
Fig. 1. The SMEG pipeline.
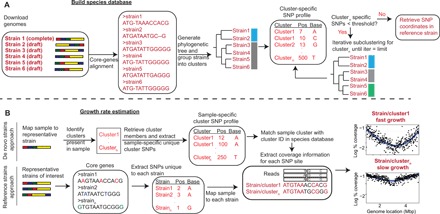
(A) Building the species database. Complete and draft-quality genomes of member strains available for a species are used to build the species database. Core genes are extracted, and multiple sequence alignment is conducted to generate a phylogenetic tree. Strains are grouped into clusters, and SMEG then generates cluster-specific unique SNPs, which are SNPs shared between a given proportion of cluster members but absent in strains from other clusters. Clusters having insufficient number of unique SNPs are iteratively subclustered to re-generate a unique SNPs profile, or “SNP coordinate set.” (B) Growth rate estimation. SMEG estimates growth rate using either a de novo or reference-based approach. In the de novo approach, SMEG maps reads to the representative strain to detect clusters in a sample. SMEG then re-generates a sample-specific, unique SNP profile using only the detected clusters, increasing the number of discriminatory SNPs usable to calculate growth rate. Then, nucleotide coverage is determined at each cluster’s unique SNP site across a variable sliding window to calculate the peak-to-trough ratio (PTR).