

Abstract
The deep terrestrial subsurface remains an environment where there is limited understanding of the extant microbial metabolisms. At Olkiluoto, Finland, a deep geological repository is under construction for the final storage of spent nuclear fuel. It is therefore critical to evaluate the potential impact microbial metabolism, including sulfide generation, could have upon the safety of the repository. We investigated a deep groundwater where sulfate is present, but groundwater geochemistry suggests limited microbial sulfate-reducing activity. Examination of the microbial community at the genome-level revealed microorganisms with the metabolic capacity for both oxidative and reductive sulfur transformations. Deltaproteobacteria are shown to have the genetic capacity for sulfate reduction and possibly sulfur disproportionation, while Rhizobiaceae, Rhodocyclaceae, Sideroxydans, and Sulfurimonas oxidize reduced sulfur compounds. Further examination of the proteome confirmed an active sulfur cycle, serving for microbial energy generation and growth. Our results reveal that this sulfide-poor groundwater harbors an active microbial community of sulfate-reducing and sulfide-oxidizing bacteria, together mediating a sulfur cycle that remained undetected by geochemical monitoring alone. The ability of sulfide-oxidizing bacteria to limit the accumulation of sulfide was further demonstrated in groundwater incubations and highlights a potential sink for sulfide that could be beneficial for geological repository safety.
Subject terms: Microbial ecology, Biogeochemistry, Environmental sciences, Ecosystem ecology
Introduction
The terrestrial subsurface harbors a significant proportion of the Earth’s microbial cells [1, 2]. In the terrestrial subsurface, photosynthetically derived electron donors are in relatively low abundance and geogenic gases, H2 and CH4, provide energy sources for microbial communities [3]. These energy sources support the occurrence of autotrophic methanogens, acetogens, and iron- and sulfate-reducing bacteria [4–13]. While autotrophic hydrogen-fueled microbial communities are often predominant, heterotrophic fermenters have also been shown to contribute to biogeochemical cycling in the subsurface, degrading recalcitrant natural organic matter and microbial necromass [11, 14, 15].
Sulfate-reducing bacteria are an important constituent of the terrestrial subsurface and are often the dominant metabolic group where sulfate is present [4–6, 11, 15]. Even when sulfate is limited, sulfate-reducing bacteria are often detected [9, 14, 16]. Electron acceptors with a higher reduction potential such as nitrite, nitrate, and oxygen are generally depleted in the deep subsurface. Despite limited availability, the metabolic potential for nitrate reduction has been identified in groundwater from Finland [11, 17] and an active community of sulfur-driven denitrifiers were identified in fluids from the Witwatersrand Basin, South Africa [18].
When hydrogeochemical end products or reaction intermediates are undetected or remain at steady state, the importance of the corresponding process may be overlooked. In particular, cryptic cycles, where elements are rapidly recycled, can be active under those conditions. This was demonstrated in a meromictic lake, where despite the low iron concentration, iron turnover rates were high and a cryptic iron cycle was proposed [19]. Similarly, isotopic and genomic studies have revealed cryptic sulfur cycling in marine oxygen minimum zones [20, 21], marine-, freshwater-, and salt marsh sediments [22–25].
The activity of sulfate-reducing bacteria in subsurface groundwaters at Olkiluoto, an island on the south-west coast of Finland, is of particular interest as it is the site of a deep geological repository for the final storage of spent nuclear fuel. The generation of sulfide, the metabolic product of sulfate reduction, can potentially compromise the longevity of copper canisters used to store spent nuclear fuel. It is therefore crucial to understand the energy sources fueling sulfate reduction at Olkiluoto.
The geological and environmental history of the island of Olkiluoto has resulted in the stratification of different groundwater types [26]. Sulfate-reducing bacteria are detected where sulfate-rich groundwater mixes with deeper methane-rich groundwater (~250–350 m depth) and the activity of sulfate-reducing bacteria is evident by elevated concentrations of sulfide (~0.1–0.4 mM) and isotopic sulfate signatures enriched in 34S (up to 40‰ CDT) [26]. The stable isotopic signature of sulfate (~25‰ CDT) in shallower groundwater (<300 m) suggests that sulfate reduction is presently limited. This is surprising, given that numerous studies have highlighted the presence of sulfate-reducing bacteria [27–30] able to gain energy from the oxidation of geogenic hydrogen and organic carbon produced from fermentation and acetogenesis [11, 31], in sulfate-rich groundwaters at Olkiluoto. However, at this depth, electron donors are limited. Organic carbon is neither replenished from the surface nor produced in situ by acetogenesis due to the limited availability of hydrogen in shallower anoxic sulfate-rich groundwaters [26].
We investigated a groundwater at Olkiluoto where sulfate is relatively abundant (~3 mM), the sulfate concentration is stable long term, and sulfide is not detected. The stable isotopic signature of sulfur in sulfate is consistent with the sulfate source, indicating that there has been no further activity of sulfate-reducing microorganisms [26]. A possible interpretation is that the lack of electron donors known to fuel sulfate reduction in deeper groundwaters could limit metabolic activity despite the presence of sulfate. Or perhaps, other hydrogeochemical or microbial constraints preclude sulfate reduction. To understand sulfur cycling at Olkiluoto, we used a metaproteogenomic approach to probe this sulfate-rich, sulfide-poor, groundwater with a sulfur isotopic signature consistent with an absence of sulfate reduction.
Materials and methods
Microbial biomass collection
Groundwater from fractured veined gneiss at 366.7–383.5 m below sea level on the island at Olkiluoto (61°14′28″N 21°29′41″E) was collected in 2016. The groundwater was filtered (~10 L per sample, 0.2 and 0.1 µm pore size) and the biomass was preserved for DNA and protein analysis. Details of sample collection (sampling, DNA extraction, DNA sequencing, 16S rRNA gene amplicon analysis, metagenome assembly, and geochemical measurements) are described in detail in Supplementary Information.
Metagenome-assembled genomes (MAGs)
Six metagenomes were generated from DNA extracted from biomass collected on a 0.2 µm pore size filter (in 2016 sampling months; June (one metagenome), September (three metagenomes), and November (two metagenomes)) and one metagenome was generated from biomass collected on a 0.1 µm pore size filter (sampling month; November 2016). 16S rRNA gene amplicon libraries were generated for each of the metagenome sequenced samples.
Assembled metagenomes were binned using CONCOCT [32]. The resulting bins were assessed for contamination and completeness using CheckM [33]. High quality (>95% completeness, <5% contamination) and good quality (>75% completeness, <10% contamination) bins were retained and considered MAGs. Genes were annotated with KEGG Orthology using GhostKOALA [34] and eggNOG [35]. TIGRFAMs were annotated using WebMGA [36] and hydrogenases were classified using HydDB [37]. Metabolic pathways were constructed using KEGG Mapper [38] and MetaCyc [39]. Pathway completeness was determined with KEGG Decoder [40].
Metaproteomics
Two sample months (September and November 2016) were chosen for paired metagenomic and metaproteomic analyses. Protein was extracted at the Oak Ridge National Laboratory (Oak Ridge, TN, USA). A detailed description can be found in [11]. A protein database was constructed from protein sequences predicted from the assembled genomes. All collected MS/MS spectra were searched against protein databases using Myrimatch v2.2 [41]. Peptides were identified and assembled into proteins using IDPicker v3.1 [42] with a minimum of two distinct peptides per protein and a false discovery rate of <1% at the peptide level. Each sample was run in triplicate.
Single amplified genomes (SAGs)
Genomes of individual cells were sequenced to augment MAGs and uncover novel and/or rare diversity. Cells were collected in November 2016. A shotgun library was constructed for 138 SAGs (Supplementary Information). In total, 75 SAGs were recovered with >10% completeness (Table S1). Thus, SAGs were used in support of the MAGs by confirming the presence of genes using both sequencing methods, but metabolism usually could not be determined from SAGs alone. Average nucleotide identity between SAGs and MAGs was determined with FastANI [43]. Two SAGs were recovered with greater than 60% completeness and were included in our metabolic analysis.
Microbial enrichments
Biomass was collected for enrichments (2018) by pumping groundwater directly into sterile, N2 flushed, 1 L serum bottles. Bottles were filled so that there was no headspace and stored at 4 °C. Bottles were then transported in coolers from the sampling site in Finland to the laboratory in Switzerland. Within 1 week of sampling, groundwater was transferred into sterile, anoxic 120 mL Wheaton glass serum bottles, using an N2 flushed syringe. The groundwater was amended with an electron donor (Na2S; 0.40 mM), electron acceptor (NaNO3; 0.64 mM or NaNO2; 0.80 mM), and a nitrogen source (NH4Cl; 1 mM) from sterile anoxic stock solutions. Control incubations were prepared in the same way and subject to one cycle of autoclaving. All incubations were conducted in triplicate. Serum bottles were incubated at 15 °C and periodically subsampled with an N2 flushed syringe. Subsamples were mixed with zinc acetate (1% final concentration) to fix hydrogen sulfide, and stored at −20 °C.
Results and discussion
Groundwater chemistry
The groundwater was temperate (10.1 °C), of circumneutral pH (pH value 7.7), hypoxic (<10.5 ppb O2, measured upon sampling at the surface), and reducing (oxidation reduction potential ~−150 mV). As pumping commenced and groundwater was drawn from within the fracture, an initial change in hydrogeochemistry was observed, followed by stabilization as pumping continued (Table 1); total dissolved solids decreased from 5.20 to ~4.72 g/L, chloride decreased from 79.26 to 71.36 mM, and sulfate decreased from 3.54 to 2.95 mM.
Table 1.
Groundwater chemistry.
| Sample date | Sulfate (mM) | Sulfide (µM) | Iron(total) (µM) | Iron(II) (µM) | Sodium (mM) | Chloride (mM) | TDS (mg/L) |
|---|---|---|---|---|---|---|---|
| 08/02/2016 | 3.54 | <0.62 | 3.94 | 3.76 | 60.90 | 79.26 | 5.20 |
| 14/03/2016 | 3.38 | <0.62 | 10.39 | 10.21 | 58.72 | 76.44 | 4.99 |
| 26/04/2016 | 3.12 | 0.94 | 2.87 | 2.69 | 57.85 | 72.21 | 4.76 |
| 30/05/2016 | 3.06 | 1.87 | 2.33 | 1.97 | 57.85 | 72.21 | 4.73 |
| 27/06/2016 | 2.95 | 1.25 | 4.12 | 1.07 | 56.11 | 71.36 | 4.72 |
| 01/08/2016 | 2.90 | 4.05 | 1.47 | 1.07 | 57.42 | 72.77 | 4.79 |
| 19/09/2016 | 2.88 | 4.37 | 1.68 | 1.97 | 56.11 | 69.95 | 4.64 |
| 31/10/2016 | 2.94 | 0.94 | 1.34 | 1.43 | 56.55 | 71.36 | 4.72 |
TDS total dissolved solids.
Sulfate was abundant, and the concentration was relatively stable for long term (~3 mM measured from 2014 to 2018). Comparison of the sulfate concentration to total sulfur indicates that there are no other abundant sulfur species in the groundwater (Table S2). Sulfide was initially below detection limit (<0.62 µM) and was detected at ≤4.37 µM as pumping continued (Table 1). Sulfate in Olkiluoto brackish groundwater originates from the Littorina Sea, which infiltrated 2500–8000 years ago, and has a sulfate sulfur stable isotopic composition (δ34SSO4) of ~25‰ VCDT [26]. The sulfur isotopic composition of sulfate in the sampled fracture was 25.19 ± 0.82‰ VCDT (Table S2). Sulfate reduction generates 34S-enriched sulfate, as sulfate-reducing bacteria preferentially utilize the light isotope, 32S. The sulfur isotopic composition of sulfate is not depleted in 32S relative to sulfate with the same source at Olkiluoto, suggesting that there has been no further microbial sulfate reduction.
Reconstruction of subsurface genomes
In total, 65 MAGs were recovered from the seven metagenomes; 45 were high quality and 20 were good quality (Table S3). Dereplication resulted in 26 MAGs of different taxa (Table S3). Eight of the different MAGs contained a 16S rRNA gene sequence that covered the V4 region and could be directly compared with operational taxonomic units (OTUs) from our 16S rRNA gene amplicon dataset. MAGs that were missing a 16S rRNA gene sequence covering the V4 region were related back to 16S rRNA gene amplicons indirectly based on the phylogeny determined for that MAG (Table S3).
The first SAG recovered, Candidatus Omnitrophica, shared 100% 16S rRNA sequence identity with an Omnitrophicaeota OTU (OTU 476), which has an average relative abundance of 0.02% in 16S rRNA gene amplicon libraries (Table S4). The second SAG, Candidatus Levybacterales, did not contain a 16S rRNA gene sequence, but the same lineage was detected by 16S rRNA gene amplicon sequencing at 0.01% relative abundance. These taxa were also low abundance in the metagenome, highlighting the ability of single-cell sequencing to detect rare taxa that would not be recovered by metagenomic binning due to their low abundance in the environment.
Abundance of sulfate-reducing Deltaproteobacteria
Analysis of 16S rRNA gene amplicon libraries showed an abundance of putative sulfate-reducing Deltaproteobacteria (Fig. 1). As hydrogeochemical evidence for sulfate reduction was minimal, the abundance of sulfate-reducing genera from the Deltaproteobacteria was unexpected. Evidence for dissimilatory sulfur metabolism was sought by probing the metagenome for the genes necessary to perform dissimilatory sulfate reduction; sulfate adenylyltransferase (sat), adenylylsulfate reductase alpha, and beta subunits (aprAB), dissimilatory sulfite reductase alpha and beta subunits (dsrAB), and dissimilatory sulfite reductase D (dsrD). The presence of dsrD was considered necessary for a MAG to be classified as a sulfate reducer as this gene is absent in microorganisms that use reverse dissimilatory sulfite reductase for sulfur oxidation [44–46]. It is important to note, however, that there are exceptions to this rule. For instance, Desulfurivibrio alkaliphilus has also been reported to oxidize sulfide, despite having the complete genetic make-up for dissimilatory sulfate reduction [47]. In addition, Desulfocapsa sulfexigens, which was isolated from marine sediments, is unable to respire sulfate despite having the complete set of genes known to be involved in dissimilatory sulfate reduction [48]. Thus, dissimilatory sulfate reduction cannot be determined definitively from genomic features alone.
Fig. 1. Operational taxonomic units (OTUs) identified by 16S rRNA gene amplicon sequencing.
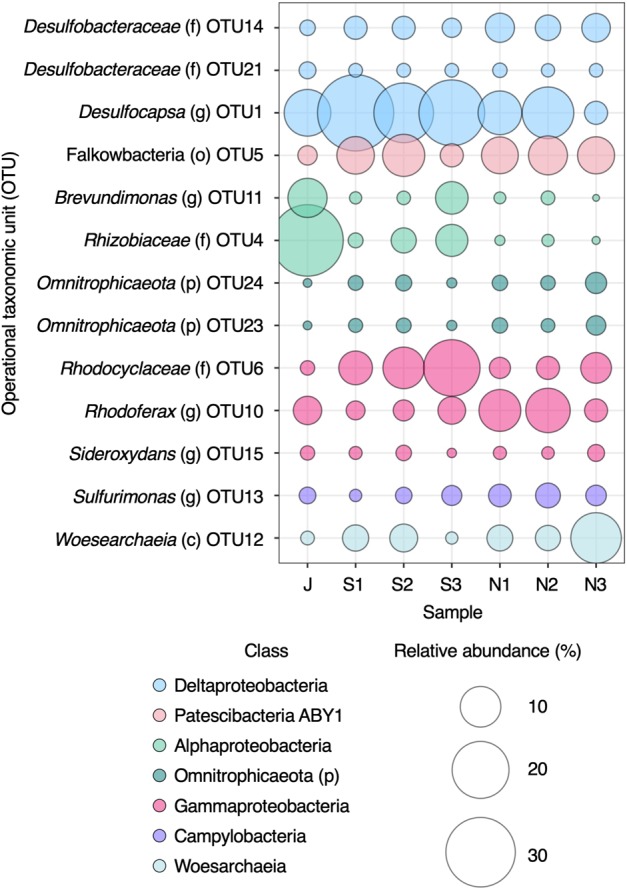
For clarity, only abundant OTUs (≥1% average relative abundance) are shown. Letters in parentheses following OTU names denote the taxonomy level the OTU was assigned to; p phylum, c class, o order, f family, g genus. All OTUs are provided in Table S4. Samples are named after collection month in 2016: J June; S1, S2, and S3 = three sampling times in September; N1, N2, and N3 = three sampling times in November.
Seven Deltaproteobacteria MAGs were recovered with the complete genetic capacity for dissimilatory sulfate reduction. These MAGs also encoded the two membrane complexes QmoABC and DsrMKJOP involved in electron transfer as well as the sulfur relay protein DsrC (Fig. 2). Five of the seven Deltaproteobacteria expressed genes for dissimilatory sulfate reduction in their proteome (Fig. 2), providing evidence that these microorganisms are active.
Fig. 2. Metabolic analysis of metagenome-assembled genomes (MAGs) and single amplified genomes (SAGs) uncovered from Olkiluoto groundwater.

Percent (%) reads mapped to MAG is the mean read coverage from separate assemblies (Table S3). Percent (%) of proteins assigned to MAG is the mean number of proteins identified per MAG as a percentage of the total metaproteome (Table S3). Subunit completeness refers to the presence of genes encoding all subunits for a given protein. Abbreviations: Disproportionation (Disp), S metabolism (S met.), oxidative phosphorylation (Ox P), hydrogen oxidation (H Ox.), organic carbon (Org. C), central carbon metabolism (CCM), motility (Mot.), dissimilatory nitrate reduction to ammonia (DNRA). Refer to Tables S5 and S6 for further detail of genes included in the metabolic analysis and Supplementary Information for additional discussion on carbon metabolism.
Acetyl-CoA synthetase and alcohol dehydrogenase were detected in the proteome of Desulfocapsa (Fig. 2), suggesting that the acetyl-CoA pathway could also be used for the oxidation of small organic compounds. All genomes of Deltaproteobacteria contained [NiFe] group 1 respiratory H2-uptake hydrogenases (Fig. 2), which can couple hydrogen oxidation to sulfate respiration. The large subunit (hyaB) was found in the proteome of Desulfobacula, suggesting this sulfate reducer utilizes hydrogen as an electron donor.
Deltaproteobacteria genomes also had the metabolic capacity for the disproportionation of intermediate oxidation state sulfur compounds (Fig. 2). Thiosulfate disproportionation is a trait common amongst sulfate-reducing Deltaproteobacteria [49, 50], and was determined for the MAGs by the presence of thiosulfate reductase (phsA). The genes for the disproportionation of thiosulfate and elemental sulfur (polysulfides) are annotated together with the same KEGG ortholog (K08352; EC:1.8.5.5). In both the Desulfocapsa and Desulfurivibrio genomes, the catalytic subunit of thiosulfate reductase (phsA)/polysulfide reductase (psrA) was adjacent to a 4Fe–4S ferredoxin annotated as tetrathionate reductase subunit B (ttrB), which has a high similarity to the same subunit of polysulfide reductase (psrB) [51]. PsrAB also requires an integral membrane subunit (psrC). This subunit is from the NrfD-type family that includes tetrathionate, DMSO, polysulfide, and nitrite reductases [51]. Both genomes encoded a gene annotated as a delta subunit of nitrate reductase adjacent to the putative psrAB, as well as multiple polysulfide reductase (nrfD) elsewhere in the genome. These genes (phsA/psrA, ttrB, and nrfD) may be used for the dissimilatory reduction of thiosulfate, polysulfide, or tetrathionate with an electron donor such as hydrogen. Alternatively, some Desulfocapsa and Desulfurivibrio species are known to disproportionate elemental sulfur [52–54]. Thus, we speculate that these genes could be involved in the disproportionation of sulfur in Desulfocapsa and Desulfurivibrio.
Under standard conditions, sulfur disproportionation is endergonic, and requires the sulfide to be reoxidized or precipitated with metal ions to be thermodynamically favorable [55, 56]. The concentration of sulfide in the groundwater is low (<5 µM). Iron and manganese (oxyhydr)oxides are not expected to occur in deep bedrock at Olkiluoto, although iron silicates (biotite and chlorite) in the rock matrix could include microscopic iron (oxyhydr)oxides [26, 57]. Dissolved iron(II) and manganese are both detected in the groundwater (~2 and ~5 µM, respectively) and could provide a sink for sulfide via abiotic precipitation. We propose that dissolved iron(II) may be produced by the metabolic activity of Rhodoferax. This MAG encoded the Mtr pathway for metal reduction (Fig. 2). The decaheme c-type cytochrome (MtrC) and decaheme-associated outer membrane protein (MtrB) were detected in the proteome, suggesting that this microorganism has the capability to respire extracellular electron acceptors such as iron(III).
Uncovering a sulfur cycle
The oxidation of sulfide by sulfide-oxidizing microorganisms could also account for the lack of sulfide in a groundwater where sulfate-reducing and sulfur-disproportionating bacteria are active. Genes for oxidative sulfur cycling were therefore sought within the genomes recovered from the groundwater. Indeed, ten MAGs contained genes for oxidative sulfur cycling (Fig. 2). The metabolic activity of sulfide-oxidizing bacteria was further confirmed by the presence of peptides from sulfide-oxidizing genes in the proteome (Fig. 2), indicating that sulfide is maintained at low concentrations in this groundwater at least partly by the activity of sulfide-oxidizing bacteria.
The first sulfide oxidation pathway considered was the reverse sulfite reduction pathway, including the genes dsrABEFH and dsrC. Deconvoluting whether the dissimilatory sulfite reductase (encoded by dsrAB) is a reverse- or a forward-acting isoform relies on the presence of genes encoding the subunits DsrEFH, which are essential for the reverse function, or that of the gene encoding DsrD, needed for the forward action [44, 58]. Peptides from the reverse-acting dissimilatory sulfite reductase were detected in the proteome of Rhodocyclaceae and Sideroxydans, along with peptides from DsrEFH and DsrC (Fig. 2). In sulfur oxidizers, DsrEFH and DsrC are involved in sulfur transfer to DsrAB acting in the oxidative direction, i.e., in the reverse direction of the reaction catalyzed in sulfate reducers [59]. Evidence for the expression of both these proteins in addition to DsrAB thus points to active oxidation of sulfur, an intermediate formed during the oxidation of sulfide and thiosulfate [60, 61]. In addition, sulfite generated by reverse dissimilatory sulfite reductase can be oxidized to sulfate via adenylylsulfate reductase and sulfate adenylyltransferase (aprBA-sat) operating in the reverse direction or by the cytoplasmic sulfite oxidizing enzyme soeABC. Rhodocyclaceae and Sideroxydans contained both aprBA-sat and soeABC in their genome (Fig. 2). Rhodocyclaceae expressed AprBA-Sat indicating this sulfide oxidizer reduces sulfite to sulfate via the two-step (sulfite to APS and APS to sulfate) pathway.
The Sox pathway, a multienzyme system that oxidizes the sulfur compound stepwise, finally producing sulfate [58, 62], is also a prominent form of sulfide oxidation. Rhodocyclaceae, Sideroxydans, and Rhizobiaceae all harbored genes for the oxidation of reduced sulfur compounds catalyzed by the Sox pathway. The complete Sox pathway was expressed in the proteome of Rhizobiaceae (Fig. 2) indicating that Rhizobiaceae produces sulfate. Rhodocyclaceae and Sideroxydans harbor an incomplete Sox pathway (soxXAYZB) and lack the sulfur dehydrogenase (soxCD), as observed for other Betaproteobacteriales [63]. In organisms where sulfur dehydrogenase (soxCD) is absent, elemental sulfur deposits accumulate, which can be subsequently oxidized via the reverse dissimilatory sulfate reduction pathway, generating sulfite [64]. There was no proteomic evidence for active Sox-dependent sulfide oxidation by these two bacteria, only genome-based potential.
Sulfite oxidation can also be catalyzed directly by the sulfite:cytochrome c oxidorectase (sorAB) [65]. Both subunits of this gene were detected in Sulfurimonas MAG, but they were not expressed in the proteome (Fig. 2). Sulfurimonas did express sulfide:quinone oxidoreductase (sqr), which catalyzes the oxidation of sulfide to elemental sulfur. The produced sulfur can be stored intracellularly or deposited outside of the cell [66]. This gene was also detected in eight other MAGs (Fig. 2), seven of which contained no other sulfide-oxidizing genes. In some species that harbor only sqr, such as the Propionicimonas MAG, sulfide may be oxidized to sulfur either as a strategy to detoxify sulfide, or as a sulfur assimilation strategy [67, 68].
Desulfurivibrio alkaliphilus can grow by coupling sulfide oxidation to dissimilatory reduction of nitrate to ammonium [47]. In D. alkaliphilus, the only known gene associated with sulfide oxidation detected in the genome was sulfide:quinone oxidoreductase (sqr) [47]. Similar to D. alkaliphilus, the Desulfurivibrio MAG uncovered here also contained sqr as well as the gene repertoire for dissimilatory sulfate reduction (Fig. 2). Thus, it is possible that Desulfurivibrio may also be involved in oxidative sulfur cycling here, although this cannot be unambiguously distinguished from genomic features.
The detection of multiple pathways catalyzing sulfur transformations in the metaproteome (Figs. 2 and 3) suggests that the sulfur isotopic signature of the groundwater reflects isotope fractionation controlled by reduction, disproportionation, and oxidation. Like sulfate reduction, the disproportionation of sulfur also generates 34S-enriched sulfate whereby 32S is preferentially used for the formation of sulfide and 34S is preferentially used for sulfate production [69], although the extent of the fractionation can be variable depending on the enzymatic pathway and environmental conditions [70, 71]. While the fractionation associated with sulfide oxidation can be relatively small [71], the oxidation of sulfide to sulfate by sulfide-oxidizing bacteria would transfer the light isotopic signature of sulfide produced by sulfate-reducing bacteria back to the sulfate pool [24]. Over 2 years of sampling (2016–2018), there was no isotopic enrichment of sulfur in sulfate. The oxidative and reductive cycling of sulfur compounds may explain why the expected isotopic depletion of 32S associated with sulfate reduction was not readily detected. An alternative interpretation is that the rate of activity (both oxidative and reductive) may be extremely low. In the marine subsurface, mean generation times can reach thousands of years and nutrient turnover may take hundreds of years [72]. This could also make it difficult to detect activity by geochemical and isotopic methods.
Fig. 3. Sulfur cycling in Olkiluoto groundwater.

Solid colored arrows indicate oxidation/reduction reactions that were expressed in the proteome. Dashed colored arrows indicate that only genomic potential for that reaction was found. Genes involved in each reaction are indicated for each MAG. Putative processes are indicated by a dashed gray line. SCFA short chain fatty acids.
Linking nitrogen and sulfur cycles
The sulfide-oxidizing bacteria identified are facultative anaerobes. Their genomes contained terminal oxidases for oxygen reduction and reductases for nitrate reduction to N2, nitrite, (Fig. 3) or ammonium (Fig. 2). Nitrate reductase (napAB) was detected in the proteome of Rhodocyclaceae as was a cbb3-type oxidase, suggesting Rhodocyclaceae is actively respiring nitrate and oxygen. This periplasmic nitrate reductase (nap) has a high affinity for nitrate [73] and may be advantageous in a nitrate-limited environment, like Olkiluoto. The presence of high-affinity cbb3-type oxidase suggests that electrons from the oxidation of reduced sulfur compounds could be utilized to reduce dissolved oxygen, if trace concentrations were present in the groundwater. However, groundwaters at Olkiluoto with long residence times are highly reducing and there is no evidence for oxygen penetration [26]. Cbb3-oxidases can also be expressed during anaerobic biological processes such as nitrogen fixation [74] and denitrification [75]. The cbb3-type oxidase has also been suggested to scavenge oxygen in species not known to utilize oxygen as an electron acceptor, preventing oxidative stress [76, 77]. Desulfocapsa expressed nitrate reductase (napA) and nitrite reductase (nrfA), which together catalyze the reduction of nitrate to ammonia (Fig. 2). Many sulfate-reducing bacteria encode NapA and can use nitrate as an alternative terminal electron acceptor [78]. The nitrite reductase NrfAH is commonly found among sulfate-reducing bacteria and is proposed to detoxify nitrite, suppressing the inhibition of sulfate reduction by nitrite [79, 80].
The detection of nitrate and nitrite reductases in the proteome indicates that nitrogen compounds must be cycled at concentrations below detection limit, as neither nitrate nor nitrite were detected in the groundwater (limit of quantification 8 and 11 µM, respectively). The source of nitrate/nitrite in the groundwater is unknown but given that the groundwater is not meteorically recharged, nitrogen compounds must be generated in situ. Ammonium is detected in the groundwater (~5 µM) and could possibly be oxidized through coupling to the reduction of sulfate, iron, or manganese [81–83]. The produced oxidized nitrogen compounds would then be rapidly utilized and thus not accumulate in the groundwater. However, the enzymes required for this anaerobic ammonium oxidation are not well understood and we could not determine this metabolism from any genomes recovered. There also was no evidence for anammox (nitrite-dependent anaerobic ammonium oxidation).
Nitrate-reducing and denitrifying bacteria have been detected elsewhere in the Fennoscandian Shield [14, 17] and in other subsurface environments where nitrate appears to be limited [18, 84]. Anaerobic ammonium oxidation was proposed as a possible source of nitrate in subseafloor sediments from the Peru margin, where transcripts encoding nitrate reductases were detected but nitrate was not [84]. Active sulfide-oxidizing denitrifying bacteria were found to dominate a deep groundwater from the Witwatersrand Basin, South Africa, where only trace concentrations of nitrate were detected (0.3 µM) [18]. In that case, the source was proposed to be either radiolytic oxidation of ammonium in the rock formation porewater, or paleometeoric recharge.
Carbon fixation in deep groundwater
Dissolved inorganic carbon is relatively high in the groundwater (2.6 ± 0.1 mM) and can provide a carbon source for autotrophic microorganisms. The key enzymes of the reductive acetyl-CoA (Wood–Ljungdahl) pathway, anaerobic carbon monoxide dehydrogenase (cooS), and acetyl-CoA synthase (acsB) were detected in all Deltaproteobacteria MAGs, and corresponding peptides were detected in the proteomes of Desulfocapsa and Desulfobacula (Fig. 2). Deltaproteobacteria were missing the genes of the CO2-reducing formate dehydrogenase (fdhAB, EC:1.17.1.10), which catalyzes the two-electron reduction of CO2 to formate, the first step in the methyl branch of the Wood–Ljungdahl pathway, but analogous subunits from another formate dehydrogenase were present (fdoG, EC:1.17.1.9).
Two carbon fixation pathways were identified among sulfide-oxidizing MAGs, the reductive pentose phosphate pathway and the reductive tricarboxylic acid cycle (Fig. 2). Peptides from key enzymes of the reductive pentose phosphate pathway, ribulose-1,5-biphosphate carboxylase (RubisCO) and phosphoribulokinase, were detected in the proteomes of Rhodocyclaceae, Sideroxydans, and Rhizobiaceae (Fig. 2). Sulfurimonas harbored the reductive tricarboxylic acid cycle and the key enzyme ATP-citrate lyase (aclAB) was detected in the proteome (Fig. 2). These microorganisms are thus inferred to fix inorganic carbon.
Sulfurimonas oxidizes sulfide to sulfur
Metaproteogenomics revealed active autotrophic sulfide-oxidizers using oxidized nitrogen species as electron acceptors in a sulfide-poor environment (Figs. 2 and 3). To provide direct evidence for this metabolism, groundwater was amended with sulfide and stoichiometric amounts of nitrate or nitrite in the laboratory under anoxic conditions. Within 10 days of incubation, the amended sulfide was completely oxidized (Fig. 4a, b). DAPI (4′6-diamidino-2-phenylindole) stained cell images from 14 days incubation confirmed enrichment in biomass. To further enrich sulfide-oxidizing bacteria, the incubations were spiked with sulfide and nitrate or nitrite again after 27 days of incubation. Sulfide was again completely oxidized (Fig. 4a, b). After 49 days of incubation, a total of 1.01 ± 0.11 mM sulfide had been oxidized and 0.40 ± 0.07 mM nitrate reduced (Fig. 4a). The stoichiometry indicates that sulfide is oxidized to sulfur when coupled to denitrification (Eq. 1). Sulfate did not accumulate in the enrichments, consistent with the oxidation of sulfide to an intermediate oxidation state species.
| 1 |
Fig. 4. Enrichment of sulfide-oxidizing bacteria from Olkiluoto groundwater.

Groundwater amended with sulfide (HS−) and nitrate (NO3−) (a) or nitrite (NO2−) (b). Abundant genera enriched following incubation are shown relative to in situ abundance (c). Error bars show standard deviation (SD) (N = 3). All OTUs are provided in Table S8.
In incubations amended with nitrite, a total of 0.88 ± 0.07 mM sulfide was oxidized and 1.07 mM ± 0.08 mM nitrite reduced (Fig. 4b). Sulfate also did not accumulate in incubations with nitrite (0.16 ± 0.09 mM increase in sulfate) showing that the sulfide is not fully oxidized to sulfate. However, a greater concentration of nitrite was consumed than the requirement to oxidize sulfide to sulfur (Eq. 2), and it was neither consistent with the oxidation of sulfide to sulfite nor to thiosulfate.
| 2 |
The concentration of nitrite continues to decrease after sulfide is depleted, suggesting a secondary process is occurring. If sulfide is oxidized to elemental sulfur, this may fuel further nitrite consumption via denitrification. Or perhaps there is another sink for nitrite, such as abiotic reaction with iron(II) [85], although the iron(II) concentration in the groundwater was low (<1 µM) and nitrite was not reduced in sterile control incubations (Table S7).
16S rRNA gene amplicon libraries generated from the enrichments showed an abundance of the genus Sulfurimonas, accounting for ~80% of the amplicon relative abundance (Fig. 4c). Multiple OTUs of Sulfurimonas were detected in the enrichments (Table S8). The same OTUs were detected by 16S rRNA gene amplicon sequencing of the groundwater in 2016 and 2018 (Table S8). There was no 16S rRNA gene sequence in the Sulfurimonas MAG recovered in 2016 so it is not possible to conclude if the MAG corresponds to one of the Sulfurimonas OTUs detected.
The genus Sulfurimonas consists of a group of sulfur-oxidizing bacteria that can grow with a broad range of reduced sulfur compounds [76, 86, 87]. Published genomes of Sulfurimonas isolates encode sulfide:quinone oxidoreductase (sqr), which catalyzes sulfide oxidation, as well as the Sox multienzyme complex (soxXYZAB) that oxidizes reduced sulfur compounds to sulfate [65]. The Sulfurimonas MAG recovered in 2016 encoded three sulfide:quinone oxidoreductases (sqr) but lacked the Sox gene cluster (Fig. 2), suggesting it is unable to completely oxidize sulfide to sulfate. Experimental data indicate that in enrichments dominated by Sulfurimonas, sulfide is oxidized primarily to sulfur under nitrate- and nitrite-reducing conditions (Fig. 4a–c). This is consistent with the genetic capacity of the Sulfurimonas MAG recovered.
The Sulfurimonas MAG encoded the nitrate reductase napAB, which has a high affinity for nitrate, and may represent an adaptation to low nitrate environments [65]. This would be advantageous in Olkiluoto groundwater where nitrate is below the detection limit. Unusually, the Sulfurimonas MAG lacked genes for complete denitrification (Fig. 2) and the only nitrite reductase found was an assimilatory type, despite being clearly enriched in incubations with nitrite (Fig. 4c). One explanation is that the genes for nitrite reduction were not binned, although no nitrite reductases belonging to Campylobacteria were found in the metagenome. An alternative explanation is that different genotypes of Sulfurimonas exist in the groundwater and the MAG recovered in 2016 does not represent the Sulfurimonas OTUs detected by 16S rRNA gene amplicon sequencing in groundwater incubations from 2018.
The concentrations of sulfide and nitrate/nitrite added to these groundwater incubations exceed the concentration that would be encountered in situ, nevertheless it demonstrates the metabolic capacity for active coupling of sulfur and nitrogen cycles in Olkiluoto groundwater. Clearly, these incubations artificially enrich a single genus (Fig. 4c) and sulfide is incompletely oxidized to sulfur. Under natural conditions, intermediate oxidation state sulfur compounds generated by one genus can be further oxidized to sulfate by other genera (perhaps unable to grow in laboratory conditions) with the metabolic capacity for complete oxidation or provide an energy source for disproportionating sulfate reducers (Fig. 3).
Conclusion
The deep terrestrial subsurface remains an environment where there is a limited understanding of the extant metabolisms, despite the reported large contribution of the deep subsurface to the overall biomass on this planet. The hydrological isolation of this environment suggests that extensive elemental (re)cycling should occur in order to support microbial processes. Here we show that, even in the absence of hydrogeochemical evidence for sulfur and nitrogen transformation, an extensive sulfur cycle coupled to nitrate reduction can be unveiled by a combination of metagenomic and metaproteomic approaches. The main driver of the sulfur cycle is microbial sulfate reduction primarily by Deltaproteobacteria, which ultimately produces sulfide. Due to the lack of the expected metabolic end product and a stable isotopic signature that was not depleted in 32S, active sulfate reduction was only revealed by probing the metagenome and the metaproteome. This also revealed the activity of autotrophic, sulfide-oxidizing bacteria, which are able to recycle sulfide making sulfate reduction difficult to detect. Metagenomic and metaproteomic evidence of disproportionation and experimental evidence for sulfide oxidation to sulfur also indicate that intermediate oxidation state sulfur species, which are also below detection limit in the groundwater, may also play an important role in the biogeochemical cycling of sulfur in Olkiluoto groundwater.
The results have implications in terms of nuclear waste repository construction at this site. The generation of sulfide is a concern as it has the potential to corrode copper canisters used to store spent nuclear fuel. The activity of sulfide oxidizers could therefore provide a beneficial sink for sulfide in Olkiluoto groundwater, which could limit the accumulation of sulfide after repository closure. Presently, the rate of sulfate reduction may be relatively low due to limited electron donor availability. Similarly, sulfide oxidation is presumably limited by the availability of nitrate but still sufficient to maintain a low concentration of sulfide in the groundwater. These results shed light on sulfur cycling in the terrestrial deep subsurface and show that limited hydrogeochemical evidence for sulfate reduction need not be interpreted as a lack of microbial sulfur cycling.
Supplementary information
Acknowledgements
The authors would like to thank Maarit Yli-Kaila and Raila Viitala for their support and assistance conducting field work, Louise Balmer and Guillaume Sommer for their help collecting samples, and Petteri Pitkänen for valuable discussions. This research was funded by a grant from Posiva Oy to RBL (2015-06-17). Additional funding for metagenomic sequencing was provided by The Census of Deep Life within the Deep Carbon Observatory and the U.S. Department of Energy Joint Genome Institute (JGI). The work conducted by the JGI, a DOE Office of Science User Facility, is supported under contract no. DE-AC02-05CH11231. The metagenomic computations were performed on resources provided by SNIC through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX).
Data availability
16S rRNA gene amplicon data are available at the National Centre for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under BioProject PRJNA472445. Metagenomic data were deposited at the NCBI SRA; accession numbers PRJNA404452, PRJNA404453, PRJNA444021, and PRJNA472439. MAGs are available under accessions SAMN12221356–SAMN12221381. SAGs are available at the JGI Genome Portal, GOLD Study ID’s Ga0214289 and Gs0127568.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41396-020-0602-x) contains supplementary material, which is available to authorized users.
References
- 1.Mcmahon S, Parnell J. Weighing the deep continental biosphere. FEMS Microbiol Ecol. 2014;87:113–20. doi: 10.1111/1574-6941.12196. [DOI] [PubMed] [Google Scholar]
- 2.Magnabosco C, Lin L-H, Dong H, Bomberg M, Ghiorse W, Stan-Lotter H, et al. The biomass and biodiversity of the continental subsurface. Nat Geosci. 2018;11:707–17. [Google Scholar]
- 3.Pedersen K. Microbial life in deep granitic rock. FEMS Microbiol Rev. 1997;20:399–414. [Google Scholar]
- 4.Bagnoud A, de Bruijn I, Andersson AF, Diomidis N, Leupin OX, Schwyn B, et al. A minimalistic microbial food web in an excavated deep subsurface clay rock. FEMS Microbiol Ecol. 2016;92:fiv138. doi: 10.1093/femsec/fiv138. [DOI] [PubMed] [Google Scholar]
- 5.Bagnoud A, Chourey K, Hettich RL, De Bruijn I, Andersson AF, Leupin OX, et al. Reconstructing a hydrogen-driven microbial metabolic network in Opalinus Clay rock. Nat Commun. 2016;7:12770. doi: 10.1038/ncomms12770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boylan AA, Perez-Mon C, Guillard L, Burzan N, Loreggian L, Maisch M, et al. H2-fuelled microbial metabolism in Opalinus Clay. Appl Clay Sci. 2019;174:69–76. [Google Scholar]
- 7.Ino K, Hernsdorf AW, Konno U, Kouduka M, Yanagawa K, Kato S, et al. Ecological and genomic profiling of anaerobic methane-oxidizing archaea in a deep granitic environment. ISME J. 2018;12:31–47. doi: 10.1038/ismej.2017.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chivian D, Brodie EL, Alm EJ, Culley DE, Dehal PS, DeSantis TZ, et al. Environmental genomics reveals a single-species ecosystem deep within earth. Science. 2008;322:275–8. doi: 10.1126/science.1155495. [DOI] [PubMed] [Google Scholar]
- 9.Hernsdorf AW, Amano Y, Miyakawa K, Ise K, Suzuki Y, Anantharaman K, et al. Potential for microbial H2 and metal transformations associated with novel bacteria and archaea in deep terrestrial subsurface sediments. ISME J. 2017;11:1915–29. doi: 10.1038/ismej.2017.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haveman SA, Pedersen K. Distribution of culturable microorganisms in Fennoscandian Shield groundwater. FEMS Microbiol Ecol. 2002;39:129–37. doi: 10.1111/j.1574-6941.2002.tb00914.x. [DOI] [PubMed] [Google Scholar]
- 11.Bell E, Lamminmäki T, Alneberg J, Andersson AF, Qian C, Xiong W, et al. Biogeochemical cycling by a low-diversity microbial community in deep groundwater. Front Microbiol. 2018;9:2129. doi: 10.3389/fmicb.2018.02129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nyyssönen M, Hultman J, Ahonen L, Kukkonen I, Paulin L, Laine P, et al. Taxonomically and functionally diverse microbial communities in deep crystalline rocks of the Fennoscandian Shield. ISME J. 2014;8:126–38. doi: 10.1038/ismej.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pedersen K. Metabolic activity of subterranean microbial communities in deep granitic groundwater supplemented with methane and H2. ISME J. 2013;7:839–49. doi: 10.1038/ismej.2012.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Purkamo L, Bomberg M, Nyyssönen M, Kukkonen I, Ahonen L, Itävaara M. Heterotrophic communities supplied by ancient organic carbon predominate in deep fennoscandian bedrock fluids. Micro Ecol. 2015;69:319–32. doi: 10.1007/s00248-014-0490-6. [DOI] [PubMed] [Google Scholar]
- 15.Wu X, Holmfeldt K, Hubalek V, Lundin D, Åström M, Bertilsson S, et al. Microbial metagenomes from three aquifers in the Fennoscandian Shield terrestrial deep biosphere reveal metabolic partitioning among populations. ISME J. 2016;10:1192–203. doi: 10.1038/ismej.2015.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Purkamo L, Bomberg M, Kietäväinen R, Salavirta H, Nyyssönen M, Nuppunen-Puputti M, et al. Microbial co-occurrence patterns in deep Precambrian bedrock fracture fluids. Biogeosciences. 2016;13:3091–108. [Google Scholar]
- 17.Rajala P, Bomberg M, Kietäväinen R, Kukkonen I, Ahonen L, Nyyssönen M, et al. Rapid reactivation of deep subsurface microbes in the presence of C-1 compounds. Microorganisms. 2015;3:17–33. doi: 10.3390/microorganisms3010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lau MCY, Kieft TL, Kuloyo O, Linage-Alvarez B, van Heerden E, Lindsay MR, et al. An oligotrophic deep-subsurface community dependent on syntrophy is dominated by sulfur-driven autotrophic denitrifiers. Proc Natl Acad Sci USA. 2016;113:E7927–36. doi: 10.1073/pnas.1612244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berg JS, Michellod D, Pjevac P, Martinez-Perez C, Buckner CRT, Hach PF, et al. Intensive cryptic microbial iron cycling in the low iron water column of the meromictic Lake Cadagno. Environ Microbiol. 2016;18:5288–302. doi: 10.1111/1462-2920.13587. [DOI] [PubMed] [Google Scholar]
- 20.Canfield DE, Stewart FJ, Thamdrup B, De Brabandere L, Dalsgaard T, Delong EF, et al. A cryptic sulfur cycle in oxygen-minimum–zone waters off the Chilean Coast. Science. 2010;330:1375–8. doi: 10.1126/science.1196889. [DOI] [PubMed] [Google Scholar]
- 21.Callbeck CM, Lavik G, Ferdelman TG, Fuchs B, Gruber-Vodicka HR, Hach PF, et al. Oxygen minimum zone cryptic sulfur cycling sustained by offshore transport of key sulfur oxidizing bacteria. Nat Commun. 2018;9:1729. doi: 10.1038/s41467-018-04041-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holmkvist L, Ferdelman TG, Jørgensen BB. A cryptic sulfur cycle driven by iron in the methane zone of marine sediment (Aarhus Bay, Denmark) Geochim Cosmochim Acta. 2011;75:3581–99. [Google Scholar]
- 23.Hansel CM, Lentini CJ, Tang Y, Johnston DT, Wankel SD, Jardine PM. Dominance of sulfur-fueled iron oxide reduction in low-sulfate freshwater sediments. ISME J. 2015;9:2400–12. doi: 10.1038/ismej.2015.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mills JV, Antler G, Turchyn AV. Geochemical evidence for cryptic sulfur cycling in salt marsh sediments. Earth Planet Sci Lett. 2016;453:23–32. [Google Scholar]
- 25.Berg JS, Jézéquel D, Duverger A, Lamy D, Laberty-Robert C, Miot J. Microbial diversity involved in iron and cryptic sulfur cycling in the ferruginous, low-sulfate waters of Lake Pavin. PLoS ONE. 2019;14:e0212787. doi: 10.1371/journal.pone.0212787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Posiva Oy. Olkiluoto site description. Posiva report 2011-02. 2013. http://www.posiva.fi/en/databank/posiva_reports/olkiluoto_site_description_2011.1871.xhtml.
- 27.Pedersen K, Arlinger J, Eriksson S, Hallbeck A, Hallbeck L, Johansson J. Numbers, biomass and cultivable diversity of microbial populations relate to depth and borehole-specific conditions in groundwater from depths of 4-450m in Olkiluoto, Finland. ISME J. 2008;2:760–75. doi: 10.1038/ismej.2008.43. [DOI] [PubMed] [Google Scholar]
- 28.Bomberg M, Nyyssönen M, Pitkänen P, Lehtinen A, Itävaara M. Active microbial communities inhabit sulphate-methane interphase in deep bedrock fracture fluids in Olkiluoto, Finland. Biomed Res Int. 2015;2015:1–17. doi: 10.1155/2015/979530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nyyssönen M, Bomberg M, Kapanen A, Nousiainen A, Pitkänen P, Itävaara M. Methanogenic and sulphate-reducing microbial communities in deep groundwater of crystalline rock fractures in Olkiluoto, Finland. Geomicrobiol J. 2012;29:863–78. [Google Scholar]
- 30.Pedersen K, Bengtsson AF, Edlund JS, Eriksson LC. Sulphate-controlled diversity of subterranean microbial communities over depth in deep groundwater with opposing gradients of sulphate and methane. Geomicrobiol J. 2014;31:617–31. [Google Scholar]
- 31.Miettinen H, Bomberg M, Vikman M. Acetate activates deep subsurface fracture fluid microbial communities in Olkiluoto, Finland. Geosciences. 2018;8:399. [Google Scholar]
- 32.Alneberg J, Bjarnason BS, De Bruijn I, Schirmer M, Quick J, Ijaz UZ, et al. Binning metagenomic contigs by coverage and composition. Nat Methods. 2014;11:1144–6. doi: 10.1038/nmeth.3103. [DOI] [PubMed] [Google Scholar]
- 33.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol. 2016;428:726–31. doi: 10.1016/j.jmb.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 35.Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter MC, et al. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016;44:D286–93. doi: 10.1093/nar/gkv1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu S, Zhu Z, Fu L, Niu B, Li W. WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genom. 2011;12:444. doi: 10.1186/1471-2164-12-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Søndergaard D, Pedersen CNS, Greening C. HydDB: a web tool for hydrogenase classification and analysis. Sci Rep. 2016;6:34212. doi: 10.1038/srep34212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–14. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caspi R, Billington R, Fulcher CA, Keseler IM, Kothari A, Krummenacker M, et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018;46:D633–9. doi: 10.1093/nar/gkx935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Graham ED, Heidelberg JF, Tully BJ. Potential for primary productivity in a globally-distributed bacterial phototroph. ISME J. 2018;12:1861–6. doi: 10.1038/s41396-018-0091-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tabb DL, Fernando CG, Chambers MC. MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J Proteome Res. 2007;6:654–61. doi: 10.1021/pr0604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma ZQ, Dasari S, Chambers MC, Litton MD, Sobecki SM, Zimmerman LJ, et al. IDPicker 2.0: improved protein assembly with high discrimination peptide identification filtering. J Proteome Res. 2009;8:3872–81. doi: 10.1021/pr900360j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9:5114. doi: 10.1038/s41467-018-07641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rabus R, Venceslau SS, Wöhlbrand L, Voordouw G, Wall JD, Pereira IAC. A post-genomic view of the ecophysiology, catabolism and biotechnological relevance of sulphate-reducing prokaryotes. Adv Micro Physiol. 2015;66:55–321. doi: 10.1016/bs.ampbs.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 45.Mizuno N, Voordouw G, Miki K, Sarai A, Higuchi Y. Crystal structure of dissimilatory sulfite reductase D (DsrD) protein—possible interaction with B- and Z-DNA by its winged-helix motif. Structure. 2003;11:1133–40. doi: 10.1016/s0969-2126(03)00156-4. [DOI] [PubMed] [Google Scholar]
- 46.Hittel DS, Voordouw G. Overexpression, purification and immunodetection of DsrD from Desulfovibrio vulgaris Hildenborough. Antonie Van Leeuwenhoek. 2000;77:271–80. doi: 10.1023/a:1002449227469. [DOI] [PubMed] [Google Scholar]
- 47.Thorup C, Schramm A, Findlay AJ, Finster KW, Schreiber L. Disguised as a sulfate reducer: growth of the Deltaproteobacterium Desulfurivibrio alkaliphilus by sulfide oxidation with nitrate. MBio. 2017;8:e00671–17. doi: 10.1128/mBio.00671-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finster KW, Kjeldsen KU, Kube M, Reinhardt R, Mussmann M, Amann R, et al. Complete genome sequence of Desulfocapsa sulfexigens, a marine deltaproteobacterium specialized in disproportionating inorganic sulfur compounds. Stand Genom Sci. 2013;8:58–68. doi: 10.4056/sigs.3777412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krämer M, Cypionka H. Sulfate formation via ATP sulfurylase in thiosulfate- and sulfite-disproportionating bacteria. Arch Microbiol. 1989;151:232–7. [Google Scholar]
- 50.Jørgensen BB, Bak F. Pathways and microbiology of thiosulfate transformations and sulfate reduction in a marine sediment (Kattegat, Denmark) Appl Environ Microbiol. 1991;57:847–56. doi: 10.1128/aem.57.3.847-856.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jormakka M, Yokoyama K, Yano T, Tamakoshi M, Akimoto S, Shimamura T, et al. Molecular mechanism of energy conservation in polysulfide respiration. Nat Struct Mol Biol. 2008;15:730–7. doi: 10.1038/nsmb.1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Janssen PH, Schuhmann A, Bak F, Liesack W. Disproportionation of inorganic sulfur compounds by the sulfate-reducing bacterium Desulfocapsa thiozymogenes gen, nov., sp. nov. Arch Microbiol. 1996;166:184–92. [Google Scholar]
- 53.Finster KW, Kjeldsen KU, Kube M, Reinhardt R, Mussmann M, Amann R, et al. Complete genome sequence of Desulfocapsa sulfexigens, a marine deltaproteobacterium specialized in disproportionating inorganic sulfur compounds. Stand Genom Sci. 2013;8:58–68. doi: 10.4056/sigs.3777412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Melton ED, Sorokin DY, Overmars L, Chertkov O, Clum A, Pillay M, et al. Complete genome sequence of Desulfurivibrio alkaliphilus strain AHT2T, a haloalkaliphilic sulfidogen from Egyptian hypersaline alkaline lakes. Stand Genom Sci. 2016;11:67. doi: 10.1186/s40793-016-0184-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Finster K. Microbiological disproportionation of inorganic sulfur compounds. J Sulfur Chem. 2008;29:281–92. [Google Scholar]
- 56.Poser A, Lohmayer R, Vogt C, Knoeller K, Planer-Friedrich B, Sorokin D, et al. Disproportionation of elemental sulfur by haloalkaliphilic bacteria from soda lakes. Extremophiles. 2013;17:1003–12. doi: 10.1007/s00792-013-0582-0. [DOI] [PubMed] [Google Scholar]
- 57.Wersin P, Alt-Epping P, Pitkänen P, Román-Ross G, Trinchero P, Molinero J, et al. Sulphide fluxes and concentrations in the spent nuclear fuel repository at Olkiluoto. Posiva Report 2014-01. Posiva, Olkiluoto, Finland: 2014.
- 58.Dahl C, Friedrich C, Kletzin A. Sulfur oxidation in prokaryotes. In: Encyclopedia of life sciences. John Wiley & Sons, Ltd, Chichester, UK; 2008.
- 59.Venceslau SS, Stockdreher Y, Dahl C, Pereira IAC. The “bacterial heterodisulfide” DsrC is a key protein in dissimilatory sulfur metabolism. Biochim Biophys Acta. 2014;1837:1148–64. doi: 10.1016/j.bbabio.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 60.Dahl C, Engels S, Pott-Sperling AS, Schulte A, Sander J, Lübbe Y, et al. Novel genes of the dsr gene cluster and evidence for close interaction of Dsr proteins during sulfur oxidation in the phototrophic sulfur bacterium Allochromatium vinosum. J Bacteriol. 2005;187:1392–404. doi: 10.1128/JB.187.4.1392-1404.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pott AS, Dahl C. Sirohaem sulfite reductase and other proteins encoded by genes at the dsr locus of Chromatium vinosum are involved in the oxidation of intracellular sulfur. Microbiology. 1998;144:1881–94. doi: 10.1099/00221287-144-7-1881. [DOI] [PubMed] [Google Scholar]
- 62.Friedrich CG, Bardischewsky F, Rother D, Quentmeier A, Fischer J. Prokaryotic sulfur oxidation. Curr Opin Microbiol. 2005;8:253–9. doi: 10.1016/j.mib.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 63.Watanabe T, Kojima H, Fukui M. Complete genomes of freshwater sulfur oxidizers Sulfuricella denitrificans skB26 and Sulfuritalea hydrogenivorans sk43H: genetic insights into the sulfur oxidation pathway of betaproteobacteria. Syst Appl Microbiol. 2014;37:387–95. doi: 10.1016/j.syapm.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 64.Anantharaman K, Duhaime MB, Breier JA, Wendt KA, Toner BM, Dick GJ. Sulfur oxidation genes in diverse deep-sea viruses. Science. 2014;344:757–60. doi: 10.1126/science.1252229. [DOI] [PubMed] [Google Scholar]
- 65.Han Y, Perner M. The globally widespread genus Sulfurimonas: versatile energy metabolisms and adaptations to redox clines. Front Microbiol. 2015;6:989. doi: 10.3389/fmicb.2015.00989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schütz M, Maldener I, Griesbeck C, Hauska G. Sulfide-quinone reductase from Rhodobacter capsulatus: requirement for growth, periplasmic localization, and extension of gene sequence analysis. J Bacteriol. 1999;181:6516–23. doi: 10.1128/jb.181.20.6516-6523.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marcia M, Ermler U, Peng G, Michel H. The structure of Aquifex aeolicus sulfide:quinone oxidoreductase, a basis to understand sulfide detoxification and respiration. Proc Natl Acad Sci USA. 2009;106:9625–30. doi: 10.1073/pnas.0904165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xia Y, Lü C, Hou N, Xin Y, Liu J, Liu H, et al. Sulfide production and oxidation by heterotrophic bacteria under aerobic conditions. ISME J. 2017;11:2754–66. doi: 10.1038/ismej.2017.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Canfield DE, Thamdrup B, Fleischer S. Isotope fractionation and sulfur metabolism by pure and enrichment cultures of elemental sulfur-disproportionating bacteria. Limnol Oceanogr. 1998;43:253–64. [Google Scholar]
- 70.Habicht KS, Canfield DE, Rethmeier J. Sulfur isotope fractionation during bacterial reduction and disproportionation of thiosulfate and sulfite. Geochim Cosmochim Acta. 1998;62:2585–95. [Google Scholar]
- 71.Poser A, Vogt C, Knöller K, Ahlheim J, Weiss H, Kleinsteuber S, et al. Stable sulfur and oxygen isotope fractionation of anoxic sulfide oxidation by two different enzymatic pathways. Environ Sci Technol. 2014;48:9094–102. doi: 10.1021/es404808r. [DOI] [PubMed] [Google Scholar]
- 72.Jørgensen BB, Marshall IPG. Slow Microbial Life in the Seabed. Annu Rev Mar Sci. 2015;8:311–32. doi: 10.1146/annurev-marine-010814-015535. [DOI] [PubMed] [Google Scholar]
- 73.Potter LC, Millington P, Griffiths L, Thomas GH, Cole JA. Competition between Escherichia coli strains expressing either a periplasmic or a membrane-bound nitrate reductase: does Nap confer a selective advantage during nitrate-limited growth? Biochem J. 1999;344:77–84. [PMC free article] [PubMed] [Google Scholar]
- 74.Ekici S, Pawlik G, Lohmeyer E, Koch H-G, Daldal F. Biogenesis of cbb(3)-type cytochrome c oxidase in Rhodobacter capsulatus. Biochim Biophys Acta. 2012;1817:898–910. doi: 10.1016/j.bbabio.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hamada M, Toyofuku M, Miyano T, Nomura N. cbb3-type cytochrome c oxidases, aerobic respiratory enzymes, impact the anaerobic life of Pseudomonas aeruginosa PAO1. J Bacteriol. 2014;196:3881–9. doi: 10.1128/JB.01978-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sievert SM, Scott KM, Klotz MG, Chain PSG, Hauser LJ, Hemp J, et al. Genome of the epsilonproteobacterial chemolithoautotroph Sulfurimonas denitrificans. Appl Environ Microbiol. 2008;74:1145–56. doi: 10.1128/AEM.01844-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Grote J, Schott T, Bruckner CG, Glockner FO, Jost G, Teeling H, et al. Genome and physiology of a model Epsilonproteobacterium responsible for sulfide detoxification in marine oxygen depletion zones. Proc Natl Acad Sci USA. 2012;109:506–10. doi: 10.1073/pnas.1111262109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marietou A. Nitrate reduction in sulfate-reducing bacteria. FEMS Microbiol Lett. 2016;363:fnw155. doi: 10.1093/femsle/fnw155. [DOI] [PubMed] [Google Scholar]
- 79.Greene EA, Hubert C, Nemati M, Jenneman GE, Voordouw G. Nitrite reductase activity of sulphate-reducing bacteria prevents their inhibition by nitrate-reducing, sulphide-oxidizing bacteria. Environ Microbiol. 2003;5:607–17. doi: 10.1046/j.1462-2920.2003.00446.x. [DOI] [PubMed] [Google Scholar]
- 80.Pereira IAC, Ramos AR, Grein F, Marques MC, da Silva SM, Venceslau SS. A comparative genomic analysis of energy metabolism in sulfate reducing bacteria and archaea. Front Microbiol. 2011;2:69. doi: 10.3389/fmicb.2011.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gilson ER, Huang S, Jaffé PR. Biological reduction of uranium coupled with oxidation of ammonium by Acidimicrobiaceae bacterium A6 under iron reducing conditions. Biodegradation. 2015;26:475–82. doi: 10.1007/s10532-015-9749-y. [DOI] [PubMed] [Google Scholar]
- 82.Zhou G-W, Yang X-R, Li H, Marshall CW, Zheng B-X, Yan Y, et al. Electron shuttles enhance anaerobic ammonium oxidation coupled to iron(III) reduction. Environ Sci Technol. 2016;50:9298–307. doi: 10.1021/acs.est.6b02077. [DOI] [PubMed] [Google Scholar]
- 83.Bao P, Li G-X. Sulfur-driven iron reduction coupled to anaerobic ammonium oxidation. Environ Sci Technol. 2017;51:6691–8. doi: 10.1021/acs.est.6b05971. [DOI] [PubMed] [Google Scholar]
- 84.Orsi WD, Edgcomb VP, Christman GD, Biddle JF. Gene expression in the deep biosphere. Nature. 2013;499:205–8. doi: 10.1038/nature12230. [DOI] [PubMed] [Google Scholar]
- 85.Jamieson J, Prommer H, Kaksonen AH, Sun J, Siade AJ, Yusov A, et al. Identifying and quantifying the intermediate processes during nitrate-dependent iron(II) oxidation. Environ Sci Technol. 2018;52:5771–81. doi: 10.1021/acs.est.8b01122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Takai K, Campbell BJ, Cary SC, Suzuki M, Oida H, Nunoura T, et al. Enzymatic and genetic characterization of carbon and energy metabolisms by deep-sea hydrothermal chemolithoautotrophic isolates of Epsilonproteobacteria. Appl Environ Microbiol. 2005;71:7310–20. doi: 10.1128/AEM.71.11.7310-7320.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sikorski J, Munk C, Lapidus A, Djao ODN, Lucas S, del Rio TG, et al. Complete genome sequence of Sulfurimonas autotrophica type strain (OK10 T) Stand Genom Sci. 2010;3:194–202. doi: 10.4056/sigs.1173118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
16S rRNA gene amplicon data are available at the National Centre for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under BioProject PRJNA472445. Metagenomic data were deposited at the NCBI SRA; accession numbers PRJNA404452, PRJNA404453, PRJNA444021, and PRJNA472439. MAGs are available under accessions SAMN12221356–SAMN12221381. SAGs are available at the JGI Genome Portal, GOLD Study ID’s Ga0214289 and Gs0127568.