
Fig. 1.
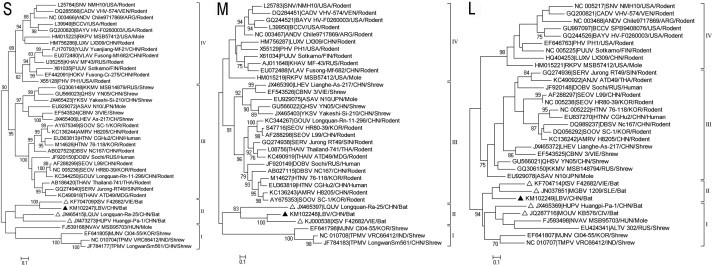
Phylogenetic trees were constructed according to partial S (positions 500–1458), M (positions 1985–2647) and L (positions 2948–3284) stretches of the hantaviral genomic segments, selected from the limited availability of sequences of most known bat-borne hantaviruses (see Table 1), fragment positions refer to complete sequences of HTNV strain 76–118 (Genbank accession Nos: NC_005218, NC_005219, NC_005222). MEGA5.0 was used to construct the phylogenetic trees with the maximum-likelihood method. Numbers above or below branches are bootstrap values of 1000 replicates. Filled triangles indicate the sequences of LBV, while open triangles indicate the sequences of other known bat hantaviruses. The scale bars indicate nucleotide substitutions per site. The accession number is prefixed to the name of each virus strain used.