
Fig. 3.
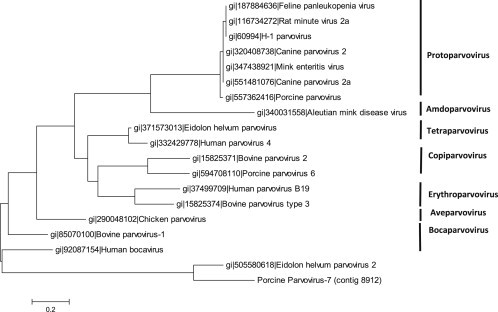
Phylogenetic tree of the putative new porcine parvovirus (PPV7) The evolutionary history was inferred by using the Maximum Likelihood method based on the JTT matrix-based model [35]. The tree with the highest log likelihood (−1514.4280) is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model, and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 19 partial amino acid sequences of the putative protein NS1. All positions containing gaps and missing data were eliminated. There were a total of 66 positions in the final dataset. Evolutionary analyses were conducted in MEGA6 [36]. The tree is labeled according to the proposed new genus names within the Parvoviridae family [37].