

Abstract
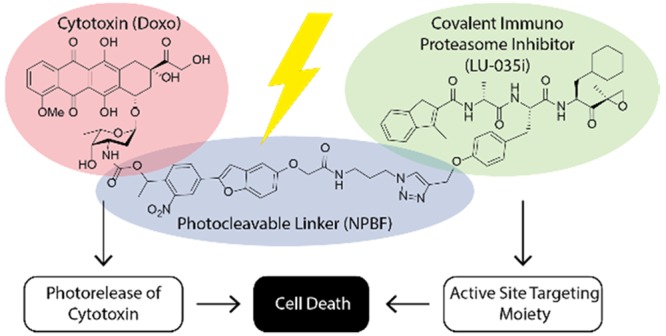
Proteasome inhibitors are established therapeutic agents for the treatment of hematological cancers, as are anthracyclines such as doxorubicin. We here present a new drug targeting approach that combines both drug classes into a single molecule. Doxorubicin was conjugated to an immunoproteasome-selective inhibitor via light-cleavable linkers, yielding peptide epoxyketone–doxorubicin prodrugs that remained selective and active toward immunoproteasomes. Upon cellular uptake and immunoproteasome inhibition, doxorubicin is released from the immunoproteasome inhibitor through photoirradiation. Multiple myeloma cells in this way take a double hit: immunoproteasome inhibition and doxorubicin-induced toxicity. Our strategy, which entails targeting of a cytotoxic agent, through a covalent enzyme inhibitor that is detrimental to tumor tissue in its own right, may find use in the search for improved anticancer drugs.
The selective delivery of cytotoxic agents to tumor cells is an important strategy to increase efficacy and to reduce side effects.1−3 By rendering the drug inactive and attaching it to a targeting element—often through a cleavable linker—toxicity is controlled and drugs can be used at higher doses. There are multiple ways to dissociate the drug from the carrier: release through local (bio)chemical conditions, such as the activity of specific enzymes,4 pH,5 or changes in redox potential,6,7 offers one approach. Alternatively, exogenous triggers can be used to initiate release of the drug from the construct, including bioorthogonal click-to-release8−10 and photochemical deprotection.11−13 In current strategies, the cytotoxic agents are normally conjugated to antibodies, peptides,12 or carbohydrates13 with high affinity and selectivity for cell surface proteins residing exclusively or predominantly (in comparison to healthy tissue) on tumor cells. It is, however, known that many hematological malignancies are also marked by the upregulation of very specific intracellular enzyme activities. We hypothesized that targeting such enzyme activities through covalent and irreversible inhibitors carrying a cytotoxic agent would present an alternative to the above cell-surface-targeting conjugation strategies (Figure 1). In such a scheme, the conjugate is allowed to accumulate in malignant cells with high enzyme activity, after which the cytotoxic cargo is released through one of the aforementioned exogenous triggers to fulfill its function.
Figure 1.
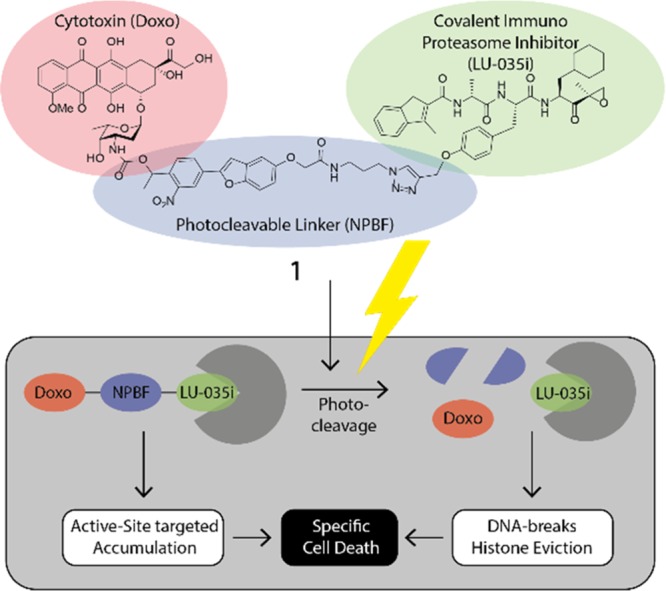
Structure of photocleavable doxorubicin–proteasome inhibitor conjugate (1), and the schematic strategy for dual targeting. First, the probe is targeted to cells containing immunoproteasome core particles. Then, after photocleavage, doxorubicin is released, leading to DNA breakage and histone eviction, leading to cell death.
Multiple myeloma poses such a target. It is a hematological malignancy characterized by a distinct protease activity profile. It expresses predominantly immunoproteasomes rather than constitutive proteasomes, unlike healthy B-cells, which have both proteasome types at about equal amounts.14 Multiple myeloma patients are currently treated with covalent and irreversible proteasome inhibitors, such as bortezomib and carfilzomib (CFZ). These agents block both constitutive proteasomes and immunoproteasomes.15 Resistance to these inhibitors is common, and new treatment strategies are needed.16 Therefore, we selected a multiple myeloma cell line for testing the validity of our “drug targeting through enzyme inhibitor” scheme (Figure 1).
All cells contain constitutive proteasomes, while those of myeloid lineage (including multiple myeloma cells) harbor immunoproteasomes.17 We therefore selected our immunoproteasome-selective, covalent, irreversible proteasome inhibitor LU-035i (Figure 1, green fragment) as the targeting agent.18 LU-035i has high selectivity for the chymotryptic sites (β5i) of immunoproteasomes over the corresponding chymotryptic sites (β5c) of constitutive proteasomes. This selectivity is conferred by the cyclohexylalanyl moiety within the peptide epoxyketone sequence. We have previously shown that functionalization of the phenolic alcohol of the tyrosine moiety in LU-035i does not affect immunoproteasome β5i inhibition activity. For this reason, but also based on our previous findings that β5i inhibition alone does not lead to cell death (combined β5c/β5i inhibition is minimally required for this),14 we selected this site in LU-035i for conjugation of the cytotoxic agent. We elected to use a non-cytotoxic proteasome inhibitor as our targeting device to allow for unambiguous establishment whether release of toxic payload leads to cell death. We selected doxorubicin as the cytotoxic agent due to its clinical pedigree and the previously reported potential clinical synergy with proteasome inhibition (for which in the future broad-spectrum immunoproteasome inhibitors might be utilized). Its use is limited by its severe side-effect profile, particularly to cardiac tissue.19 Recent studies that demonstrate synergistic effect of proteasome inhibitors and doxorubicin when applied to tumor tissue further supported our conjugate design.20 Furthermore, N-acylation renders doxorubicin inactive (and non-toxic), allowing its use in targeting conjugates.21
We here opted to link doxorubicin (Figure 1, red fragment 14) to LU-035i via two photolabile linkers: the oft-used bis-functionalized 6-nitroveratryloxycarbonyl moiety22 and one based on the recently reported 2-(4-nitrophenyl)benzofuran chromophore (Figure 1, blue fragment, Scheme S1).23 The latter was chosen for its favorable photochemical properties (ε = 13 × 103 M–1 cm–1, λmax = 360 nm), its modest size, and its compatibility with two-photon irradiation (δu = 20.7 GM at 740 nm) that allows deeper tissue penetration. Altogether this led to the design of the two doxorubicin–LU-035i conjugates 1 and 2 (Figure 1, Scheme S2).
The synthesis of conjugate 1 (Scheme 1) was accomplished in a convergent fashion to yield the product as a mixture of stereoisomers, allowing for variation of both bio-active agents as well as the nature of the linker moiety. (See SI for synthetic, analytical, and data, as well as for the synthesis of peptide epoxyketone 14 and conjugate 2.) Briefly, p-methoxyphenol 3 was converted into benzofuran 6, which was then lithiated prior to treatment with triisopropylborate to yield boronic acid 7. Suzuki cross-coupling with aryl bromide 8 provided, after desilylation (9 to 10), ketone reduction and reaction of the generated phenolic OH with tert-butyl bromoacetate and then trifluoroacetic acid, key photocage 11 amenable for functionalization with two bio-active agents.
Scheme 1. Synthesis of Conjugate 1.

Reagents and conditions: (a) bromoacetaldehyde diethyl acetal, KOH, NMP, 70 °C, 15 h, 90%; (b) PPA, toluene, 111 °C, 16 h, 33%; (c) (i) 1 M BBr3 in DCM, DCM, −78 °C (1 h) → r.t. (1 h), 89%, (ii) TBS-Cl, imidazole, DMF, 1 h, r.t., 70%; (d) (i) 1.6 M n-BuLi in hexanes, THF, −78 °C, 1 h, (ii) triisopropyl borate, −78 °C (0.5 h) → r.t. (0.5 h); (e) 8, Pd(PPh3)4, K2CO3, THF/H2O, 75 °C, 18 h, 75%; (f) HF·pyridine, THF, r.t., 2 h, 93%; (g) (i) NaBH4, MeOH, 0 °C, 1 h, quant., (ii) tert-butyl bromoacetate, K2CO3, DMF, 70 °C, 3 h, 72%, (iii) TFA, DCM, r.t., 3 h, quant.; (h) 12, HCTU, DiPEA, DCM, r.t. protected from UV, 18 h, quant.; (i) (i) N,N′-disuccinimidyl carbonate, Et3N, DMF, (ii) 14, DMF, r.t., protected from UV, 59%.
Conjugates 1 and 2 were next evaluated for their ability to inhibit constitutive proteasome and immunoproteasome active sites in live cells. To this end, AMO-1 cells, a human B cell lymphoma cell line that expresses both proteasome subtypes, were incubated with an increasing concentration of conjugates 1 and 2. Ensuing lysis and treatment with our set of three activity-based proteasome probes that combined report on the six active sites of constitutive proteasomes and immunoproteasomes16 (for structures, see Figure S3) and SDS-PAGE resolution allows for detection of inhibited proteasome active sites by fluorescence scanning of the wet gel slabs. As can be seen (Figure 2A), both conjugates 1 effectively and selectively block β5i (β5i IC50 = 0.53 μM, β5i/β5c = 18) and 2 (β5i IC50 = 0.42 μM, β5i/β5c = 21) and thus resemble the activity of the parent compound, LU-035i (β5i IC50 = 0.37 μM, β5i/β5c = 500).18 Compounds 1 and 2 were shown to be cell penetrable, with 90% of the maximum signal reduction of the targeted proteasome subunit observed within 40 min of incubation (Figures S1 and S2). At this time point, about equal inhibition is observed of proteasomes in lysates and in living AMO-1 cells (Figure S1).
Figure 2.
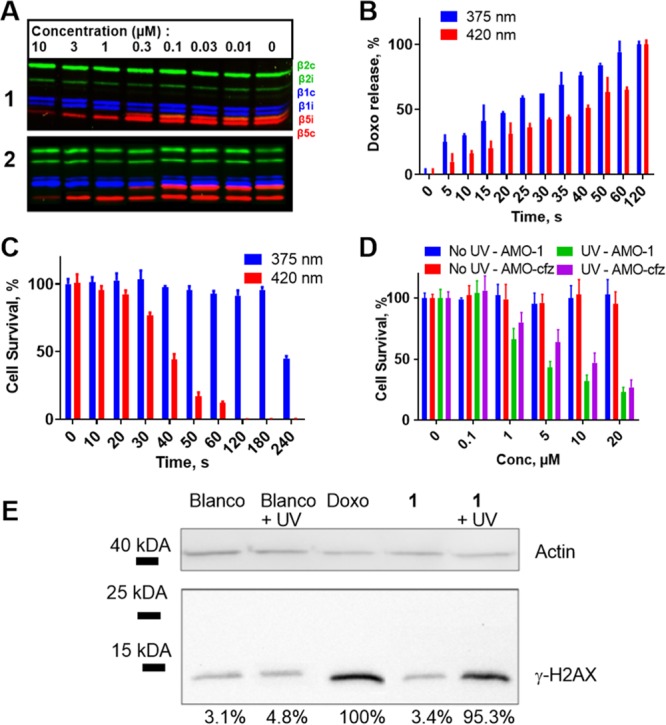
Biological evaluation of conjugates 1 and 2. (A) Competitive ABPP of compounds 1 and 2 with established proteasome subunit-selective activity based probes in AMO-1 cells. (B) Release of doxorubicin from 1 (10 μM) as determined by LC-MS. (C) Survival of AMO-1 cells after irradiation with 375 or 420 nm. (D) Cell survival of CFZ-resistant AMO-1 cells by treating with various concentrations of compound 1 and consequent irradiation with 375 nm for 60 s. (E) Western blot of γ-H2AX response after treatment of AMO-1 cells with 10 μM of compound followed with irradiation at 375 nm for 60 s.
With the aim to establish optimal photocleavage conditions for releasing doxorubicin within cells following immunoproteasome β5i inhibition, we investigated the amount of doxorubicin formed as a function of irradiation time (Figures 2B, S3, and S4). As much as 50% doxorubicin was released when compound 1 was irradiated for 30 s at 375 nm (3 mW/cm2), whereas 50 s of irradiation with 420 nm light (13 mW/cm2) was required to yield the same percentage of the free anthracycline. Quantitative release was observed after 1 min irradiation of 1 at 375 nm and after 2 min irradiation at 420 nm. Construct 2 showed insufficient uncaging yields to warrant further investigation (Figures S5 and S6). Phototoxicity studies on cells exposed to either of the two wavelengths yielded a surprising result (Figures 2C and S7). Photolytic cell death reached 50% after cells were exposed for 40 s to irradiation at 420 nm, while no significant decrease in cell viability was observed after 180 s of irradiation at the higher energetic wavelength (375 nm). We therefore conducted further experiments at 375 nm to exclude phototoxic effects.
We next explored the ability of conjugate 1 to induce apoptosis in AMO-1 cells, both those with an acquired resistance for CFZ and CFZ-sensitive cells (Figures 2D and S8). After incubation with conjugate 1, the cells were washed and irradiated for 60 s at 375 nm. Three days later cell survival was determined. Although CFZ-resistant AMO-1 cells displayed minor resistance toward conjugate 1 when compared to non-resistant AMO-1 cells, 50% of both cell variants were killed when exposed to 5 μM of the compound after irradiation.
As a control, we established that photocaged doxorubicin without the proteasome-targeting moiety, combined with irradiation, did not lead to significant cell death over background (Figures S9–S11 depict the doxorubicin conjugate 46 we synthesized for this purpose as well as the doxorubicin release as measured by LCMS and cytotoxicity assay). We also assessed the toxicity of construct 1 in a cell line devoid of immunoproteasome (HeLa S3). Pleasingly, no toxicity was observed in this experiment over background either (Figure S12).
If doxorubicin is released, compound 1 should induce DNA double-stranded breaks, as assessed by γ-H2AX. Indeed, irradiation of the cells induced γ-H2AX labeling as detected by Western blots illustrating the release of active doxorubicin by irradiation (Figure 2E).24
In conclusion, two probes that target the chymotrypsin-like activity of immunoproteasomes have been synthesized and used to deliver a cytotoxic payload to AMO-1 cells. One of the conjugates (2) linked via an 6-nitroveratryloxycarbonyl did not release the drug within a reasonable time frame. However, the more photolabile NPBF-linked conjugate 1 yielded quantitative release of the toxic payload after 1 min of irradiation at 375 nm, and after 2 min at 420 nm. Incubation with 1 followed by irradiation with 375 nm light induced apoptosis in both regular and CFZ-resistant AMO-1 cells in a concentration-dependent manner. Based on these results, we believe that the combined effect of proteolytic stress and poisoning topoisomerase II yielding DNA damage may overcome the resistance that cells can develop toward proteasome inhibitors. Although photodynamic therapy with photosensitizers is already applied in cancer therapy for esophageal and lung cancer,25 extending this approach with new cytotoxic drug combinations and alternative linkers may expand the clinical opportunities. Finally, substitution of either of the two pharmacological agents, for instance MMAE (a compound that cannot be used systemically due to its high toxicity and that is often used in antibody–drug conjugates)26 for doxorubicin, or the proteasome inhibitor for other mechanism-based hydrolase inhibitors, may expand the scope of our approach toward other diseases.
Acknowledgments
We are thankful to Prof. Dr. Sylvestre Bonnet and Dr. Andrea Pannwitz for assistance in the photochemical characterization. H.S.O. was funded by the Nederland Genomics Initiative (Zenith). S.I.v.K. was funded by the European Research Council (ERC-2014-StG-639005).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b11969.
Supplementary Schemes S1–S6 and Figures S1–S13, all experimental procedures, and spectra (PDF)
Author Contributions
∥ E.M. and M.J.v.d.G. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Nicolaou K. C.; Rigol S. The Role of Organic Synthesis in the Emergence and Development of Antibody-Drug Conjugates as Targeted Cancer Therapies. Angew. Chem., Int. Ed. 2019, 58, 11206. 10.1002/anie.201903498. [DOI] [PubMed] [Google Scholar]
- Bargh J. D.; Isidro-Llobet A.; Parker J. S.; Spring D. R. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 2019, 48, 4361. 10.1039/C8CS00676H. [DOI] [PubMed] [Google Scholar]
- van Kasteren S. I.; Neefjes J.; Ovaa H. Creating molecules that modulate immune responses. Nat. Rev. Chem. 2018, 2, 184. 10.1038/s41570-018-0023-9. [DOI] [Google Scholar]
- Lu J.; Jiang F.; Lu A.; Zhang G. Linkers having a crucial role in antibody-drug conjugates. Int. J. Mol. Sci. 2016, 17, 561. 10.3390/ijms17040561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trail P.; Willner D.; Lasch S.; Henderson A.; Hofstead S.; Casazza A.; Firestone R.; Hellstrom I.; Hellstrom K. Cure of xenografted human carcinomas by BR96-doxorubicin immunoconjugates. Science 1993, 261, 212. 10.1126/science.8327892. [DOI] [PubMed] [Google Scholar]
- Kellogg B. A.; Garrett L.; Kovtun Y.; Lai K. C.; Leece B.; Miller M.; Payne G.; Steeves R.; Whiteman K. R.; Widdison W.; Xie H.; Singh R.; Chari R. V.; Lambert J. M.; Lutz R. J. Disulfide-Linked Antibody-Maytansinoid Conjugates: Optimization of In Vivo Activity by Varying the Steric Hindrance at Carbon Atoms Adjacent to the Disulfide Linkage. Bioconjugate Chem. 2011, 22, 717. 10.1021/bc100480a. [DOI] [PubMed] [Google Scholar]
- Spangler B.; Kline T.; Hanson J.; Li X.; Zhou S.; Wells J. A.; Sato A. K.; Renslo A. R. Toward a Ferrous Iron-Cleavable Linker for Antibody-Drug Conjugates. Mol. Pharmaceutics 2018, 15, 2054. 10.1021/acs.molpharmaceut.8b00242. [DOI] [PubMed] [Google Scholar]
- Rossin R.; van Duijnhoven S. M. J.; ten Hoeve W.; Janssen H. M.; Kleijn L. H. J.; Hoeben F. J. M.; Versteegen R. M.; Robillard M. S. Triggered Drug Release from an Antibody-Drug Conjugate Using Fast “Click-to-Release”Chemistry in Mice. Bioconjugate Chem. 2016, 27, 1697. 10.1021/acs.bioconjchem.6b00231. [DOI] [PubMed] [Google Scholar]
- Versteegen R. M.; Rossin R.; ten Hoeve W.; Janssen H. M.; Robillard M. S. Click to Release: Instantaneous Doxorubicin Elimination upon Tetrazine Ligation. Angew. Chem., Int. Ed. 2013, 52, 14112. 10.1002/anie.201305969. [DOI] [PubMed] [Google Scholar]
- Rossin R.; Versteegen R. M.; Wu J.; Khasanov A.; Wessels H. J.; Steenbergen E. J.; ten Hoeve W.; Janssen H. M.; van Onzen A. H. A. M.; Hudson P. J.; Robillard M. S. Chemically triggered drug release from an antibody-drug conjugate leads to potent antitumour activity in mice. Nat. Commun. 2018, 9, 1484. 10.1038/s41467-018-03880-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klán P.; Šolomek T.; Bochet C. G.; Blanc A.; Givens R.; Rubina M.; Popik V.; Kostikov A.; Wirz J. Photoremovable protecting groups in chemistry and biology: reaction mechanisms and efficacy. Chem. Rev. 2013, 113, 119. 10.1021/cr300177k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Cheetham A. G.; Angacian G.; Su H.; Xie L.; Cui H. Peptide-drug conjugates as effective prodrug strategies for targeted delivery. Adv. Drug Delivery Rev. 2017, 110–111, 112. 10.1016/j.addr.2016.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnier P.; Wang X.-T.; Robinson M. A.; van Kasteren S.; Perkins A. C.; Frier M.; Fairbanks A. J.; Davis B. G. Lectin-directed enzyme activated prodrug therapy (LEAPT): Synthesis and evaluation of rhamnose-capped prodrugs. J. Drug Target. 2010, 18, 794. 10.3109/1061186X.2010.529909. [DOI] [PubMed] [Google Scholar]
- Besse A.; Besse L.; Kraus M.; Mendez-Lopez M.; Bader J.; Xin B.-T.; de Bruin G.; Maurits E.; Overkleeft H. S.; Driessen C. Proteasome Inhibition in Multiple Myeloma: Head-to-Head Comparison of Currently Available Proteasome Inhibitors. Cell Chem. Biol. 2019, 26, 340. 10.1016/j.chembiol.2018.11.007. [DOI] [PubMed] [Google Scholar]
- Kisselev A. F.; van der Linden W. A.; Overkleeft H. S. Proteasome inhibitors: an expanding army attacking a unique target. Chem. Biol. 2012, 19, 99. 10.1016/j.chembiol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin G.; Xin B. T.; Kraus M.; van der Stelt M.; van der Marel G. A.; Kisselev A. F.; Driessen C.; Florea B. I.; Overkleeft H. S. A set of activity-based probes to visualize human (immuno) proteasome activities. Angew. Chem., Int. Ed. 2016, 55, 4199. 10.1002/anie.201509092. [DOI] [PubMed] [Google Scholar]
- Rouette A.; Trofimov A.; Haberl D.; Boucher G.; Lavallée V.-P.; D’Angelo G.; Hébert J.; Sauvageau G.; Lemieux S.; Perreault C. Expression of immunoproteasome genes is regulated by cell-intrinsic and-extrinsic factors in human cancers. Sci. Rep. 2016, 6, 34019. 10.1038/srep34019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin G.; Huber E. M.; Xin B.-T.; van Rooden E. J.; Al-Ayed K.; Kim K.-B.; Kisselev A. F.; Driessen C.; van der Stelt M.; van der Marel G. A.; Groll M.; Overkleeft H. S. Structure-based design of β1i or β5i specific inhibitors of human immunoproteasomes. J. Med. Chem. 2014, 57, 6197. 10.1021/jm500716s. [DOI] [PubMed] [Google Scholar]
- Singal P. K.; Iliskovic N. Doxorubicin-Induced Cardiomyopathy. N. Engl. J. Med. 1998, 339, 900. 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- Ashley J. D.; Quinlan C. J.; Schroeder V. A.; Suckow M. A.; Pizzuti V. J.; Kiziltepe T.; Bilgicer B. Dual Carfilzomib and Doxorubicin-Loaded Liposomal Nanoparticles for Synergistic Efficacy in Multiple Myeloma. Mol. Cancer Ther. 2016, 15, 1452. 10.1158/1535-7163.MCT-15-0867. [DOI] [PubMed] [Google Scholar]
- Schoener C. A.; Carillo-Conde B.; Hutson H. N.; Peppas N. A. An inulin and doxorubicin conjugate for improving cancer therapy. J. Drug Delivery Sci. Technol. 2013, 23, 111. 10.1016/S1773-2247(13)50018-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teague S. J. Facile synthesis of a o-nitrobenzyl photolabile linker for combinatorial chemistry. Tetrahedron Lett. 1996, 37, 5751. 10.1016/0040-4039(96)01175-6. [DOI] [Google Scholar]
- Komori N.; Jakkampudi S.; Motoishi R.; Abe M.; Kamada K.; Furukawa K.; Katan C.; Sawada W.; Takahashi N.; Kasai H.; Xue B.; Kobayashi T. Design and synthesis of a new chromophore, 2-(4-nitrophenyl) benzofuran, for two-photon uncaging using near-IR light. Chem. Commun. 2016, 52, 331. 10.1039/C5CC07664A. [DOI] [PubMed] [Google Scholar]
- Pang B.; Qiao X.; Janssen L.; Velds A.; Groothuis T.; Kerkhoven R.; Nieuwland M.; Ovaa H.; Rottenberg S.; van Tellingen O.; Janssen J.; Huijgens P.; Zwart W.; Neefjes J. Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat. Commun. 2013, 4, 1908. 10.1038/ncomms2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.; Pennathur A.; Luketich J. D. Role of photodynamic therapy in unresectable esophageal and lung cancer. Lasers Surg. Med. 2006, 38, 396. 10.1002/lsm.20364. [DOI] [PubMed] [Google Scholar]
- Dosio F.; Brusa P.; Cattel L. Immunotoxins and anticancer drug conjugate assemblies: the role of the linkage between components. Toxins 2011, 3, 848. 10.3390/toxins3070848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.