

Abstract
Fragile X syndrome (FXS) is a very frequent cause of inherited intellectual disability (ID) and autism. Most FXS patients have an expansion over 200 repeats of (CGG)n sequence (“full mutation” (FM)) located in the 5′UTR of the FMR1 gene, resulting in local DNA methylation (methylated “full mutation” (MFM)) and epigenetic silencing. The absence of the FMRP protein is responsible for the clinical phenotype of FXS. FM arises from a smaller maternal allele with 56–200 CGG repeats (“premutation” (PM)) during maternal meiosis. Carriers of PM alleles, which are typically unmethylated, can manifest other clinical features (primary ovarian insufficiency (POI) or FXS-associated tremor-ataxia syndrome (FXTAS)), known as fragile X-related disorders. In FXS families, rare males who have inherited an unmethylated “full mutation” (UFM) have been described. These individuals produce enough FMRP to allow normal intellectual functioning. Here we report the rare case of three sisters with a completely methylated PM of around 140 CGGs and detail their neuropsychological function. X inactivation analysis confirmed that the three sisters have a random inactivation of the X chromosome, suggesting that the PM allele is always methylated also when residing on the active X. We propose that in exceptional cases, just as the FM may be unmethylated, also a PM allele may be fully methylated. To our knowledge, females with a methylated PM allele and a mild impairment have reported only once. The study of these atypical individuals demonstrates that the size of the CGG expansion is not as tightly coupled to methylation as previously thought.
Subject terms: Disease genetics, DNA methylation
Introduction
Fragile X syndrome (FXS, OMIM #300624) is the leading cause of inherited intellectual disability (ID) and autism with an estimated prevalence of 1 in 7000 males and 1:11,000 females [1], the highest within the group of X-linked ID conditions [2]. The syndrome is almost exclusively caused by the expansion of a CGG trinucleotide repeat in the 5′UTR of the FMR1 gene beyond 200 units (“full mutation” (FM); structural variation), followed by methylation of cytosines in the repeat itself and in the CpG island upstream of the gene’s promoter (methylated “full mutation” (MFM); epigenetic modification) [3]. The epigenetic changes (DNA methylation and histone modifications) eventually block transcription, preventing the production of the FMRP protein (loss-of-function effect), even though the coding sequence of the FMR1 gene remains intact. FMRP is an RNA-binding protein that associates with polyribosomes, playing an important role in neuronal protein synthesis and RNA metabolism [4], and its absence causes the clinical phenotype of FXS.
FM arises from a smaller maternal allele with 56–200 CGG repeats known as “premutation” (PM) during maternal meiosis. PM alleles are typically unmethylated, and carriers do not have FXS but may present with other clinical features such as primary ovarian insufficiency (FXPOI; OMIM #311360) in females or FXS-associated tremor-ataxia syndrome (FXTAS; OMIM #300623) in both males and females. These conditions are also known as FMR1-related disorders or FRAXopathies [5, 6]. While FXS results from the FMR1 loss-of-function, FXPOI and FXTAS are caused by the gain-of-function effect of the PM, that is typically unmethylated and transcriptionally active [7]. PM alleles (particularly those with 100–200 CGGs) show increased levels of transcription and result in somewhat reduced FMRP levels [8]. The overall picture is further complicated by the existence of rare unmethylated “full mutation” (UFM) alleles, identified in males with apparently normal intelligence belonging to FXS families [9–11]. These individuals carry expansions with more than 200 CGG repeats, completely devoid of cytosine methylation. These rare alleles likely represent the status of FXS cells before “full mutations” are epigenetically silenced, at around 11 weeks of gestation [12]. Despite the great amount of data accumulated since the discovery of the FMR1 gene in 1991, there is still limited knowledge of the events that lead to CGG instability and to FMR1 silencing. Expansion and epigenetic silencing coexist in the same locus, but appear to be determined by different mechanisms as testified by the occurrence of UFM males.
PM alleles show small expansions through paternal transmission and male carriers of PM are not considered at risk of transmitting an FM allele to their children [13], while large expansions into FM range seem to be transmitted exclusively through maternal meiosis [14]. The number of AGG interruptions within the CGG repeat tract represents the main risk factor for instability. Unstable alleles probably arise from stable ones after the loss of AGG interruptions [15]. In small maternal PM alleles (with ~70–90 CGGs) the presence of two interspersed AGGs associates with a 60% risk reduction of transmitting an FM to the next generation [16]. Contractions, on the other hand, do not seem to be influenced by the presence of AGG interruptions [17]. It has been proposed that the expansion mechanism depends on the interaction between components of the base excision repair, mismatch repair, and transcription-coupled repair pathways [18]. Secondary structures, which form on DNA strands of the CGG repeat, are the likely substrates upon which the expansion process acts [19]. Recent works suggest that secondary structures formed by the expanded (CGG)n sequence also play a crucial role in the epigenetic silencing. DNA–RNA hybrids (or R-loops) form during transcription of the CGG repeat sequence [20], and bidirectional transcription of the FMR1 locus may induce the formation of double R-loops [21]. According to this model, the FMR1 mRNA hybridizes to the complementary CGG repeat of the FMR1 gene, forming an RNA:DNA duplex [22]. Disrupting the interaction of the mRNA with the CGG repeat seems to prevent silencing of the promoter, supporting a model of RNA-directed gene silencing. However, it should be noted that this mechanism does not explain the existence of UFM individuals, who preserve FMR1 transcription in the presence of the CGG expansion. Different mechanisms, one associated with DNA repair and the other with epigenetic machinery, acting on the secondary structures formed by the CGG tract, may contribute to expansion and silencing of the FMR1 gene.
Here we describe three sisters, carriers of completely methylated PM alleles of ~140 CGGs. The X-chromosome inactivation (XCI) analysis, performed through the study of the methylation-sensitive androgen receptor (AR) locus, was found to be nearly random in all three sisters, demonstrating that the methylation of the PM allele was not limited to the inactive X chromosome. This peculiar status of the FMR1 locus, never described previously, correlated with slight intellectual impairment, as measured through neuropsychological evaluation. Finally, segregation analysis of flanking FMR1 markers indicated that this PM was stably inherited from their father. Overall, these findings further confirmed that CGG expansion and epigenetic silencing result from the action of different mechanisms.
Patients and methods
Patients
Participants provided written informed consent for clinical and molecular analysis performed in this study. The study protocol was approved by the Ethic Committee of the Catholic University of Rome (prot. N. 9917/15 and prot.cm 10/15). The pedigree is reported in Fig. 1, in which all family members evaluated for neuropsychological function, molecular analysis, and segregation study are identified. Neuropsychological evaluation consisted of IQ measure through WAIS-R (Wechsler Adult Intelligence Scale-Revised), which allowed us to calculate performance IQ, verbal IQ, and global IQ. Four female (II-3, II-4, II-6, and III-6) carriers of expanded FMR1 alleles were evaluated through cognitive testing with standardized IQ measure. Test scoring was performed according to [23], classifying an IQ score ≥130 as clearly superior, 120–129 superior, 110–109 high-average, 90–109 average, 80–89 low-average, 70–79 borderline, and ≤69 as ID.
Fig. 1.

Pedigree of the family and haplotype segregation within the FMR1 locus. Black square (IV-4) or circles (III-6 and III-9) indicate carriers of MFM. The gray square (III-1) indicates a relative with ID of unknown origin. Individuals tested for the FMR1 expansion are indicated by a dash above their symbol, while the boxed haplotypes refer to the X chromosome with an expanded CGG (PM/FM). On the left side the polymorphic markers (CGG, DXS548, and FRAXAC1) are reported. Asterisks indicate females who were tested for XCI and evaluated with neuropsychological tests
Southern blot
Blood was drawn from various family members, as indicated in the pedigree (Fig. 1). Genomic DNA was isolated from 10 ml of peripheral blood leukocytes using the salting-out protocol and 10 μg of DNA was digested with HindIII and/or the methylation-sensitive enzyme EagI (New England Biolabs). Digested DNA and a size marker were run on a 0.8% agarose gel with 1× TAE buffer, blotted on Amersham Hybond N + nylon membrane, and hybridized with radioactive Ox1.9 probe [24]. After overnight hybridization and subsequent washing, radioactive filters were exposed to films at −80 °C with reinforcing screens before development.
CGG sizing
CGG sizing was assessed by the AmplideX®PCR assay (Asuragen, Austin, TX, USA), a sensitive and efficient amplification method [25]. This protocol uses a three-primer CGG repeat primed PCR and fragment sizing on a Genetic Analyzer (Thermo Fisher), allowing detection of AGG interruptions within the FMR1 allele. PCR reagents include gene-specific and CGG primers, a polymerase mix buffer for amplification of the CGG repeat region in the FMR1 gene and a ROX 1000 Size Ladder for sizing by capillary electrophoresis. Amplified products were visualized by capillary electrophoresis (3500xL Genetic Analyzer, Thermo Fisher) and separated using POP-7TM polymer (Thermo Fisher), following the manufacturer’s instructions. The size of the PCR products has been converted to the estimated number of CGG repeats using size and mobility conversion factors. The reference sequence of the FMR1 gene used for sizing the CGG repeat is LRG_762 (identical to NG_007529.2).
Haplotype analysis
The segregation study was performed on all family members identified in Fig. 1. PCR amplification of the polymorphic markers DXS548 (forward 5′-AGA GCT TCA CTA TGC AAT GGA ATC and reverse 5′FAM-GTA CAT TAG AGT CAC CTG TGG TGC) and FRAXAC1 (forward 5′-GAT CTA ATC AAC ATC TAT AGA CTT TAT T and reverse 5′VIC-AGG CTT GGA GTG CAG TGG GCA ATC T) was carried out with fluorescent primers. The DXS548 marker is located ~150 kb upstream of the CGG repeats, while the FRAXAC1 marker is in intron 1. Amplifications were performed as follows: 30 cycles (1 min at 95 °C; 1 min at 55 °C; 1 min at 72 °C) with 200 μM of dNTPs, 2.0 mM of MgCl2, 1 U of Taq polymerase, and 1 pmol of each primer in a final volume of 20 µl. Alleles were resolved on capillary gel electrophoresis using an ABI3130 sequencer and analyzed with GeneMapper v4.0 (Thermo Fisher).
X inactivation analysis
The XCI assay was carried out on genomic DNA extracted from peripheral blood of the females indicated in the pedigree (Fig. 1), using primers flanking the CAG repeat in exon 1 of the human AR gene (Xq11–12), adapting the protocol of [26]. Approximately 200 ng of DNA from each individual was divided into two separate tubes, one was single digested with DdeI and the other was double digested with DdeI and methylation-sensitive enzyme HpaII. Twenty nanograms of digested DNA was amplified in a total volume of 20 μl using the forward unlabeled primer, AR-F (5′-GCT GTG AAG GTT GCT GTT CCT CAT), and the reverse FAM-conjugated primer, AR-R (5′-TCC AGA ATC TGT TCC AGA GCG TGC). Following PCR, 1 μl of the amplified product and GeneScanTM 500 LIZ size standard (Thermo Fisher) were separated through gel capillary electrophoresis using an ABI3130 sequencer and fragments were analyzed with GeneMapper v4.0 (Thermo Fisher). XCI was calculated for informative heterozygous females as the ratio of the peak area of two alleles of the highly polymorphic CAG repeat of the AR gene both in single DdeI-digested and in double DdeI-HpaII-digested sample. The active proportion of the maternally inherited chromosome was determined by the ratio p/(p + m), where p and m are the areas of the peaks corresponding to the paternal and maternal AR alleles, respectively. The range of the XCI ratio can vary from 50:50 (random XCI) to 100:0 (completely skewed XCI) [27]. One female sample with the known skewed XCI ratio (99:1) and one male sample (that of the unaffected brother II-2) were used as controls.
FMR1-mRNA quantification
Total RNA was extracted from peripheral blood cells of II-3, II-4, and II-6 as well as three normal females by the standard Trizol protocol (Life Biotechnologies). RNA concentration and purity were checked on 1.5% agarose gel and by a UV spectrophotometer. Afterward, 500 ng of total RNA were retro-transcribed into cDNA by the SensiFAST cDNA Synthesis kit (Bioline), according to the manufacturer’s instructions. For a relative quantification of FMR1 transcript using ABI7900HT (Life Technologies), the following pre-developed TaqMan® assays were employed: FMR1 (Hs.PT.58.2579089, IDT) and GAPDH (glyceraldehyde-3-phosphate-dehydrogenase) (Hs.PT.39a.22214836, IDT), the latter being constitutively expressed in every cell and thus used as endogenous control. The cycle parameters were 2 min at 50 °C and 10 min at 95 °C, followed by 40 cycles with 15 s at 95 °C (denaturation) and 1 min at 60 °C (annealing/extension). The relative quantification of target transcript vs. endogenous transcript was calculated as follows: 2–(ΔCt(FMR1)) – ΔCt(GAPDH)) = 2–ΔΔCt, where ΔCt is the difference (Ct(FMR1) – Ct(GAPDH)) and Ct is the cycle at which the detected fluorescence overcomes the threshold.
Results
The proband of this family was a 17-year-old boy (IV-4), who was born at term by vaginal delivery. Perinatal period was unremarkable, except for mild signs of hypotonia (i.e., artificial feeding for weak sucking). At 8 months of age his parents noted swinging movements. He walked at 2 years of age and language development was delayed with first words at 3 years. Metabolic screening, sleeping EEG, and brain MRI were all normal. Family history includes a 40-year-old man with mild ID of unknown origin (III-1), who was the son of a maternal aunt of the proband mother. For his psychomotor delay, the proband IV-4 has been studied for chromosomes analysis and FXS. Karyotype resulted normal (46,XY). FXS molecular test was performed by Southern blot and displayed a mosaicism of methylated FM alleles with an expansion between 0.7 and 2.7 kb, corresponding to g.5061_5063[(260–930)]│gom (data not shown). After the diagnosis of FXS in the proband, molecular analysis for the FMR1 locus was extended to at-risk members of the family with PCR for sizing (Fig. 2) and Southern blot to estimate methylation status (Fig. 3). The proband’s mother (III-6) was found to carry an MFM with an expansion of 1.3–1.75 kb (g.5061_5063[(460–610)]│gom) (Figs. 2 and 3); his unaffected sister (IV-3) had two normal FMR1 alleles (not shown). The molecular analysis performed through PCR on the maternal grandmother (II-3) showed a normal allele (g.5061_5063[29] corresponding to 29 triplets) and a PM (g.5061_5063[135] corresponding to 135 triplets) and a tiny amount of FM (Fig. 2). Southern blot analysis of II-3 with methylation-sensitive enzyme EagI shows only a completely methylated PM without any sign of FM (Fig. 3). Both sisters of II-3, II-4 and II-6, were heterozygous for a normal and a PM allele; the normal alleles were found to be g.5061_5063[28] and g.5061_5063[29], respectively, indirectly confirming that the normal alleles were inherited from their mother I-2, while the PM alleles had a similar size to that of II-3 being g.5061_5063[143] and g.5061_5063[140] in II-4 and II-6, respectively. As in their sister II-3, a tiny peak in the range of FM was detected by PCR (Fig. 2). As observed in II-3, also the PM allele of her sisters II-4 and II-6 was found to be completely methylated as shown by Southern blot (Fig. 3), which shows no sign of the small amount of FM, undetectable by this method. None of the three sisters (II-3, II-4, and II-6) developed FXPOI: menopause occurred at the age of 42 years for II-3 and II-6, and at 55 years for II-4. They had apparently no signs of tremor or movement disorder suggestive of FXTAS. Presently, they are 69 (II-3), 68 (II-4), and 60 (II-6) years old, respectively. The offspring of II-6 were also studied by Southern blot analysis: III-9 was mosaic for an unmethylated PM (g.5061_5063[(115)]│met = ) and an MFM (g.5061_5063[(310_560)]│gom); her sister and brother had both alleles in the normal range (data not shown).
Fig. 2.

CGG sizing through PCR. Capillary electrophoresis of the three sisters and of the proband mother. In each panel CGG size of peaks is calculated. In the three sisters, two alleles were detected, one in the normal range and one expanded. The expanded allele was in the PM range, but note that a tiny peak above 200 CGGs in the full mutation range was detected in all of three females. III-6 was heterozygous for normal allele (g.5061_5063[19]) of paternal origin and an FM (inherited by her mother II-3). Bars on the capillary electrophoresis indicated the presence of interspersed AGGs. The normal allele of the three sisters had two AGG interruptions, while the 19 CGG allele of III-6 had only one interspersed AGG, as usually observed
Fig. 3.
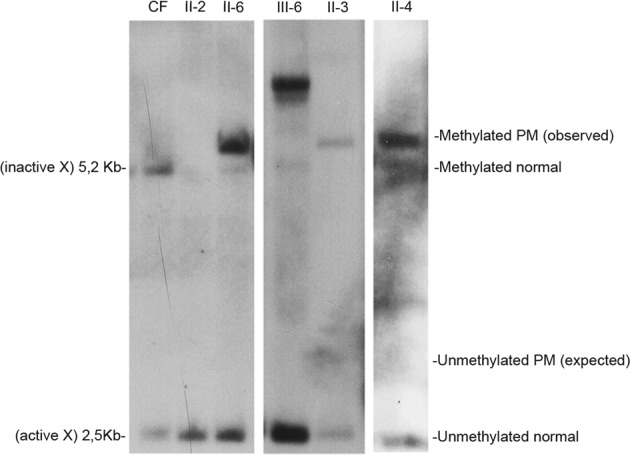
Southern blot analysis of genomic DNA digested with methylation-sensitive enzyme. The normal alleles on the active (2.5 kb) and inactive (5.2 kb) X chromosomes are shown in lane 1 (normal control female, CF). Unaffected brother II-2 was loaded in lane 2. The PM alleles of II-3, II-4, and II-6 appear to be completely methylated (all bands above 5.2 kb). No signal in the PM range was noted above 2.5 kb corresponding to the unmethylated X chromosome. Finally, III-6 (daughter of III-3) turned out to carry a normal and an MFM alleles (note the band well above 5.2 kb)
Segregation analysis with CGG sizing and two polymorphic markers flanking FMR1 locus (DXS548 and FRAXAC1) was also performed and the results are reported in Fig. 1. Comparing the FXS proband IV-4 with the unaffected male II-2, we deduced that the X chromosome carrying the FMR1 expansion was transmitted by I-1, in accordance with the relatively stable transmission to his daughters in the second generation. Since FMR1 expanded alleles may be methylated either by X inactivation or because the CGG expansion induces local DNA methylation, XCI analysis was performed using a methylation assay of the AR locus in order to exclude that the PM in the three sisters was preferentially located on the inactive X chromosome. Results are summarized in Table 1 and electropherograms are shown in Fig. 4. All three sisters had nearly random XCI, suggesting that the observed methylation of the PM allele was not due to skewed XCI. Transcription quantification through real-time PCR of the FMR1-mRNA showed no significant difference in FMR1 transcription between the three sisters and normal control females (not shown).
Table 1.
Results of XCI expressed as ratio of the areas of two alleles before and after methylation-sensitive enzyme HpaII digestion
| Name | Ratio before HpaII digestion | Ratio after HpaII digestion |
|---|---|---|
| II-3 | 60:40 | 17:83 |
| II-4 | 57:43 | 40:60 |
| II-6 | 57:43 | 20:80 |
| III-6 | 60:40 | 10:90 |
| Skewed female (control) | 61:39 | 1:99 |
Fig. 4.

Electropherograms of the polymorphic CAG triplets in the AR locus to study XCI. II-3 female presented two polymorphic alleles of 282 bp and 297 bp, that after methylation-sensitive HpaII digestion (upper panel) passed from 60:40 (lower panel) to 17:83, respectively. Her daughter III-6 was heterozygous for a 282 and 297 bp allele, which after digestion (upper panel) resulted 10:90, while before were 60:40 (lower panel). Middle panels showed results of II-4 and II-6: both heterozygous 282/297 bp and 285/297 bp, respectively. II-4 remained with a ratio between two alleles after HpaII digestion of 40:60, while her sister II-6 sister arrived at 20:80 (upper panels). In the bottom panels controls were reported: II-2 unaffected male was used as control for digestion (note the absence of PCR after HpaII digestion, in the bottom); on the right a skewed control female revealed an heterozygous genotype with 264/288 bp, with the first allele disappearing after methylation-sensitive enzyme digestion (ratio 1:99 from 61:39). Orange peaks are those of the size standard
Finally, a neuropsychological evaluation was performed for the three PM sisters by the administration of WAIS-R. In II-3 the verbal IQ was 77 and the performance IQ was 86, with a global IQ of 79 (borderline); II-4 had verbal IQ of 85, performance IQ of 85 and global IQ of 85 (low-average level); II-6 had a verbal IQ of 69, performance IQ of 66 and global IQ of 65 (mild ID). MFM carrier III-6 was also tested and found to have a verbal IQ of 65, performance IQ of 66 and global IQ of 62 (mild ID). The presence of a methylated PM allele is the likely cause of these findings; however, it should also be noted that the three sisters had a low level of schooling.
Discussion
Three sisters from the same FXS family were found to be carriers of completely methylated PM alleles. Methylation was found to be independent of XCI, at least in peripheral blood leukocytes. This epigenotype is likely to be the main cause of the slight intellectual impairment observed in these females. In fact, if the PM of II-3, II-4, and II-6 is methylated also on the active X chromosome, it will be silenced just as an FM allele and will not be transcribed as a PM usually does. Segregation analysis in this family suggested that the CGG expansion was most likely transmitted by the male ancestor I-1 and remained substantially stable (~140 CGGs) in the three carrier females of the second generation (individuals II-1 and II-5 could not be studied). Traces of FM detected by PCR in the three sisters can be attributed to postzygotic somatic instability [28, 29], but were undetectable by Southern blot. Indeed, DNA methylation did not seem to preserve the PM alleles from instability, as observed in the sisters themselves (mitotic instability) but also in III-6 and III-9 (meiotic instability), whose mutant FMR1 alleles were more expanded compared with those of their mothers (II-3 and II-6, respectively). However, it should be noted that III-9 harbors not only an MFM but also a PM smaller than that of her mother (II-6), suggesting that post-zygotic contractions of the CGG repeat also occured. It should be noted that the three sisters appear to have two AGG interruptions each, detected using the AmplideX® PCR assay; the presence of AGG interruptions within the CGG repeat is known to have a stabilizing effect preventing further expansion of the repeat [16, 17]. Although the PCR approach described by Hayward et al. [30] would have allowed positioning the AGG interruptions on either the normal (maternal) or the PM (paternal) allele, we assume that the two interspersed AGGs are located on the normal allele, since they are usually lost in PM. Clinical and molecular findings in this family suggest that CGG instability and DNA methylation are determined by different molecular mechanisms (as discussed in the Introduction). Methylation of PM in mosaic individuals harboring both PM and FM alleles has sometimes been reported [31, 32]; however, PM methylated alleles are typically larger (150–180 CGGs) than those detected in the three sisters. FMR1-mRNA levels in their peripheral blood leukocytes was similar to that of normal control females, indirectly confirming that PM alleles are silenced by DNA methylation providing a possible explanation for the absence of FXTAS or FXPOI in the three cases reported here. In fact, none of the sisters had premature menopause and movement disorder was not apparent at the respective age of 69 (II-3), 68 (II-4), and 60 (II-6).
Previous studies tried to correlate the molecular and clinical findings in PM female carriers: FMR1-mRNA expression significantly correlated with CGG repeat number [33] and with white matter microstructure observed through diffusion-weighted imaging at brain MRI [34]. Furthermore, Alvarez-Mora et al. [35] hypothesized that in PM females with FXTAS, the non-expanded (normal) X chromosome might be more frequently inactivated and showed that FXTAS women preferentially inactivate the normal X chromosome and transcribe the PM allele, whereas women without FXTAS preferentially inactivate the expanded X chromosome. In the three sisters XCI appears to be random in peripheral blood leukocytes, while PM alleles appear to be inactivated by DNA methylation in all cells. Willemsen et al. [12] showed that the timing of XCI and full mutation FMR1 allele inactivation was different, i.e., XCI occurs earlier during development than inactivation of the full mutation. Therefore, we speculate that during embryogenesis our PM sisters first randomly inactivated one X chromosome and then methylated the expanded FMR1 allele derived from their father.
To our knowledge, this represents the first report of completely methylated PM alleles with ~140 CGGs in females who present with a borderline/mild ID. In this family we cannot exclude the presence of a familial variant in one of the genes coding for the epigenetic machinery which could explain the lowering of the DNA methylation threshold. Until now, methylated PM have been occasionally reported in males with FXS and/or FXS-like phenotypes [31, 32, 36]. Description of a phenotype similar to FXS in methylated PM males is quite intuitive albeit rare, while a variable phenotype could be expected in females with a methylated PM. Two such females (belonging to two different FXS families) were found to be partly or completely methylated by bisulphite sequencing in some of the analysed clones [37]; unfortunately, the authors did not present clinical details of these individuals. However, the three cases reported in this study and the above mentioned reports [31, 32, 36, 37] suggest that the “threshold” of 200 CGG repeats needed for DNA methylation is not an absolute boundary.
In conclusion, a wide range of molecular findings and clinical phenotypes can be observed when characterizing members of an FXS family. The unexpected finding of a methylated PM allele in three sisters, with a borderline/mild ID phenotype, provides further evidence that CGG instability and the epigenetic inactivation of the FMR1 gene are independently regulated and that the threshold for transcriptional silencing is not rigidly set at ~200 CGG repeats.
Acknowledgements
We gratefully acknowledge family members, who spontaneously participate supporting our research.
Funding
This work was supported by Telethon Foundation grant (GGP15257A) and PRIN (Prot. 201789LFKB) to E.T., and by the Italian Association for FXS.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hunter J, Rivero-Arias O, Angelov A, Kim E, Fotheringham I, Leal J. Epidemiology of fragile X syndrome: a systematic review and meta-analysis. Am J Med Genet A. 2014;164:1648–58. doi: 10.1002/ajmg.a.36511. [DOI] [PubMed] [Google Scholar]
- 2.Neri G, Schwartz CE, Lubs HA, Stevenson RE. X-linked intellectual disability update 2017. Am J Med Genet A. 2018;176:1375–88. doi: 10.1002/ajmg.a.38710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–14. doi: 10.1016/0092-8674(91)90397-H. [DOI] [PubMed] [Google Scholar]
- 4.Bagni C, Oostra BA. Fragile X syndrome: from protein function to therapy. Am J Med Genet A. 2013;161:2809–21. doi: 10.1002/ajmg.a.36241. [DOI] [PubMed] [Google Scholar]
- 5.Usdin K, Kumari D. Repeat-mediated epigenetic dysregulation of the FMR1 gene in the fragile X-related disorders. Front Genet. 2015;6:192. doi: 10.3389/fgene.2015.00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pirozzi F, Tabolacci E, Neri G. The FRAXopathies: definition, overview, and update. Am J Med Genet A. 2011;155:1803–16. doi: 10.1002/ajmg.a.34113. [DOI] [PubMed] [Google Scholar]
- 7.Hagerman PJ, Hagerman RJ. The fragile-X premutation: a maturing perspective. Am J Hum Genet. 2004;74:805–16. doi: 10.1086/386296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Primerano B, Tassone F, Hagerman RJ, Hagerman P, Amaldi F, Bagni C. Reduced FMR1 mRNA translation efficiency in fragile X patients with premutations. RNA. 2002;8:1482–8. [PMC free article] [PubMed] [Google Scholar]
- 9.Smeets HJ, Smits AP, Verheij CE, Theelen JP, Willemsen R, van de Burgt I, et al. Normal phenotype in two brothers with a full FMR1 mutation. Hum Mol Genet. 1995;4:2103–8. doi: 10.1093/hmg/4.11.2103. [DOI] [PubMed] [Google Scholar]
- 10.Pietrobono R, Tabolacci E, Zalfa F, Zito I, Terracciano A, Moscato U, et al. Molecular dissection of the events leading to inactivation of the FMR1 gene. Hum Mol Genet. 2005;14:267–77. doi: 10.1093/hmg/ddi024. [DOI] [PubMed] [Google Scholar]
- 11.Tabolacci E, Moscato U, Zalfa F, Bagni C, Chiurazzi P, Neri G. Epigenetic analysis reveals a euchromatic configuration in the FMR1 unmethylated full mutations. Eur J Hum Genet. 2008;16:1487–98. doi: 10.1038/ejhg.2008.130. [DOI] [PubMed] [Google Scholar]
- 12.Willemsen R, Bontekoe CJ, Severijnen LA, Oostra BA. Timing of the absence of FMR1 expression in full mutation chorionic villi. Hum Genet. 2002;110:601–5. doi: 10.1007/s00439-002-0723-5. [DOI] [PubMed] [Google Scholar]
- 13.Sullivan AK, Crawford DC, Scott EH, Leslie ML, Sherman SL. Paternally transmitted FMR1 alleles are less stable than maternally transmitted alleles in the common and intermediate size range. Am J Hum Genet. 2002;70:1532–44. doi: 10.1086/340846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oberlé I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, et al. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252:1097–102. doi: 10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]
- 15.Chiurazzi P, Genuardi M, Kozak L, Giovannucci-Uzielli ML, Bussani C, Dagna-Bricarelli F, et al. Fragile X founder chromosomes in Italy: a few initial events and possible explanation for their heterogeneity. Am J Med Genet. 1996;64:209–15. doi: 10.1002/(SICI)1096-8628(19960712)64:1<209::AID-AJMG38>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 16.Yrigollen CM, Durbin-Johnson B, Gane L, Nelson DL, Hagerman R, Hagerman PJ, et al. AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with fragile X syndrome. Genet Med. 2012;14:729–36. doi: 10.1038/gim.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nolin SL, Glicksman A, Ersalesi N, Dobkin C, Brown WT, Cao R, et al. Fragile X full mutation expansions are inhibited by one or more AGG interruptions in premutation carriers. Genet Med. 2015;17:358–64. doi: 10.1038/gim.2014.106. [DOI] [PubMed] [Google Scholar]
- 18.Zhao XN, Usdin K. Ups and downs: mechanisms of repeat instability in the fragile X-related disorders. Genes (Basel). 2016;7:pii: E70. 10.3390/genes7090070. [DOI] [PMC free article] [PubMed]
- 19.Mirkin SM. DNA structures, repeat expansions and human hereditary disorders. Curr Opin Struct Biol. 2006;16:351–8. doi: 10.1016/j.sbi.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 20.Loomis EW, Sanz LA, Chédin F, Hagerman PJ. Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 2014;10:e1004294. doi: 10.1371/journal.pgen.1004294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reddy K, Schmidt MH, Geist JM, Thakkar NP, Panigrahi GB, Wang YH, et al. Processing of double-R-loops in (CAG)·(CTG) and C9orf72 (GGGGCC)·(GGCCCC) repeats causes instability. Nucleic Acids Res. 2014;42:10473–87. doi: 10.1093/nar/gku658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colak D, Zaninovic N, Cohen MS, Rosenwaks Z, Yang WY, Gerhardt J, et al. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science. 2014;343:1002–5. doi: 10.1126/science.1245831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orsini A, Laicardi C WAIS-R. Contribution to Italian calibration. Firenze (Florence)-Italy: Giunti Psychometrics; 2001.
- 24.Nakahori Y, Knight SJL, Holland J, Schwartz C, Roche A, Tarleton J, et al. Molecular heterogeneity of the fragile X syndrome. Nucleic Acids Res. 1991;19:4355–9. doi: 10.1093/nar/19.16.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Filipovic-Sadic S, Sah S, Chen L, Krosting J, Sekinger E, Zhang W, et al. A novel FMR1 PCR method for the routine detection of low abundance expanded alleles and full mutations in fragile X syndrome. Clin Chem. 2010;56:399–408. doi: 10.1373/clinchem.2009.136101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet. 1992;51:1229–39. [PMC free article] [PubMed] [Google Scholar]
- 27.Amos-Landgraf JM, Cottle A, Plenge RM, Friez M, Schwartz CE, Longshore J, et al. X chromosome-inactivation patterns of 1,005 phenotypically unaffected females. Am J Hum Genet. 2006;79:493–9. doi: 10.1086/507565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alvarez-Mora MI, Guitart M, Rodriguez-Revenga L, Madrigal I, Gabau E, Milà M. Paternal transmission of a FMR1 full mutation allele. Am J Med Genet A. 2017;173:2795–7. doi: 10.1002/ajmg.a.38384. [DOI] [PubMed] [Google Scholar]
- 29.Zeesman S, Zwaigenbaum L, Whelan DT, Hagerman RJ, Tassone F, Taylor SA. Paternal transmission of fragile X syndrome. Am J Med Genet A. 2004;129:184–9. doi: 10.1002/ajmg.a.30191. [DOI] [PubMed] [Google Scholar]
- 30.Hayward BE, Kumari D, Usdin K. Recent advances in assays for the fragile X-related disorders. Hum Genet. 2017;136:1313–27. doi: 10.1007/s00439-017-1840-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pretto DI, Mendoza-Morales G, Lo J, Cao R, Hadd A, Latham GJ, et al. CGG allele size somatic mosaicism and methylation in FMR1 premutation alleles. J Med Genet. 2014;51:309–18. doi: 10.1136/jmedgenet-2013-102021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiraanont P, Kumar M, Tang HT, Espinal G, Hagerman PJ, Hagerman RJ, et al. Size and methylation mosaicism in males with Fragile X syndrome. Expert Rev Mol Diagn. 2017;17:1023–32. doi: 10.1080/14737159.2017.1377612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiraanont P, Sweha SR, Al Olaby RR, Silva M, Tang HT, Durbin-Johnson B, et al. Clinical and molecular correlates in fragile X premutation females. eNeurologicalSci. 2017;7:49–56. doi: 10.1016/j.ensci.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shelton AL, Cornish KM, Godler D, Bui QM, Kolbe S, Fielding J. White matter microstructure, cognition, and molecular markers in fragile X premutation females. Neurology. 2017;88:2080–8. doi: 10.1212/WNL.0000000000003979. [DOI] [PubMed] [Google Scholar]
- 35.Alvarez-Mora MI, Rodriguez-Revenga L, Feliu A, Badenas C, Madrigal I, Milà M, et al. Inactivation in Women Carrying the FMR1 Premutation and Its Relation with Fragile-X-Associated Tremor/Ataxia Syndrome. Neurodegener Dis. 2016;16:290–2. doi: 10.1159/000441566. [DOI] [PubMed] [Google Scholar]
- 36.Fernández E, Gennaro E, Pirozzi F, Baldo C, Forzano F, Turolla L, et al. FXS-Like Phenotype in Two Unrelated Patients Carrying a Methylated Premutation of the FMR1 Gene. Front Genet. 2018;9:442. doi: 10.3389/fgene.2018.00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Genç B, Müller-Hartmann H, Zeschnigk M, Deissler H, Schmitz B, Majewski F, et al. Methylation mosaicism of 5′-(CGG)(n)-3′ repeats in fragile X, premutation and normal individuals. Nucleic Acids Res. 2000;28:2141–52. doi: 10.1093/nar/28.10.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]