

Summary
In the phase 3 study RERISE, patients with newly diagnosed chronic myeloid leukaemia in chronic phase demonstrated significantly faster and higher rates of major molecular response (MMR) with twice‐daily radotinib 300 mg (n = 79) or 400 mg (n = 81) than with once‐daily imatinib 400 mg (n = 81) after 12 months. With ≥48 months’ follow‐up, MMR was higher with radotinib 300 mg (86%) or 400 mg (83%) than with imatinib (75%). Among patients with BCR‐ABL1 ≤ 10% at three months, MMR and molecular response 4·5 (MR4·5) were achieved within 48 months by more radotinib‐treated patients (300 mg: 84% and 52%, respectively; 400 mg: 74% and 44%, respectively) than imatinib‐treated patients (71% and 44%, respectively). Estimated overall and progression‐free survival rates at 48 months were not significantly different between imatinib (94% and 94%, respectively) and radotinib 300 mg (99% and 97%, respectively) or 400 mg (95% and 93%, respectively). The treatment failure rate was significantly higher with imatinib (19%) than with radotinib 300 mg (6%; P = 0·0197) or 400 mg (5%; P = 0·0072). Safety profiles were consistent with previous reports; most adverse events occurred within 12 months. Radotinib continues to demonstrate robust, deep molecular responses, suggesting that treatment‐free remission may be attainable.
Keywords: chronic myeloid leukaemia, imatinib, newly diagnosed, long‐term data, radotinib
BCR‐ABL1–selective tyrosine kinase inhibitors (TKIs) have dramatically improved the prognosis for patients with chronic myeloid leukaemia (CML). With 10‐year survival rates >80% (Hochhaus et al., 2017), CML has been converted to a chronic disease for the majority of patients. Given that patients may remain on therapy indefinitely, the cost becomes an important consideration. A number of studies have shown that TKIs can safely be discontinued in approximately half of the patients who achieve durable deep molecular responses (Jabbour & Kantarjian, 2018; Cortes et al., 2019). Specifically, these treatment‐free remission (TFR) studies reported one‐year TFR rates of 49–68% with the second‐generation TKIs dasatinib and nilotinib (Imagawa et al., 2015; Takahashi et al., 2018) and a TFR rate of 41% with imatinib (Mahon et al., 2010). The second‐generation TKIs have been associated with deeper and more durable responses than imatinib, potentially increasing the likelihood of patients being eligible to discontinue treatment and able to experience TFR (Jabbour & Kantarjian, 2018).
Radotinib (SupectTM; IL‐YANG Pharm. Co. Ltd., Seoul, South Korea) is a second‐generation oral BCR‐ABL1 TKI that has a mutant sensitivity profile similar to that of nilotinib (Kim et al., 2014; Zabriskie et al., 2015); it is approved in South Korea as first‐line therapy for patients with CML in the chronic phase (CML‐CP) (Kwak et al., 2017). Approval as first‐line therapy was based on 12‐month results from a phase 3 study (RERISE; NCT01511289) that showed superiority of radotinib over imatinib in major molecular response (MMR) by 12 months (the primary endpoint), as well as all other efficacy endpoints evaluated (Kwak et al., 2017). Two twice‐daily (BID) doses of radotinib, 300 and 400 mg, were evaluated in this study. The lower dose was found to be the more effective of the two because of the increased toxicity associated with the higher dose, which led to more early treatment discontinuations due to adverse events (AEs) (Kwak et al., 2017). Consequently, only the 300‐mg BID dose was approved as first‐line therapy. Here we present updated results from RERISE based on a minimum follow‐up of 48 months, aiming to evaluate treatment discontinuation eligibility (with the goal of TFR) after first‐line radotinib treatment.
Patients and methods
Patients
Patients aged ≥18 years with an Eastern Cooperative Oncology Group performance status of 0–2 and Philadelphia chromosome (Ph)–positive CML‐CP diagnosed within the previous three months were eligible. Adequate organ function was a requirement. A diagnosis of Ph‐negative CML, use of imatinib for ≥8 days prior to study entry, use of targeted anticancer therapy (except hydroxycarbamide and/or anagrelide), impaired cardiac function and confirmed central nervous system involvement were criteria for exclusion. The transcript types were e13a2 and/or e14a2 in all patients except for one in each treatment group.
Study design and treatment
Details of the design and conduct of this study have been published previously (Kwak et al., 2017). In brief, this was an open‐label phase 3 study conducted at 24 sites in South Korea, Thailand, the Philippines and Indonesia (NCT01511289). Eligible patients were stratified by Sokal risk score and randomly assigned 1:1:1 to radotinib 300 mg BID, radotinib 400 mg BID or imatinib 400 mg once daily (QD). Patients received radotinib or imatinib treatment for ≤4 years until disease progression or lack of continued benefit. One‐ or two‐dose reductions of radotinib were allowed for toxicity to a minimum dose of 200 mg BID. The minimum dose allowed for imatinib was 300 mg QD. Dose escalation was not permitted for either drug. However, following dose reduction, re‐escalation to the previous dose was permitted if the severity of the relevant toxicity was not >grade 2 for 28 days with the reduced dose.
An institutional review board approved the study protocol and its amendments at each site. The trial was performed in accordance with the Declaration of Helsinki and the International Conference on Harmonisation Guidelines for Good Clinical Practice. Written informed consent was provided by all patients who participated in the study. Database lock occurred on 10 November 2017, after the last visit of the last patient in the study.
Endpoints and assessments
MMR [≤0·1% BCR‐ABL1 based on the International Scale (IS)] by 12 months was the primary endpoint. Secondary endpoints included complete cytogenetic response (CCyR), molecular response 4·5 [MR4·5; ≥4·5 log reduction in BCR‐ABL1 from standardized baseline or ≤0·0032% BCR‐ABL1 IS] and disease progression by 12 months, as well as MMR at 12 months. Additional endpoints were progression‐free survival (PFS) and overall survival (OS) among patients who participated in the extension phase. Internationally standardized real‐time polymerase chain reaction performed in a central laboratory was used to measure BCR‐ABL1 at screening and every three months on treatment. Disease progression was defined as progression to accelerated phase (AP) or blast phase (BP) and PFS as the date of death from any cause or progression to AP/BP minus the treatment initiation date +1. PFS events included death from any cause or disease progression. Efficacy assessments were performed using peripheral blood samples, bone marrow aspiration, and/or biopsy. Safety was evaluated by performing physical examinations and laboratory tests. AEs were graded using Common Terminology Criteria for Adverse Events version 3.0 (2016, http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf).
Statistical analysis
The efficacy‐ and safety‐analysis populations comprised all patients who received at least one dose of study medication. Cumulative incidence of MMR and MR4·5 with competing risk was calculated. Competing risk analysis excluded patients with atypical transcripts (n = 1 in each treatment group), treatment failure or suboptimal response, disease progression or death which were considered as competing risk events. Efficacy variables were summarised using descriptive statistics; 95% confidence intervals were determined using the normal approximation and Kaplan–Meier methods. The Cochran–Mantel–Haenszel test was used to compare rates of MMR by 12 months, with patients stratified by Sokal risk group, and Hochberg’s step‐up method was used to control for multiple testing. Comparison of the cumulative incidence of MMR and MR4·5 accounting for competing risk was compared across groups used Gray’s test (Kim, 2017). Time‐to‐event endpoints were estimated using the Kaplan–Meier method and analysed using a stratified log‐rank test. Differences in AE incidence between groups were analysed using the Fisher exact test.
Results
Patients
Between August 2011 and February 2014, 263 patients were screened, of whom 241 were randomised (radotinib 300 mg BID, n = 79; radotinib 400 mg BID, n = 81; imatinib 400 mg QD, n = 81; Fig 1). As reported previously (Kwak et al., 2017), the median age of patients was 43–45 years, and the majority were Asian males with relatively low body weight (median, approximately 61 kg; Table 1). A total of 25–26% of patients in each group had high Sokal risk scores.
Figure 1.
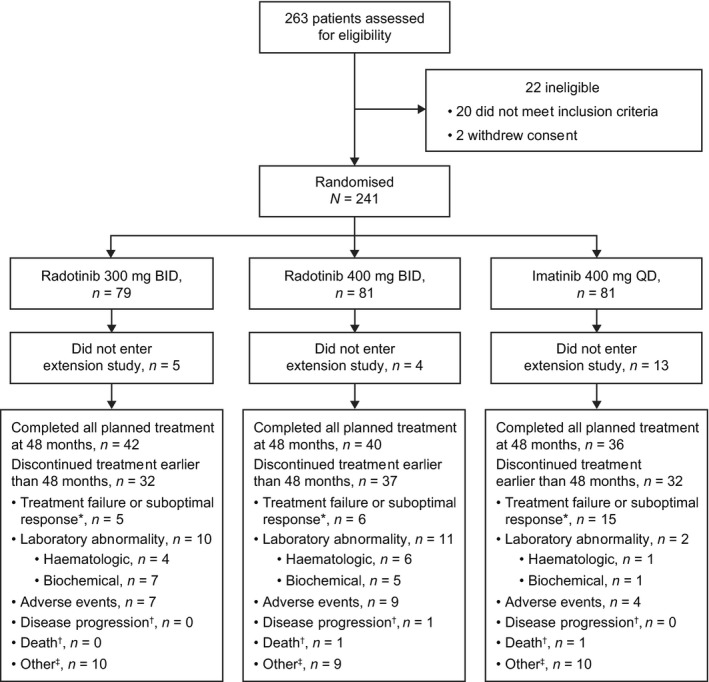
Disposition of patients treated with radotinib 300 mg BID or imatinib 400 mg QD. BID, twice daily; QD, once daily. *Assessed according to European LeukemiaNet 2009 recommendations (Baccarani et al., 2009). †Disease progression or death was only counted if this was the main reason for study discontinuation. ‡Comprises administrative problems (imatinib, n = 1), lost to follow‐up (radotinib, n = 1), protocol violations (imatinib, n = 2; radotinib, n = 1), pregnancy (imatinib, n = 1; radotinib, n = 1) and withdrawal of consent (radotinib, n = 7; imatinib, n = 6).
Table 1.
Baseline patient and disease characteristics.
| Radotinib 300 mg BID (n = 79) | Radotinib 400 mg BID (n = 81) | Imatinib 400 mg QD (n = 81) | |
|---|---|---|---|
| Age, years | |||
| Median | 45 | 43 | 45 |
| Range | 20–75 | 18–84 | 18–83 |
| Sex, n (%) | |||
| Male | 52 (66) | 47 (58) | 50 (62) |
| Female | 27 (34) | 34 (42) | 31 (38) |
| Weight, kg | |||
| Median | 61 | 60 | 62 |
| Range | 43–100 | 40–96 | 41–96 |
| ECOG performance status, n (%) | |||
| 0 | 53 (67) | 55 (68) | 51 (63) |
| 1 | 26 (33) | 25 (31) | 29 (36) |
| 2 | 0 | 1 (1) | 1 (1) |
| Additional chromosomal abnormalities, n (%) | 6 (8) | 7 (9) | 6 (7) |
| Type of BCR‐ABL1 transcript | |||
| e13a2 ± e14a2 | 78 | 80 | 80 |
| Others | 1 | 1 | 1 |
| Sokal risk, n (%) | |||
| Low | 21 (27) | 22 (27) | 22 (27) |
| Intermediate | 38 (48) | 38 (47) | 39 (48) |
| High | 20 (25) | 21 (26) | 20 (25) |
| Duration of CML, days | |||
| Median | 22 | 23 | 22 |
| Range | 7–102 | 7–66 | 6–71 |
| Prior treatment, n (%) | |||
| Hydroxycarbamide | 69 (87) | 70 (86) | 72 (89) |
| Anagrelide* | 5 (6) | 5 (6) | 8 (10) |
| Imatinib | 0 | 2 (2) | 2 (2) |
BID, twice daily; CML, chronic myeloid leukaemia; ECOG, Eastern Cooperative Oncology Group; QD, once daily.
All patients who were administered anagrelide also received hydroxycarbamide.
Sixty‐four patients (80%) in the radotinib 300 mg group, 54 patients (67%) in the radotinib 400 mg group and 53 patients (65%) in the imatinib group continued study treatment in the extension study (Fig 1). At the data cut‐off date (10 November 2017), 42 patients (53%), 40 patients (49%) and 36 patients (44%) had completed the planned 48 months of treatment in the radotinib and imatinib groups respectively. The most common reason for discontinuation of treatment at <48 months was laboratory abnormalities in both radotinib groups [300 mg: n = 10 (13%); 400 mg: n = 11 (14%)] and treatment failure or suboptimal response in the imatinib group [n = 15 (19%)]. Duration of follow‐up [safety population; median days ([range)] was similar among the groups [radotinib 300 mg: 1346 (244–1528); radotinib 400 mg: 1346 (64–1546); imatinib: 1350 (31–1554)]. Treatment dose was reduced for any reason in 54 patients (68%) in the radotinib 300 mg group, 68 patients (84%) in the radotinib 400 mg group and 19 patients (23%) in the imatinib 400 mg group. The dose of each drug was re‐escalated in 12 patients (15%), three patients (4%) and seven patients (9%), respectively.
Efficacy
With the exclusion of patients to determine the cumulative incidence of molecular response with competing risk, MMR was 85%, 83% and 75% and MR4·5 was 58%, 56% and 49% in the radotinib 300 mg, radotinib 400 mg and imatinib groups, respectively (Fig 2). The cumulative incidence of MMR and MR4·5 by 48 months in the intent‐to‐treat (ITT) population (without competing risk) is shown in Table SI.
Figure 2.
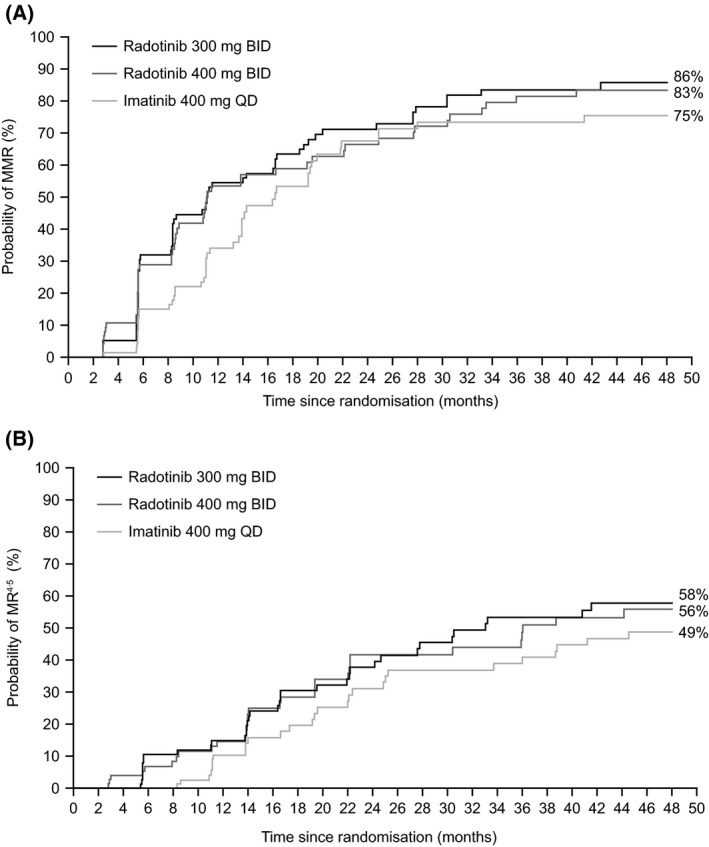
Cumulative incidences with competing risk of (A) MMR and (B) MR4·5. BID, twice daily; MMR, major molecular response; MR4·5, molecular response 4·5; QD, once daily.
Rates of BCR‐ABL1 ≤ 10% at three months and BCR‐ABL1 ≤ 1% at six months were significantly higher with radotinib 300 mg than with imatinib [86% vs. 71% (P = 0·0278) and 77% vs. 58% (P = 0·0156), respectively] (Fig 3A). These rates were also significantly higher with radotinib 400 mg (87% and 81% respectively) than with imatinib (P = 0·0154 and P = 0·0033 respectively). By 48 months, MMR and MR4·5 rates were significantly higher in evaluable patients with BCR‐ABL1 ≤ 10% than BCR‐ABL1> 10% at three months for all groups. Among patients with BCR‐ABL1 ≤ 10% at three months, more patients treated with radotinib 300 mg than with imatinib had achieved MMR or MR4·5 by 48 months (MMR, 84% vs. 71%; MR4·5, 52% vs. 44%; Fig 3B,C). In the radotinib 400 mg group, MMR and MR4·5 rates were 74% and 44%, respectively. Treatment failure or suboptimal response by 48 months was significantly lower with radotinib 300 mg (6%) or 400 mg (5%) than with imatinib (19%; P = 0·0197 and P = 0·0072, respectively; Fig 3D).
Figure 3.
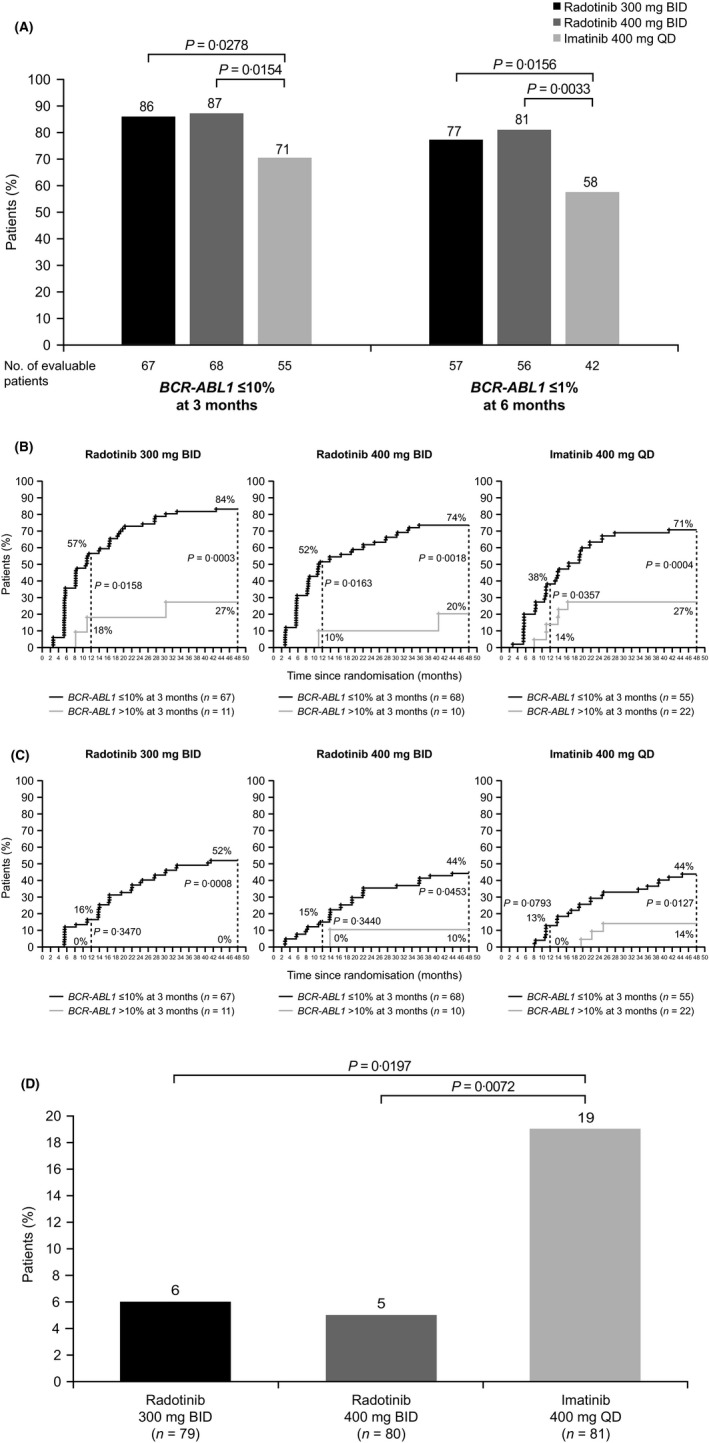
Additional efficacy analyses. BID, twice daily; MMR, major molecular response; MR4·5, molecular response 4·5; QD, once daily. (A) BCR‐ABL1 ≤ 10% at 3 months and BCR‐ABL1 ≤ 1% at 6 months. (B) MMR by 48 months according to BCR‐ABL1 status (≤10% or >10%) at 3 months. (C) MR4·5 by 48 months according to status of BCR‐ABL1 status (≤10% or >10%) at 3 months. (D) Rate of treatment failure or suboptimal response by last follow‐up. Treatment failure or suboptimal response definition is based on European LeukemiaNet 2013 recommendations [Baccarani et al., 2013]). All patients had a minimum follow‐up of 48 months.
The Kaplan–Meier estimated 48‐month OS rate was 99% with radotinib 300 mg, 95% with radotinib 400 mg and 94% with imatinib (P = 0·3224 vs. radotinib 300 mg; P = 0·6748 vs. radotinib 400 mg). One death occurred during study treatment, in the imatinib arm, due to acute respiratory failure (grade 4 on day 233 of study drug; not suspected to be treatment‐related). Four patients who discontinued the study early due to AEs died: one in the radotinib 300 mg group (for unknown reason) and three in the radotinib 400 mg group (two for unknown reasons and one due to disease progression). One patient in the radotinib 400 mg group who had been on the study drug for 518 days died due to disease progression one month after study discontinuation. Two patients in the imatinib group who completed the main study and did not enter the extension study died for unknown reasons.
Kaplan–Meier‐estimated 48‐month PFS rates were similar for radotinib 300 mg (97%, P = 0·4328 vs. imatinib), radotinib 400 mg (93%, P = 0·7069 vs. imatinib) and imatinib (94%). With at least 48 months of follow‐up, each group had patients who progressed to CML‐AP/BP [radotinib 300 mg, n = 2 (3%); radotinib 400 mg, n = 3 (4%); imatinib, n = 3 (4%)]. One imatinib‐treated patient exhibited disease progression during the study, while the other seven study patients with disease progression experienced it during follow‐up, after study treatment discontinuation.
Safety
Occurrence of most non‐laboratory AEs (any cause, any grade) reported in the first 12 months either remained the same or increased modestly at 48 months (Table 2). By 48 months, the overall incidence of the following non‐laboratory AEs increased in all three treatment groups (radotinib 300 mg, radotinib 400 mg and imatinib, respectively): nausea/vomiting (35%, 36% and 42%), constipation (6%, 16% and 2%), fatigue (20%, 26% and 14%), headache (24%, 38% and 15%), rash (38%, 35% and 22%) and pruritus (22%, 33% and 12%). Also, the incidence of four grade 3–4 AEs increased in the radotinib 300 mg group (fatigue, 9%; headache, 3%; pruritus, 3%; rash, 5%) and the radotinib 400 mg group (fatigue, 9%; headache, 2%; nausea, 6%; pruritus, 5%). All of these grade 3–4 AEs were suspected to be related to study treatment except for one episode of grade 3 rash in the radotinib 300 mg group and one episode of headache in each radotinib group.
Table 2.
Non‐laboratory AEs (any cause) occurring in ≥10% of patients.
| Patients with an AE, n (%) | Radotinib 300 mg BID (n = 79) | Radotinib 400 mg BID (n = 81) | Imatinib 400 mg QD (n = 81) | |||
|---|---|---|---|---|---|---|
| 12 months | 48 months | 12 months | 48 months | 12 months | 48 months | |
| All AEs, any cause | 78 (99) | 78 (99) | 78 (96) | 80 (99) | 80 (99) | 80 (99) |
| Gastrointestinal disorders | ||||||
| Nausea | 17 (22) | 21 (27) | 18 (22) | 25 (31) | 19 (23) | 25 (31) |
| Dyspepsia | 9 (11) | 9 (11) | 11 (14) | 15 (19) | 5 (6) | 7 (9) |
| Diarrhoea | 7 (9) | 7 (9) | 4 (5) | 9 (11) | 11 (14) | 13 (16) |
| Vomiting | 6 (8) | 7 (9) | 7 (9) | 11 (14) | 7 (9) | 9 (11) |
| Abdominal pain | 8 (10) | 10 (13) | 3 (4) | 6 (7) | 2 (2) | 5 (6) |
| Constipation | 4 (5) | 5 (6) | 10 (12) | 13 (16) | 1 (1) | 2 (2) |
| Upper abdominal pain | 3 (4) | 5 (6) | 7 (9) | 8 (10) | 3 (4) | 5 (6) |
| General disorder/administration site conditions | ||||||
| Fatigue | 12 (15) | 16 (20) | 13 (16) | 21 (26) | 8 (10) | 11 (14) |
| Facial oedema | 0 | 0 | 2 (2) | 3 (4) | 20 (25) | 22 (27) |
| Pyrexia | 5 (6) | 6 (8) | 4 (5) | 9 (11) | 3 (4) | 4 (5) |
| Asthenia | 2 (5) | 3 (4) | 5 (6) | 8 (10) | 4 (5) | 4 (5) |
| Oedema | 0 | 0 | 0 | 0 | 8 (10) | 8 (10) |
| Infection/Infestation | ||||||
| Upper respiratory tract infection | 4 (5) | 6 (8) | 7 (9) | 15 (19) | 7 (9) | 8 (10) |
| Musculoskeletal/connective tissue disorders | ||||||
| Myalgia | 11 (14) | 11 (14) | 14 (17) | 19 (23) | 23 (28) | 25 (31) |
| Back pain | 9 (14) | 12 (15) | 4 (5) | 7 (9) | 4 (5) | 6 (7) |
| Arthralgia | 4 (5) | 5 (6) | 3 (4) | 6 (7) | 8 (10) | 11 (14) |
| Pain in extremity | 1 (1) | 4 (5) | 3 (4) | 5 (6) | 8 (10) | 10 (12) |
| Muscle spasms | 1 (1) | 1 (1) | 2 (2) | 2 (2) | 7 (9) | 8 (10) |
| Nervous system disorders | ||||||
| Headache | 15 (19) | 19 (24) | 25 (31) | 31 (38) | 8 (10) | 12 (15) |
| Dizziness | 6 (8) | 11 (14) | 5 (6) | 7 (9) | 4 (5) | 7 (9) |
| Nutrition disorder | ||||||
| Decreased appetite | 12 (15) | 12 (15) | 13 (16) | 19 (23) | 5 (6) | 5 (6) |
| Respiratory, thoracic, mediastinal disorders | ||||||
| Cough | 11 (14) | 14 (18) | 9 (11) | 10 (12) | 5 (6) | 8 (10) |
| Skin and subcutaneous tissue disorders | ||||||
| Rash | 28 (35) | 30 (38) | 14 (31) | 28 (35) | 17 (21) | 18 (22) |
| Pruritus | 13 (16) | 17 (22) | 23 (28) | 27 (33) | 7 (9) | 10 (12) |
| Alopecia | 9 (11) | 9 (11) | 9 (11) | 10 (12) | 2 (2) | 2 (2) |
AE, adverse event; BID, twice daily; QD, once daily.
Most grade 3–4 haematologic laboratory abnormalities were reported during the first 12 months of the study. Over the full 48‐month study period, grade 3–4 neutropenia was the most common haematologic abnormality, reported in 15 patients (19%) treated with radotinib 300 mg, 19 patients (23%) treated with radotinib 400 mg and 26 patients (32%) treated with imatinib (Table 3). Likewise, the incidence of newly occurring or worsening biochemical laboratory abnormalities was generally low (Table SII). Grade 3–4 AEs with radotinib 300 mg, radotinib 400 mg or imatinib respectively were hyperbilirubinaemia [n = 25 (32%), n = 36 (44%) and n = 0], increased aspartate aminotransferase [AST; n = 9 (11%), n = 7 (9%) and n = 2 (2%)] increased alanine aminotransferase [ALT; n = 22 (28%), n = 26 (32%) and n = 1 (1%)], hyperglycaemia [n = 9 (11%), n = 12 (15%) and n = 4 (5%)] and hypercholesterolaemia [n = 1 (1%), n = 0 and n = 0]. Hypophosphataemia was the most frequently occurring laboratory abnormality in the imatinib group [n = 50 (62%)], with grade 3–4 events in 16 patients (20%), compared to seven patients (9%) and five patients (6%) in the radotinib 300 and 400 mg groups, respectively. No treatment‐related grade 3–4 cardiovascular AEs were reported with imatinib. Any‐cause cardiovascular AEs of interest can be seen in Table SIII. Treatment‐related cardiovascular events were reported in eight patients (10%), five patients (6%) and one patient (1%) in the radotinib 300 mg, radotinib 400 mg and imatinib groups respectively. There was one case each of grade 1 atrial tachycardia and grade 2 palpitations in the radotinib 300 mg group and one case each of grade 1 coronary artery occlusion, grade 1 peripheral arterial occlusive disease (PAOD) and grade 2 angina pectoris in the radotinib 400 mg group. The patient with PAOD (diagnosed at 39 months) had a history of atherosclerosis of arteries of the extremities and a left femorotibial bypass with reverse greater saphenous vein. Two cardiovascular AEs [acute myocardial infarction (grade 4) and atrial tachycardia (grade 1)] that led to discontinuation were reported in the radotinib 400 mg group.
Table 3.
Haematologic AEs.
| Patients with an AE, n (%) | Radotinib 300 mg BID (n = 79) | Radotinib 400 mg BID (n = 81) | Imatinib 400 mg QD (n = 81) | |||
|---|---|---|---|---|---|---|
| Any grade | Grade 3–4 | Any grade | Grade 3–4 | Any grade | Grade 3–4 | |
| Anaemia | 22 (28) | 6 (8) | 29 (36) | 10 (12) | 37 (46) | 4 (5) |
| Leucopenia | 41 (52) | 8 (10) | 32 (40) | 11 (14) | 60 (74) | 9 (11) |
| Neutropenia | 33 (42) | 15 (19) | 30 (37) | 19 (23) | 52 (64) | 26 (32) |
| Thrombocytopenia | 54 (68) | 13 (16) | 47 (58) | 12 (15) | 58 (72) | 16 (20) |
AE, adverse event; BID, twice daily; QD, once daily.
Discussion
Long‐term follow‐up of the RERISE study shows that the superiority of radotinib over imatinib for the treatment of CML‐CP seen at 12 months was maintained at 48 months. The cumulative incidence of MMR and MR4·5 with competing risk were both higher with radotinib than imatinib by 48 months. Significantly higher BCR‐ABL1 ≤10% at three months and BCR‐ABL1 ≤1% at six months were observed with radotinib, coupled with significantly lower levels of treatment failure. Taken together, these results demonstrate that earlier and deeper molecular responses were achieved with radotinib than with imatinib. By 48 months, cumulative incidence of MMR in the ITT population was significantly higher with radotinib 300 mg (76%) than with imatinib (56%; P = 0·0107), and both MMR and MR4·5 were numerically higher with radotinib (both doses) than with imatinib at all time points.
The overall efficacy results reported here were comparable to the long‐term results obtained with other second‐generation TKIs in phase 3 studies. The MMR rates by 48 months with nilotinib 300 and 400 mg BID in the front‐line setting were reported to be 76% and 73%, respectively; MR4·5 by 48 months was 40% and 37%, respectively (Hochhaus et al., 2016). With dasatinib 100 mg QD, the MMR and MR4·5 rates by 48 months were 73% and 34%, respectively (Cortes et al., 2016).
Overall, the safety profiles observed with both radotinib and imatinib at 48 months of follow‐up were consistent with those previously reported (Kwak et al., 2017). In general, haematologic abnormalities tended to occur in the first 12 months of treatment, with no increase or a modest increase over time, while biochemical abnormalities were moderately cumulative. More liver‐related abnormalities occurred with radotinib than with imatinib, whereas more haematological and other biochemical abnormalities occurred with imatinib than with radotinib. As in the earlier report, patients treated with radotinib experienced a higher frequency (≥20% incidence difference versus imatinib) of biochemical abnormalities (increased blood alkaline phosphatase, AST, hyperbilirubinaemia and hypercholesterolaemia), whereas patients had a higher frequency of hypophosphataemia and hypokalaemia with imatinib than with radotinib. Elevated liver enzymes have been observed with other BCR‐ABL1 TKIs (Larson et al., 2012; Cortes et al., 2013; Brümmendorf et al., 2015), and this response could potentially be a class effect (Pinilla‐Ibarz et al., 2015; Gambacorti‐Passerini et al., 2016). All recorded musculoskeletal/connective tissue disorders (other than back pain) and both facial and general oedema occurred at a higher frequency with imatinib versus radotinib. Occurrence of rash, decreased appetite, headache and alopecia was higher with radotinib than with imatinib. As reported earlier, most AEs were reversible and could be properly managed through dosing modifications.
Often, certain AE subsets may be more commonly associated with a specific drug or drug class. Cardiovascular AEs have been increasingly recognized as toxicities associated with TKI treatment, especially with the second‐ and third‐generation TKIs (Damrongwatanasuk & Fradley, 2017). By 36 months and compared with imatinib, low but numerically higher rates of ischaemic heart disease and PAOD were reported with nilotinib treatment (Larson et al., 2012); still higher rates were reported with nilotinib at 76 months (Damrongwatanasuk & Fradley, 2017). As noted with first‐line nilotinib in the ENESTnd study (Hochhaus et al., 2016), the incidence of cardiovascular AEs in RERISE also appears to be increasing slightly; one patient reported PAOD and angina after approximately 39 months. This trend underscores the need for ongoing cardiovascular monitoring in patients receiving radotinib, and other agents of the same class; evidence suggests that those patients who have baseline cardiovascular risk factors are often the ones who develop cardiovascular AEs during TKI treatment (Breccia et al., 2015; Hochhaus et al., 2016). Additionally, there were no reported cases of pleural effusion in this study, an adverse event of interest related to TKI (i.e. dasatinib) treatment (Cortes et al., 2017).
In summary, the 48‐month follow‐up in the RERISE study demonstrated sustained superiority of radotinib 300 mg BID or 400 mg BID compared with imatinib 400 QD as treatment for patients with newly diagnosed CML‐CP. Both doses of radotinib had a manageable safety profile, with the 300‐mg BID dose better tolerated than the 400‐mg BID dose. At a time when costs of TKIs used to treat patients with CML are considerable (Experts in Chronic Myeloid Leukemia, 2013; Jabbour & Kantarjian, 2018), radotinib potentially represents an attractive treatment option, given its lower cost compared with other on‐patent second‐generation BCR‐ABL1 TKIs (Experts in Chronic Myeloid Leukemia, 2013). However, it is important to note that, to date, this drug has been tested only in Asian patient populations. Additionally, the high level of deep molecular responses achieved with radotinib suggests that TFR may be attainable with radotinib, a possibility that warrants further investigation.
Author contributions
D‐WK designed the research study. All authors performed the research. YRD and D‐WK wrote the paper. All authors have reviewed and approved the manuscript.
Conflicts of interest
YRD has served as a consultant to Bristol‐Myers Squibb, IL‐YANG, Novartis, Pfizer and Otsuka Korea. All other authors have no competing interests.
Supporting information
Table SI. Cumulative incidence of MMR and MR4·5 (ITT population*).
Table SII. Newly occurring or worsening laboratory investigations/metabolic conditions (any cause).
Table SIII. Any‐cause cardiovascular AEs of interest.
Acknowledgements
We thank the patients and investigators for their participation in the trial and the Korea Leukemia Bank for biomaterial banking and analysis (NRF‐2013M3A9B8031236). This study was supported by funding from IL‐YANG PHARM. Co., Ltd. All authors contributed to and approved the manuscript; writing and editorial assistance was provided by Rebecca Turner, PhD, of Ashfield Healthcare Communications, funded by IL‐YANG PHARM. Co., Ltd.
References
- Baccarani, M. , Cortes, J. , Pane, F. , Niederwieser, D. , Saglio, G. , Apperley, J. , Cervantes, F. , Deininger, M. , Gratwohl, A. , Guilhot, F. , Hochhaus, A. , Horowitz, M. , Hughes, T. , Kantarjian, H. , Larson, R. , Radich, J. , Simonsson, B. , Silver, R.T. , Goldman, J. , Hehlmann, R. & European LeukemiaNet (2009) Chronic myeloid leukemia: an update of concepts and management recommendations of European LeukemiaNet. Journal of Clinical Oncology, 27, 6041–6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccarani, M. , Deininger, M.W. , Rosti, G. , Hochhaus, A. , Soverini, S. , Apperley, J.F. , Cervantes, F. , Clark, R.E. , Cortes, J.E. , Guilhot, F. , Hjorth‐Hansen, H. , Hughes, T.P. , Kantarjian, H.M. , Kim, D.W. , Larson, R.A. , Lipton, J.H. , Mahon, F.X. , Martinelli, G. , Mayer, J. , Müller, M.C. , Niederwieser, D. , Pane, F. , Radich, J.P. , Rousselot, P. , Saglio, G. , Saußele, S. , Schiffer, C. , Silver, R. , Simonsson, B. , Steegmann, J.L. , Goldman, J.M. & Hehlmann, R. (2013) European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood, 122, 872–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breccia, M. , Molica, M. , Zacheo, I. , Serrao, A. & Alimena, G. (2015) Application of systematic coronary risk evaluation chart to identify chronic myeloid leukemia patients at risk of cardiovascular diseases during nilotinib treatment. Annals of Hematology, 94, 393–397. [DOI] [PubMed] [Google Scholar]
- Brümmendorf, T.H. , Cortes, J.E. , de Souza, C.A. , Guilhot, F. , Duvillié, L. , Pavlov, D. , Gogat, K. , Countouriotis, A.M. & Gambacorti‐Passerini, C. (2015) Bosutinib versus imatinib in newly diagnosed chronic‐phase chronic myeloid leukaemia: results from the 24‐month follow‐up of the BELA trial. British Journal of Haematology, 168, 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes, J.E. , Kim, D.W. , Pinilla‐Ibarz, J. , le Coutre, P. , Paquette, R. , Chuah, C. , Nicolini, F.E. , Apperley, J.F. , Khoury, H.J. , Talpaz, M. , DiPersio, J. , DeAngelo, D.J. , Abruzzese, E. , Rea, D. , Baccarani, M. , Müller, M.C. , Gambacorti‐Passerini, C. , Wong, S. , Lustgarten, S. , Rivera, V.M. , Clackson, T. , Turner, C.D. , Haluska, F.G. , Guilhot, F. , Deininger, M.W. , Hochhaus, A. , Hughes, T. , Goldman, J.M. , Shah, N.P. , Kantarjian, H. & Investigators, P.A.C.E. (2013) A phase 2 trial of ponatinib in Philadelphia chromosome‐positive leukemias. New England Journal of Medicine, 369, 1783–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes, J.E. , Saglio, G. , Kantarjian, H.M. , Baccarani, M. , Mayer, J. , Boqué, C. , Shah, N.P. , Chuah, C. , Casanova, L. , Bradley‐Garelik, B. , Manos, G. & Hochhaus, A. (2016) Final 5‐year study results of DASISION: the dasatinib versus imatinib study in treatment‐naïve chronic myeloid leukemia patients trial. Journal of Clinical Oncology, 34, 2333–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes, J.E. , Jimenez, C.A. , Mauro, M.J. , Geyer, A. , Pinilla‐Ibarz, J. & Smith, B.D. (2017) Pleural effusion in dasatinib‐treated patients with chronic myeloid leukemia in chronic phase: identification and management. Clinical Lymphoma Myeloma and Leukemia, 17, 78–82. [DOI] [PubMed] [Google Scholar]
- Cortes, J. , Rea, D. & Lipton, J.H. (2019) Treatment‐free remission with first‐ and second‐generation tyrosine kinase inhibitors. American Journal of Hematology, 94, 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damrongwatanasuk, R. & Fradley, M.G. (2017) Cardiovascular complications of targeted therapies for chronic myeloid leukemia. Current Treatment Options in Cardiovascular Medicine, 19, 24. [DOI] [PubMed] [Google Scholar]
- Experts in Chronic Myeloid Leukemia (2013) The price of drugs for chronic myeloid leukemia (CML) is a reflection of the unsustainable prices of cancer drugs: from the perspective of a large group of CML experts. Blood, 121, 4439–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambacorti‐Passerini, C. , Aroldi, A. , Cordani, N. & Piazza, R. (2016) Chronic myeloid leukemia: second‐line drugs of choice. American Journal of Hematology, 91, 67–75. [DOI] [PubMed] [Google Scholar]
- Hochhaus, A. , Saglio, G. , Hughes, T.P. , Larson, R.A. , Kim, D.W. , Issaragrisil, S. , le Coutre, P.D. , Etienne, G. , Dorlhiac‐Llacer, P.E. , Clark, R.E. , Flinn, I.W. , Nakamae, H. , Donohue, B. , Deng, W. , Dalal, D. , Menssen, H.D. & Kantarjian, H.M. (2016) Long‐term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5‐year update of the randomized ENESTnd trial. Leukemia, 30, 1044–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochhaus, A. , Larson, R.A. , Guilhot, F. , Radich, J.P. , Branford, S. , Hughes, T.P. , Baccarani, M. , Deininger, M.W. , Cervantes, F. , Fujihara, S. , Ortmann, C.E. , Menssen, H.D. , Kantarjian, H. , O'Brien, S.G. , Druker, B.J. & Investigators, I.R.I.S. (2017) Long‐term outcomes of imatinib treatment for chronic myeloid leukemia. New England Journal of Medicine, 376, 917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imagawa, J. , Tanaka, H. , Okada, M. , Nakamae, H. , Hino, M. , Murai, K. , Ishida, Y. , Kumagai, T. , Sato, S. , Ohashi, K. , Sakamaki, H. , Wakita, H. , Uoshima, N. , Nakagawa, Y. , Minami, Y. , Ogasawara, M. , Takeoka, T. , Akasaka, H. , Utsumi, T. , Uike, N. , Sato, T. , Ando, S. , Usuki, K. , Morita, S. , Sakamoto, J. , Kimura, S. & DADI Trial Group (2015) Discontinuation of dasatinib in patients with chronic myeloid leukaemia who have maintained deep molecular response for longer than 1 year (DADI trial): a multicentre phase 2 trial. Lancet Haematology, 2, e528–e535. [DOI] [PubMed] [Google Scholar]
- Jabbour, E. & Kantarjian, H. (2018) Chronic myeloid leukemia: 2018 update on diagnosis, therapy and monitoring. American Journal of Hematology, 93, 442–459. [DOI] [PubMed] [Google Scholar]
- Kim, H.T. (2017) Cumulative incidence in competing risks data and competing risks regression analysis. Clinical Cancer Research, 13, 559–565. [DOI] [PubMed] [Google Scholar]
- Kim, S.H. , Menon, H. , Jootar, S. , Saikia, T. , Kwak, J.Y. , Sohn, S.K. , Park, J.S. , Jeong, S.H. , Kim, H.J. , Kim, Y.K. , Oh, S.J. , Kim, H. , Zang, D.Y. , Chung, J.S. , Shin, H.J. , Do, Y.R. , Kim, J.A. , Kim, D.Y. , Choi, C.W. , Park, S. , Park, H.L. , Lee, G.Y. , Cho, D.J. , Shin, J.S. & Kim, D.W. (2014) Efficacy and safety of radotinib in chronic phase chronic myeloid leukemia patients with resistance or intolerance to BCR‐ABL1 tyrosine kinase inhibitors. Haematologica, 99, 1191–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak, J.Y. , Kim, S.H. , Oh, S.J. , Zang, D.Y. , Kim, H. , Kim, J.A. , Do, Y.R. , Kim, H.J. , Park, J.S. , Choi, C.W. , Lee, W.S. , Mun, Y.C. , Kong, J.H. , Chung, J.S. , Shin, H.J. , Kim, D.Y. , Park, J. , Jung, C.W. , Bunworasate, U. , Comia, N.S. , Jootar, S. , Reksodiputro, A.H. , Caguioa, P.B. , Lee, S.E. & Kim, D.W. (2017) Phase III clinical trial (RERISE study) results of efficacy and safety of radotinib compared with imatinib in newly diagnosed chronic phase chronic myeloid leukemia. Clinical Cancer Research, 23, 7180–7188. [DOI] [PubMed] [Google Scholar]
- Larson, R.A. , Hochhaus, A. , Hughes, T.P. , Clark, R.E. , Etienne, G. , Kim, D.W. , Flinn, I.W. , Kurokawa, M. , Moiraghi, B. , Yu, R. , Blakesley, R.E. , Gallagher, N.J. , Saglio, G. & Kantarjian, H.M. (2012) Nilotinib vs imatinib in patients with newly diagnosed Philadelphia chromosome‐positive chronic myeloid leukemia in chronic phase: ENESTnd 3‐year follow‐up. Leukemia, 26, 2197–2203. [DOI] [PubMed] [Google Scholar]
- Mahon, F.X. , Réa, D. , Guilhot, J. , Guilhot, F. , Huguet, F. , Nicolini, F. , Legros, L. , Charbonnier, A. , Guerci, A. , Varet, B. , Etienne, G. , Reiffers, J. , Rousselot, P. & Intergroupe Français des Leucémies Myéloïdes Chroniques (2010) Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncology, 11, 1029–1035. [DOI] [PubMed] [Google Scholar]
- Pinilla‐Ibarz, J. , Sweet, K. , Emole, J. & Fradley, M. (2015) Long‐term BCR‐ABL1 tyrosine kinase inhibitor therapy in chronic myeloid leukemia. Anticancer Research, 35, 6355–6364. [PubMed] [Google Scholar]
- Takahashi, N. , Nishiwaki, K. , Nakaseko, C. , Aotsuka, N. , Sano, K. , Ohwada, C. , Kuroki, J. , Kimura, H. , Tokuhira, M. , Mitani, K. , Fujikawa, K. , Iwase, O. , Ohishi, K. , Kimura, F. , Fukuda, T. , Tanosaki, S. , Takahashi, S. , Kameoka, Y. , Nishikawa, H. , Wakita, H. & STAT study group (2018) Treatment-free remission after two-year consolidation therapy with nilotinib in patients with chronic myeloid leukemia: STAT2 trial in Japan. Haematologica, 103, 1835–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabriskie, M.S. , Vellore, N.A. , Gantz, K.C. , Deininger, M.W. & O'Hare, T. (2015) Radotinib is an effective inhibitor of native and kinase domain‐mutant BCR‐ABL1. Leukemia, 29, 1939–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table SI. Cumulative incidence of MMR and MR4·5 (ITT population*).
Table SII. Newly occurring or worsening laboratory investigations/metabolic conditions (any cause).
Table SIII. Any‐cause cardiovascular AEs of interest.