
Figure 3.
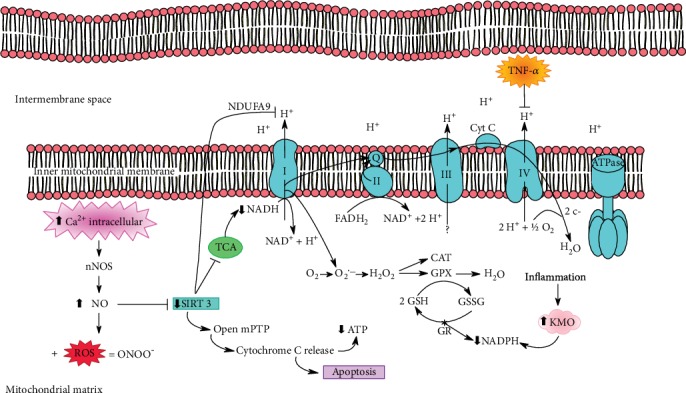
The increase of intracellular calcium activates neural nitric oxide synthase (nNOS), causing increased NO levels. This will decrease SIRT3, which acts as a key to control mitochondrial dysfunction. As SIRT3 activity decreases, mitochondrial permeability transition pores (mPTPs) open, which release cytochrome C, causing a decrease in ATP levels and inducing apoptosis. NO can also bind to ROS from the kynurenine pathway, generating peroxynitrite (ONOO), a highly unstable free radical. Reduction of SIRT3 deacetylates complex I NADH dehydrogenase, specifically in the NDUFA9 subunit, which interacts with two other ATP synthase subunits (F0 and F1). When SIRT3 is reduced, PDH activation is inadequate for the citric acid cycle, resulting in low levels of NADH and reduced activity of complex I. In addition, SIRT3 promotes deacetylation of MnSOD, an antioxidant enzyme that scavenges superoxide anion produced over the pathway. Another impaired antioxidant enzyme is GSH, because during the inflammatory process, the enzyme KMO, an enzyme dependent on NADPH, is activated, thus reducing the availability of this coenzyme for antioxidant defense systems. In parallel, TNF-α phosphorylates tyrosine 304 in subunit I of cytochrome C oxidase in complex IV, leading to further mitochondrial damage.