235):464‐467. [DOI] [PubMed] [Google Scholar]
- 12. Barrowman J, Hamblet C, George CM, Michaelis S. Analysis of prelamin a biogenesis reveals the nucleus to be a CaaX processing compartment. Mol Biol Cell. 2008;19(12):5398‐5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hennekes H, Nigg EA. The role of isoprenylation in membrane attachment of nuclear lamins. A single point mutation prevents proteolytic cleavage of the Lamin a precursor and confers membrane binding properties. J Cell Sci. 1994;107(Pt 4):1019‐1029. [DOI] [PubMed] [Google Scholar]
- 14. Kilic F, Dalton MB, Burrell SK, Mayer JP, Patterson SD, Sinensky M. In vitro assay and characterization of the farnesylation‐dependent prelamin a endoprotease. J Biol Chem. 1997;272(8):5298‐5304. [DOI] [PubMed] [Google Scholar]
- 15. Dittmer TA, Misteli T. The Lamin protein family. Genome Biol. 2011;12(5):222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gruenbaum Y, Medalia O. Lamins: the structure and protein complexes. Curr Opin Cell Biol. 2015;32:7‐12. [DOI] [PubMed] [Google Scholar]
- 17. Davies BS, Fong LG, Yang SH, Coffinier C, Young SG. The posttranslational processing of prelamin a and disease. Annu Rev Genomics Hum Genet. 2009;10:153‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barrowman J, Michaelis S. ZMPSTE24, an integral membrane zinc metalloprotease with a connection to progeroid disorders. Biol Chem. 2009;390(8):761‐773. [DOI] [PubMed] [Google Scholar]
- 19. Corrigan DP, Kuszczak D, Rusinol AE, et al. Prelamin a endoproteolytic processing in vitro by recombinant Zmpste24. Biochem J. 2005;387(Pt 1):129‐138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Young SG, Fong LG, Michaelis S, Prelamin A. Zmpste24, misshapen cell nuclei, and progeria—new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J Lipid Res. 2005;46(12):2531‐2558. [DOI] [PubMed] [Google Scholar]
- 21. Barrowman J, Wiley PA, Hudon‐Miller SE, Hrycyna CA, Michaelis S. Human ZMPSTE24 disease mutations: residual proteolytic activity correlates with disease severity. Hum Mol Genet. 2012;21(18):4084‐4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Denecke J, Brune T, Feldhaus T, et al. A homozygous ZMPSTE24 null mutation in combination with a heterozygous mutation in the LMNA gene causes Hutchinson‐Gilford progeria syndrome (HGPS): insights into the pathophysiology of HGPS. Hum Mutat. 2006;27(6):524‐531. [DOI] [PubMed] [Google Scholar]
- 23. Clark KM, Jenkins JL, Fedoriw N, Dumont ME. Human CaaX protease ZMPSTE24 expressed in yeast: structure and inhibition by HIV protease inhibitors. Protein Sci. 2017;26(2):242‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Coffinier C, Hudon SE, Farber EA, et al. HIV protease inhibitors block the zinc metalloproteinase ZMPSTE24 and lead to an accumulation of prelamin a in cells. Proc Natl Acad Sci U S A. 2007;104(33):13432‐13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Caron M, Auclair M, Donadille B, et al. Human lipodystrophies linked to mutations in A‐type lamins and to HIV protease inhibitor therapy are both associated with prelamin a accumulation, oxidative stress and premature cellular senescence. Cell Death Differ. 2007;14(10):1759‐1767. [DOI] [PubMed] [Google Scholar]
- 26. Casasola A, Scalzo D, Nandakumar V, et al. Prelamin a processing, accumulation and distribution in normal cells and laminopathy disorders. Nucleus. 2016;7(1):84‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Coffinier C, Hudon SE, Lee R, et al. A potent HIV protease inhibitor, darunavir, does not inhibit ZMPSTE24 or lead to an accumulation of farnesyl‐prelamin a in cells. J Biol Chem. 2008;283(15):9797‐9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ast T, Michaelis S, Schuldiner M. The protease Ste24 clears clogged Translocons. Cell. 2016;164(1–2):103‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kayatekin C, Amasino A, Gaglia G, et al. Translocon Declogger Ste24 protects against IAPP oligomer‐induced Proteotoxicity. Cell. 2018;173:62‐73.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hildebrandt ER, Arachea BT, Wiener MC, Schmidt WK. Ste24p mediates proteolysis of both isoprenylated and non‐prenylated oligopeptides. J Biol Chem. 2016;291:14185‐14198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fu B, Wang L, Li S, Dorf ME. ZMPSTE24 defends against influenza and other pathogenic viruses. J Exp Med. 2017;214(4):919‐929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li S, Fu B, Wang L, Dorf ME. ZMPSTE24 is downstream effector of interferon‐induced transmembrane antiviral activity. DNA Cell Biol. 2017;36(7):513‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pryor EE Jr, Horanyi PS, Clark KM, et al. Structure of the integral membrane protein CAAX protease Ste24p. Science. 2013;339(6127):1600‐1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Arachea BT, Wiener MC. Acquisition of accurate data from intramolecular quenched fluorescence protease assays. Anal Biochem. 2017;522:30‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Goblirsch BR, Arachea BT, Councell DJ, Wiener MC. Phosphoramidon inhibits the integral membrane protein zinc metalloprotease ZMPSTE24. Acta Crystallogr D Struct Biol. 2018;74(8):739‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mehmood S, Marcoux J, Gault J, et al. Mass spectrometry captures off‐target drug binding and provides mechanistic insights into the human metalloprotease ZMPSTE24. Nat Chem. 2016;8(12):1152‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Quigley A, Dong YY, Pike AC, et al. The structural basis of ZMPSTE24‐dependent laminopathies. Science. 2013;339(6127):1604‐1607. [DOI] [PubMed] [Google Scholar]
- 38. Rawlings ND, Waller M, Barrett AJ, Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014;42(Database issue):D503‐D509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cerda‐Costa N, Gomis‐Ruth FX. Architecture and function of metallopeptidase catalytic domains. Protein Sci. 2014;23(2):123‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hooper NM. Families of zinc metalloproteases. FEBS Lett. 1994;354(1):1‐6. [DOI] [PubMed] [Google Scholar]
- 41. Lopez‐Pelegrin M, Cerda‐Costa N, Martinez‐Jimenez F, et al. A novel family of soluble minimal scaffolds provides structural insight into the catalytic domains of integral membrane metallopeptidases. J Biol Chem. 2013;288(29):21279‐21294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38(Web Server issue):W545‐W549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schrodinger LLC . The AxPyMOL Molecular Graphics Plugin for Microsoft PowerPoint, Version 1.8. 2015.
- 44. Boratyn GM, Camacho C, Cooper PS, et al. BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 2013;41(Web Server issue):W29‐W33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36(Web Server):W5‐W9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal omega. Mol Syst Biol. 2011;7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li W, Cowley A, Uludag M, et al. The EMBL‐EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 2015;43(W1):W580‐W584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McWilliam H, Li W, Uludag M, et al. Analysis tool web services from the EMBL‐EBI. Nucleic Acids Res. 2013;41(Web Server issue):W597‐W600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42(Web Server issue):W320‐W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gomis‐Ruth FX, Botelho TO, Bode W. A standard orientation for metallopeptidases. Biochim Biophys Acta. 2012;1824(1):157‐163. [DOI] [PubMed] [Google Scholar]
- 51. Jongeneel CV, Bouvier J, Bairoch A. A unique signature identifies a family of zinc‐dependent metallopeptidases. FEBS Lett. 1989;242(2):211‐214. [DOI] [PubMed] [Google Scholar]
- 52. Schechter I, Berger A. On the active site of proteases. 3. Mapping the active site of papain; specific peptide inhibitors of papain. Biochem Biophys Res Commun. 1968;32(5):898‐902. [DOI] [PubMed] [Google Scholar]
- 53. de Kreij A, Venema G, van den Burg B. Substrate specificity in the highly heterogeneous M4 peptidase family is determined by a small subset of amino acids. J Biol Chem. 2000;275(40):31115‐31120. [DOI] [PubMed] [Google Scholar]
- 54. Oefner C, Roques BP, Fournie‐Zaluski MC, Dale GE. Structural analysis of neprilysin with various specific and potent inhibitors. Acta Crystallogr D. 2004;60:392‐396. [DOI] [PubMed] [Google Scholar]
- 55. Schmidt WK, Tam A, Michaelis S. Reconstitution of the Ste24p‐dependent N‐terminal proteolytic step in yeast a‐factor biogenesis. J Biol Chem. 2000;275(9):6227‐6233. [DOI] [PubMed] [Google Scholar]
- 56. Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 2012;40(Database issue):D370‐D376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Feng L, Yan H, Wu Z, et al. Structure of a site‐2 protease family intramembrane metalloprotease. Science. 2007;318(5856):1608‐1612. [DOI] [PubMed] [Google Scholar]
- 58. Suda H, Aoyagi T, Takeuchi T, Umezawa H. Letter: a thermolysin inhibitor produced by Actinomycetes: phospholamidon. J Antibiot. 1973;26(10):621‐623. [DOI] [PubMed] [Google Scholar]
- 59. Rose C, Voisin S, Gros C, Schwartz JC, Ouimet T. Cell‐specific activity of neprilysin 2 isoforms and enzymic specificity compared with neprilysin. Biochem J. 2002;363(Pt 3):697‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Spear ED, Hsu ET, Nie L, Carpenter EP, Hrycyna CA, Michaelis S. ZMPSTE24 missense mutations that cause progeroid diseases decrease prelamin a cleavage activity and/or protein stability. Dis Model Mech. 2018;11(7):ddm033670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Oda K, Takahashi T, Takada K, et al. Exploring the subsite‐structure of vimelysin and thermolysin using FRETS‐libraries. FEBS Lett. 2005;579(22):5013‐5018. [DOI] [PubMed] [Google Scholar]
- 62. Rawlings ND, Barrett AJ. Evolutionary families of metallopeptidases. Methods Enzymol. 1995;248:183‐228. [DOI] [PubMed] [Google Scholar]
- 63. Roques BP, Noble F, Dauge V, Fournie‐Zaluski MC, Beaumont A. Neutral endopeptidase 24.11: structure, inhibition, and experimental and clinical pharmacology. Pharmacol Rev. 1993;45(1):87‐146. [PubMed] [Google Scholar]
- 64. Acharya KR, Sturrock ED, Riordan JF, Ehlers MR. Ace revisited: a new target for structure‐based drug design. Nat Rev Drug Discov. 2003;2(11):891‐902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun. 1967;27(2):157‐162. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1 Multiple sequence alignment (MSA) of 58 orthologous Ste24 sequences. Sequence origins range across fungi, animal, plant, and protist. A red box highlights absolutely conserved residues. Black arrows indicate the residues compromising the conserved gluzincin “HExxH…E” motif. The locations of the progeria inducing mutations in the human ortholog of Ste24, HsSte24, are indicated by an asterisk (*)
Supplementary Figure S2 Absolutely conserved SmSte24 residues mapped onto the three‐dimensional SmSte24 structure (PDB 4IL3).33 Gray‐scaled shading and surface coloring for the Tripartite Architecture is identical to that of Figure 3C. The coloring scheme for the 38 Ste24 conserved residues is identical to that of Figure 4. For clarity, the gluzincin 297HExxH301…E390 motif residues surrounding the zinc atom (gold sphere) are shown as sticks. The locations of the remaining point mutant positions are indicated as spheres centered at the position of their alpha carbon
Supplementary Table S1 Summary of identified Ste24 family sequences used in this study. Sequences are grouped by kingdom (fungi, animal, plant, and protist), and ordered within each kingdom from highest to lowest percent identity as compared to the wild‐type SmSte24 query sequence. All identified sequences in this table are utilized in the multiple sequence alignment (MSA) presented in Figure S1