
Fig. 1.
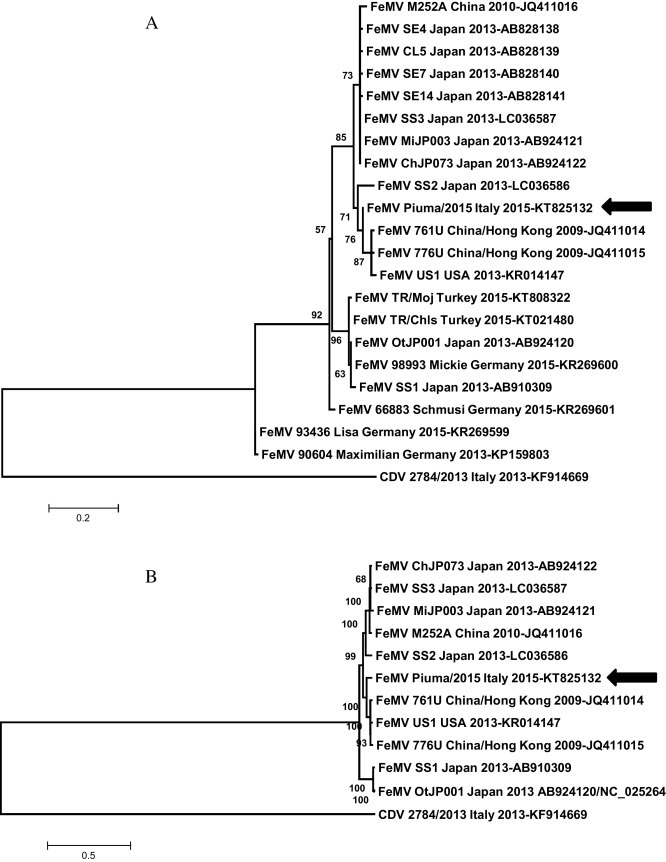
Phylogenetic analyses were inferred from partial (approximately 400 nt) L protein encoding gene of all existing FeMVs (MLpartial, 1A) and complete FeMV genomes available on line (MLcomplete, 1B). Analyses were conducted using the Maximum Likelihood (ML) method in Mega 6 (Tamura et al., 2013). The best-fit model of nucleotide substitution was identified by the Find Best DNA/protein Model available in Mega 6 as the T92 + I model for MLpartial, whereas the GTR + G + I for MLcomplete. To assess the robustness of individual nodes on the phylogenetic trees, we performed a bootstrap resampling analysis (1000 replications) using the neighbor-joining method, incorporating the ML substitution model. Canine distemper virus (CDV, Marcacci et al., 2014) served as outgroup. Bars indicate the estimated number of nt substitutions per site.