

Abstract
Disease comorbidity is a result of complex epigenetic interplay. A disease is rarely a consequence of an abnormality in a single gene; complex pathways to disease patterns emerge from gene-gene interactions and gene-environment interactions. Understanding these mechanisms of disease and comorbidity development, breaking down them into clusters and disentangling the epigenetic — actionable — components, is of utter importance from a public health perspective. With the increase in the average life expectancy, healthy aging becomes a primary objective, from both an individual (i.e. quality of life) and a societal (i.e. healthcare costs) standpoint. Many studies have analyzed disease networks based on common altered genes, on protein-protein interactions, or on shared disease comorbidites, i.e. phenotypic disease networks. In this work we aim at studying the relations between genotypic and phenotypic disease networks, using a large statewide cohort of individuals (100, 000+ from California, USA) with linked clinical and genotypic information, the Genetic Epidemiology Research on Adult Health and Aging (GERA). By comparing their phenotypic and genotypic networks, we try to disentangle the epigenetic component of disease comorbidity.
Keywords: GWAS, comorbidity network, genetic analysis
1. Introduction
Disease comorbidity is a result of complex epigenetic interplay. A disease is rarely a consequence of an abnormality in a single gene; complex pathways to disease patterns emerge from gene-gene interactions and gene-environment interactions. Understanding these mechanisms of disease and comorbidity development, breaking down them into clusters and disentangling the epigenetic — actionable — components, is of utter importance from a public health perspective. With the increase in the average life expectancy, healthy aging becomes a primary objective, from both an individual (i.e. quality of life) and a societal (i.e. healthcare costs) standpoint.
Genome-wide association studies (GWAS) are now very common due to a decrease in sequencing cost and increase in throughput, and large data bases are available publicly linking single nucleotide polymorphisms (SNPs) to diseases, e.g. the GWAS Catalog (https://www.ebi.ac.uk/gwas/). Recent studies have exploited these data bases to create high resolution SNP networks4,5. However, there is a lack of studies that analyze SNPs (single nucleotide polymorphisms) and disease comorbidities using the same data, linked at the individual level; this is the objective of our investigation, here presented.
In this work we aim at studying the relations between genotypic and phenotypic disease networks, using a large statewide cohort of 110,266 individuals from California, USA, with linked clinical and genotypic information, the Genetic Epidemiology Research on Adult Health and Aging (GERA). DNA data has been extracted from saliva samples and stored in a text file repository. To disentangle the epigenetic component of disease comorbidity, we build two types of comorbidity networks (i.e. genotypic and phenotypic) for each ethnic group in the database and for the whole patient population. The network construction workflow will be described in Section 2. We then report networks’ structural features by using standard network metrics, showing the most significant ones.
2. Methods
Study design and data sets.
The data source for this study is the Genetic Epidemiology Research on Adult Health and Aging (GERA)6, a public resource funded by the National Institutes of Health (NIH). GERA is a subset of the Kaiser Permanente’s Research Program on Genes, Environment, and Health (RPGEH). RPGEH links together comprehensive electronic medical records, data on relevant behavioral and environmental factors, and biobank data (genetic information from saliva and blood) from 500,000 consenting health plan members enrolled among the six million-member Kaiser Permanente Medical Care Plan of Northern California and Southern California. Data from over 100,000 participants from various ethnic groups, with ages from 27 to 97 years (average age: 63), are freely available in GERA, with associated genotyping information, demographics, health-related behaviors, and grouped health conditions on the basis of the International Classification of Diseases v.9 (ICD-9) ontology, from an average of 23.5 years of electronic medical records. High-density genotyping was conducted using custom designed Affymetrix Axiom arrays7,8.
The final goal, on which we are still working, is to analyze inter- and intra-ethnic group network differences and to analyze and validate the extracted knowledge with experts in the clinical domain. Instead of measuring molecular markers, we aim to categorize patients by measuring co-factors, such as existing health conditions and prescription drug use. The data analysis methods needed to achieve this aim are complex, and may have limited previous research efforts like this. Accurate prognostic classification at diagnosis remains an urgent and unmet challenge, due to confounding by screening practices and comorbid conditions in an aging population. Our endotyping effort may reveal unique clinical profiles that can help guide prognosis and treatment decisions.
Ethics Statement.
This study has been performed in accordance with the Declaration of Helsinki. The research protocol has been approved by University of Floridas Institutional Review Board. The GERA data request has been approved on April 22, 2016, and is deposited on the GERA website under Dr. Travis Gerke’s name.
Genome-wide Association Study.
For the analisys of the GERA dbGap database files we used the PLINK tool1,2, version 1.90b3.42 64-bits. In PLINK the whole GWAS pipeline is implemented in a single tool, allowing to effectively search the most significant genotypic information (i.e. SNPs) explaining the differences in the phenotypic feature set (e.g. age, gender, BMI), also called covariates. From the GERA dbGap database we obtained genotypic data, extracted at University of California San Francisco using custom designed Affymetrix Axiom arrays, for four ethnic groups: (i) AFR showing genetic similarity with African-Americans, (ii) EUR, defined, in the GERA Genotypic Data description, as Non-Hispanic White, (iii) EAS containing patients with East Asian genetic traits and (iv) LAT, with DNA belonging to the Latinos. It is worth noting that these groups were made up directly from genetic evidence and not from self-declared race memberships by individuals7. We also built an integrated dataset, called ALL, by combining those groups and we stored the race information as a feature in the phenotype covariates file. In fact, together with the genotypic data, we obtained from the GERA dbGap database a set of phenotypic files containing demographic and behavioural factors from the RPGEH (Research Program on Genes, Environment and Health) and CMHS (California Men’s Health Study) Survey. Before the GWAS analysis step, we performed a preprocessing step in which some of the phenotypic features have been categorized. For instance, the age feature has been divided into three classes (0,1,2) corresponding to the ranges (0 – 40,40 – 60,> 60).
For each of the five ethnic groups (i.e. AFR, EUR, EAS, LAT and ALL) we performed a GWAS analysis which gave us a huge set of SNPs selected by the PLINK tool by comparing the large genotypic datasets and by looking at the phenotypic features at the same time. Each of the SNPs selected by the tool is assigned a p-value. Since we are conducting a multi-class experiment we adopted the Benjamini-Hockberg (BH) correction to minimize the false positive rate, at significance level of 0.05. From the results of the GWAS experiment we built both genotypic and phenotypic networks by measuring how disease categories are related in terms of number of SNPs in common for the genotypic network and number of patients in common for the phenotypic one, as described in the following.
Genotypic network construction.
The genotypic networks have been built by analyzing the results of the GWAS experiment. For each experiment, we created a set of networks, as undirected graphs, where nodes are the GERA dbGAP disease categories and arcs between nodes have weights representing the strength of the relation between them. For genotypic networks, the arc weights are proportional to the number of SNPs in common between them. In particular, for each disease category pair (vi,vj) of the genotypic network, we calculated the Jaccard index Jγ(vi,vj), which is a similarity coefficient taking into account what is shared between the two nodes. A formal definition of Jγ is reported in equation 1:
| (1) |
where vi and vj (i ≠ j) are nodes of the genotypic network and SNP(v) is a function returning all of the significant SNPs obtained by the GWAS analysis after the Benjamini-Hockberg correction for disease v.
Figure 1 reports an example of genotypic network (on the left) for the EUR ethnic group in which the calculated Jaccard Indexes among nodes are shown as arc weights. Note also that the arcs’ line style is related to four percentile groups, starting from the lightest (i.e. dotted), associated with the 0 – 25th percentile, to the thickest line style associated with the 75 – 100th percentile. Percentiles of the arc weights have been used to extract the most relevant topological information from the network (i.e. 75 – 100th percentile). For instance, for the EUR ethnic group, we obtained the genotypic network depicted in Figure 2 (network on the left).
Fig. 1.
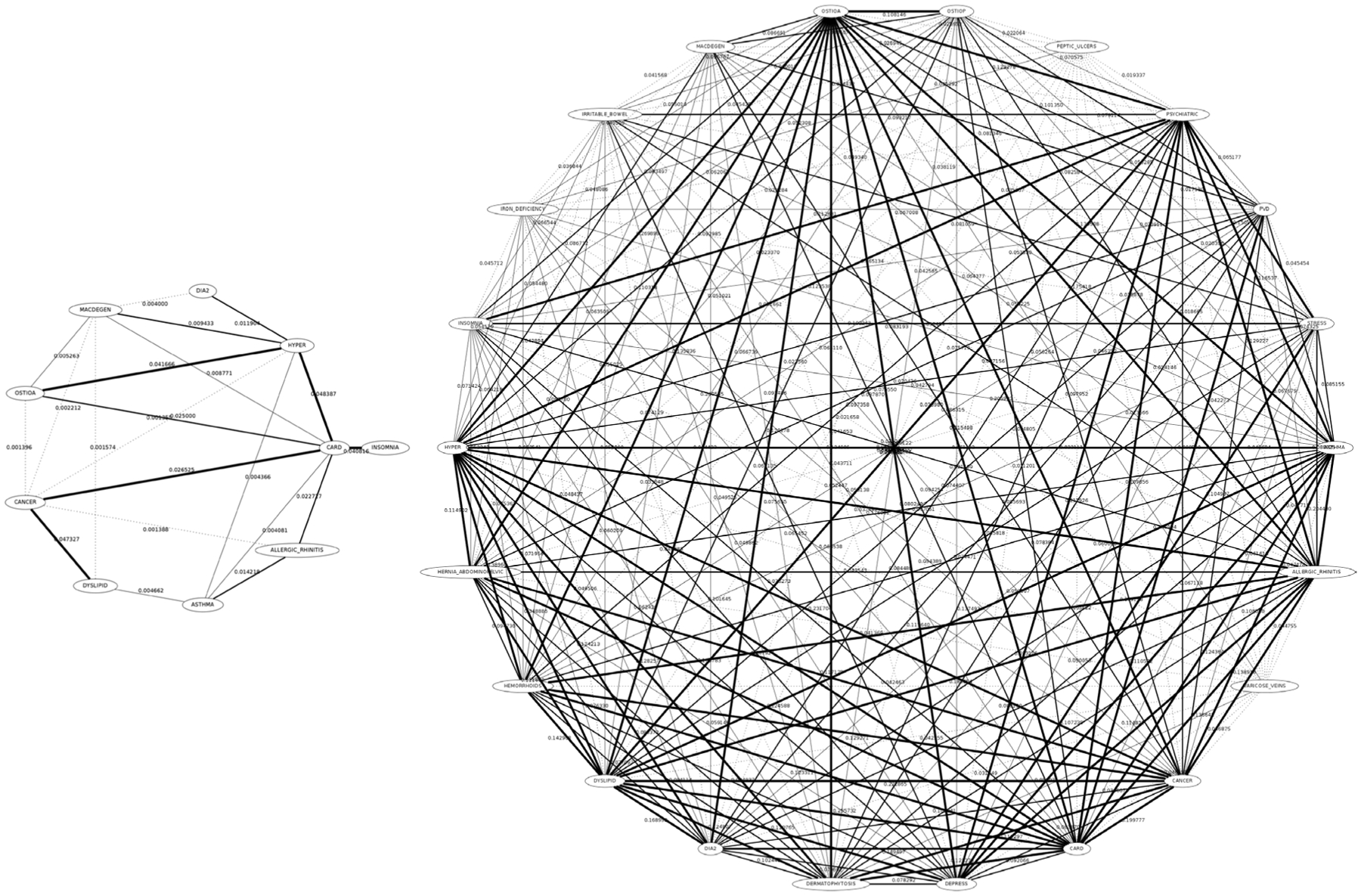
Genotypic (left) and Phenotypic (right) networks for the EUR ethnic group. Nodes are GERA dbGap disease categories (for the genotypic network) having at least one significant SNP marked by the GWAS experiment or patients (for the phenotypic network). Arcs weights are the Jaccard Index between two nodes (number of SNPs in common over the total SNPs for the genotypic or number of patients in common for the phenotypic network). Arcs line styles have been depicted according to the percentiles of the weights (dotted for the 0 – 25th percentile, thickest for the 75 – 100th percentile).
Fig. 2.

Genotypic (left) and Phenotypic (right) networks for the EUR ethnic group derived from the complete networks (see Figure 1) by selecting arcs having weights in the 75th percentile only.
Phenotypic network construction.
Similarly to the genotypic networks, a set of phenotypic networks has been built. We considered the same nodes as before (i.e. GERA dbGap disease categories). Similarly as in the genotypic networks, we created arcs between two nodes (vi,vj) with weight calculated as the Jaccard index Jϕ(vi,vj), which measures the similarity between two nodes as being proportional to the number of patients shared between them. Jϕ is described formally in equation 2:
| (2) |
where vi and vj are nodes of the genotypic network and P(v) is a function returning all of the patients affected by disease v.
Figure 1 shows the phenotypic network (on the right) built for the EUR ethnic group in which the calculated Jaccard Indexes among nodes are shown as arc weights. For this network also the arcs’ line style is related to four percentile groups: 0 − 25th percentile are the dotted arcs up to the 75 − 100th percentile which are the thickest arcs. Similarly to the genotypic network described above, also for the phenotypic ones we generated a network for each group by considering just the 75 − 100th percentile. An example for the EUR ethnic group is shown in Figure 2 (network on the right).
3. Results
From the 100,000+ population in the GERA dbGap database, we selected a total of 78479 patients having all the features needed for the analysis and also having signed the consent for genetic research studies. All data have been of course anonymized and encrypted with state of the art algorithms by the Kaiser Permanente Institute. Ethnic groups have the following patients in their respective datasets: (i) AFR with 3826 (4.9%) patients, (ii) EUR with 62313 (79.4%) patients, (iii) EAS with 5188 (6.6%) patients, (iv) LAT with 7152 (9.1%) patients.
Analysis of network correlations.
In this phase of the analysis we considered the intra-ethnic group differences between the 75th percentile genotypic networks and the corresponding phenotypic networks. As a preliminary step of the analysis, we counted the number of arcs in common between the two networks, as reported in Table 1. For instance, for the EUR ethnic group, the genotypic network (left of Figure 2) has been compared with the phenotypic network (right of Figure 2). One interesting case, which needs further investigation with the clinical domain experts, is the lack of shared arcs between the genotypic and the phenotypic networks in the LAT group. Another interesting case is about the ALL group, where both the number of nodes and arcs in common are very high, compared to the number of total nodes in both networks.
Table 1.
Structural analysis of the 75th percentile genotypic and phenotypic networks. The table reports the main structural features of both networks and the number of arcs in common for each ethnic group.
| Ethnic group | Genotypic network | Phenotypic network | nodes in common | arcs in common | ||||
|---|---|---|---|---|---|---|---|---|
| nodes | arcs | density | nodes | arcs | density | |||
| AFR | 4 | 6 | 0.29 | 16 | 58 | 0.48 | 3 (18.8%) | 3 (5.2%) |
| EUR | 6 | 6 | 0.40 | 17 | 58 | 0.43 | 6 (35.3%) | 5 (8.6%) |
| EAS | 5 | 8 | 0.29 | 16 | 58 | 0.17 | 3 (18.8%) | 2 (3.4%) |
| LAT | 3 | 2 | 0.67 | 17 | 58 | 0.43 | 2 (11.8%) | 0 (0%) |
| ALL | 14 | 26 | 0.29 | 16 | 58 | 0.48 | 13 (81.3%) | 18 (31%) |
In Figure 3 we report the distribution of the significant SNPs found for disease for each ethnic group. From our preliminary tests, the number of significant SNPs descreases when we add more covariates (e.g. gender, age). This is expected, since we are adding more constraints. However, such a trend is violated in some cases. For example for the disease group DIA2 (Diabetes II), we have a substantial increase in the significant SNPs (even after the BH correction) for both the AFR and LAT ethnic groups, and we also observe an increase in the HYPER (Hypertensive Disease) disease group for AFR and EAS. The EAS group shows and increase in the significant SNPs in the same experimental conditions for the PSYCHIATRIC disease group.
Fig. 3.
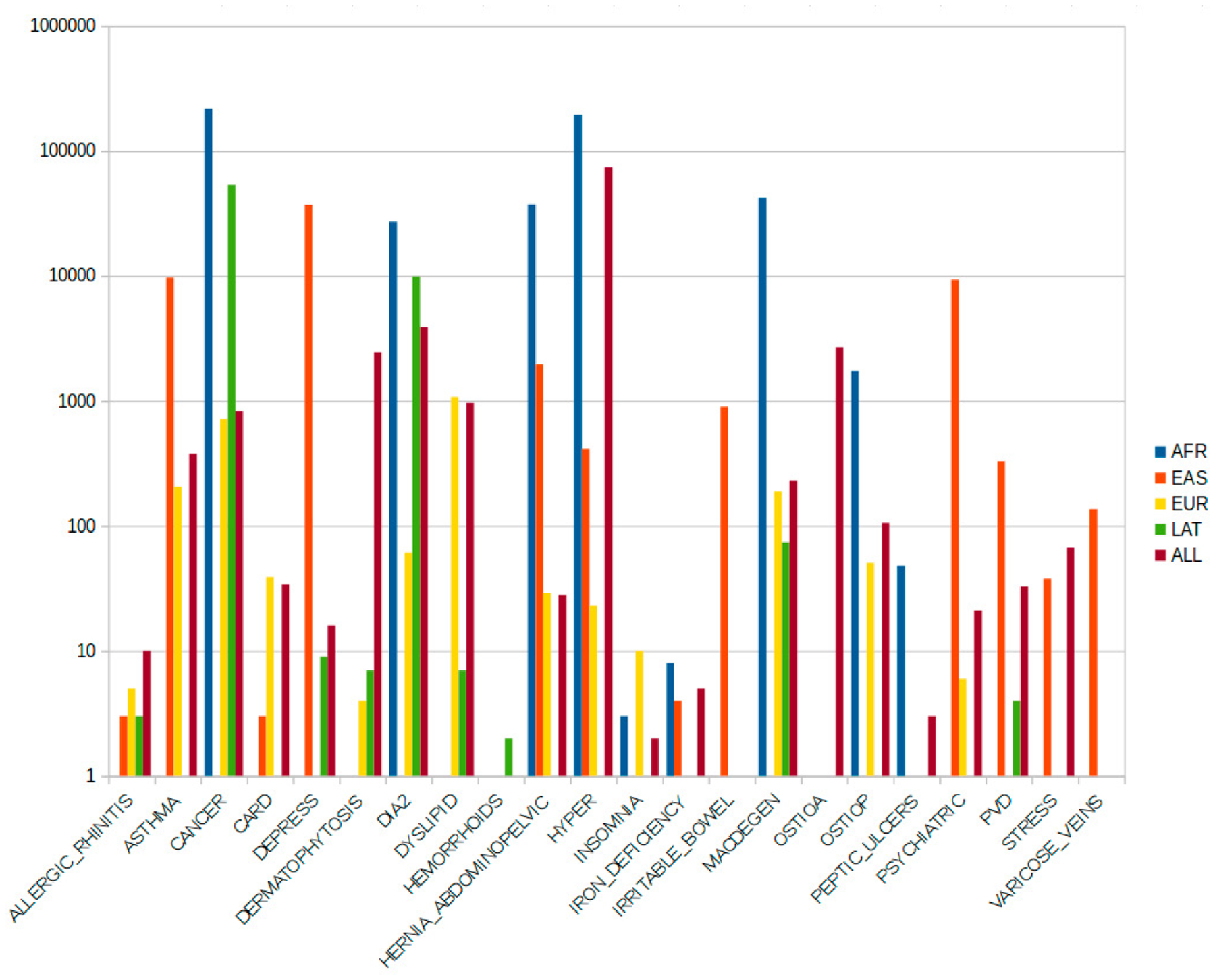
Distribution of the most significant SNPs for disease and for each ethnic group. Y axis is logarithmic.
4. Conclusions and Future Work
Network medicine, i.e. the study of disease networks is a fast-growing field or research. Both genotypic and phenotypic networks have been analyzed using large data sets, but a parallel study comparing the two with linkage at the individual level was lacking. In this study, we used the GERA dbGap cohort (100,000+ individuals) and built both a genotypic SNP network via GWAS and a phenotypic comorbidity network, using the same study population with information linked at the individual level. We then compared the two networks to measure a first similarity between the two by considering the strongest arcs in the original networks and counted the arcs present in both. We are planning to more deeply and extensively analyze the structural differences both inter- and intra- ethnic groups and we will also discuss the clinical implications with the clinical domain experts, which could eventually lead to further experiments. Furthermore we are analyzing all of the networks by using Extended Random Graph Models which give a measure of the significance of the arcs in the network allowing for a more precise evaluation of topological properties and intra-ethnic groups comparisons. We are going to discuss all of these further analyses in an extended version of this paper.
Acknowledgements
This work was funded by the University of Florida Health Cancer Center / Institute of Aging Cancer-Aging Collaborative Grant Program. Data came from a grant, the Resource for Genetic Epidemiology Research in Adult Health and Aging (RC2 AG033067; Schaefer and Risch, PIs) awarded to the Kaiser Permanente Research Program on Genes, Environment, and Health (RPGEH) and the UCSF Institute for Human Genetics.
References
- 1.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ (2015). Second-generation PLINK: rising to the challenge oflarger and richer datasets. Gigascience, 4(1), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. (2007). PLINK: a tool set for whole-genome associationand population-based linkage analyses. The American Journal of Human Genetics, 81(3), 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoffmann TJ, Zhan Y, Kvale MN, Hesselson SE, Gollub J, Iribarren C, et al. (2011). Design and coverage of high throughput genotyping arrays optimized for individuals of East Asian, African American, and Latino race/ethnicity using imputation and a novel hybrid SNP selection algorithm. Genomics, 98(6), 422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li H, Lee Y, Chen JL, Rebman E, Li J, Lussier YA (2012). Complex-disease networks of trait-associated single-nucleotide polymorphisms (SNPs) unveiled by information theory. Journal of the American Medical Informatics Association, 19(2), 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qiu J, Moore JH, Darabos C (2016). Studying the Genetics of Complex Disease With Ancestry-Specific Human Phenotype Networks: TheCase of Type 2 Diabetes in East Asian Populations. Genetic epidemiology, 40(4), 293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kvale MN, Hesselson S, Hoffmann TJ, Cao Y, Chan D, Connell S, et al. (2015). Genotyping informatics and quality control for 100,000 subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort. Genetics, 200(4), 1051–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoffmann TJ, Kvale MN, Hesselson SE, Zhan Y, Aquino C, Cao Y, et al. (2011). Next generation genome-wide association tool: design and coverage of a high-throughput European-optimized SNP array. Genomics, 98(2), 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoffmann TJ, Zhan Y, Kvale MN, Hesselson SE, Gollub J, Iribarren C, et al. (2011). Design and coverage of high throughput genotyping arrays optimized for individuals of East Asian, African American, and Latino race/ethnicity using imputation and a novel hybrid SNP selection algorithm. Genomics, 98(6), 422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]