

Abstract
Genome‐wide association studies (GWASs) in European and East Asian populations have identified more than 40 disease‐susceptibility genes in primary biliary cholangitis (PBC). The aim of this study is to computationally identify disease pathways, upstream regulators, and therapeutic targets in PBC through integrated GWAS and messenger RNA (mRNA) microarray analysis. Disease pathways and upstream regulators were analyzed with ingenuity pathway analysis in data set 1 for GWASs (1,920 patients with PBC and 1,770 controls), which included 261 annotated genes derived from 6,760 single‐nucleotide polymorphisms (P < 0.00001), and data set 2 for mRNA microarray analysis of liver biopsy specimens (36 patients with PBC and 5 normal controls), which included 1,574 genes with fold change >2 versus controls (P < 0.05). Hierarchical cluster analysis and categorization of cell type–specific genes were performed for data set 2. There were 27 genes, 10 pathways, and 149 upstream regulators that overlapped between data sets 1 and 2. All 10 pathways were immune‐related. The most significant common upstream regulators associated with PBC disease susceptibility identified were interferon‐gamma (IFNG) and CD40 ligand (CD40L). Hierarchical cluster analysis of data set 2 revealed two distinct groups of patients with PBC by disease activity. The most significant upstream regulators associated with disease activity were IFNG and CD40L. Several molecules expressed in B cells, T cells, Kupffer cells, and natural killer–like cells were identified as potential therapeutic targets in PBC with reference to a recently reported list of cell type–specific gene expression in the liver. Conclusion: Our integrated analysis using GWAS and mRNA microarray data sets predicted that IFNG and CD40L are the central upstream regulators in both disease susceptibility and activity of PBC and identified potential downstream therapeutic targets.

Abbreviations
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- APRIL
a proliferation‐inducing ligand
- AST
aspartate aminotransferase
- BAFF
B‐cell‐activating factor
- BAFFR
BAFF receptor
- BCMA
B‐cell maturation antigen
- BH
Benjamini‐Hochberg
- CD40L
CD40 ligand
- GWAS
genome‐wide association study
- HLA
human leukocyte antigen
- IFNG
interferon‐gamma
- IL
interleukin
- IPA
ingenuity pathway analysis
- mRNA
messenger RNA
- NK
natural killer
- PBC
primary biliary cholangitis
- SNP
single‐nucleotide polymorphism
- STAT3
signal transducer and activator of transcription 3
- TNF
tumor necrosis factor
- TNFRSF
tumor necrosis factor receptor superfamily
- TNFSF
tumor necrosis factor superfamily
Primary biliary cholangitis (PBC) is a rare chronic autoimmune cholestatic liver disease that predominantly affects middle‐aged females. PBC is characterized by lymphocytic cholangitis and gradual destruction of the small intrahepatic bile ducts, which leads to fibrosis, cirrhosis, and hepatic failure.( 1 , 2 ) Ursodeoxycholic acid is the established first‐line therapeutic agent for PBC. However, 10% to 20% of patients are resistant to ursodeoxycholic acid and progress to end‐stage liver disease.( 1 , 2 ) Although the beneficial effects of obeticholic acid and bezafibrate have recently been reported in patients with ursodeoxycholic acid resistance, the long‐term effects of obeticholic acid and bezafibrate have not been established.( 3 , 4 ) Therefore, there is a substantial clinical need for new treatments based on an understanding of the pathogenesis of PBC.
Identification of key disease susceptibility genes, pathways, and upstream regulators may provide important clues to the pathogenesis of PBC and the development of new therapeutic options. In addition to human leukocyte antigens (HLAs), genome‐wide association studies (GWASs) have identified more than 30 non‐HLA susceptibility regions in populations with European descents.( 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 ): MMEL1/TNFRSF14, IL12RB2/DENND1B/C11orf53, YPEL5/LBH, IL1RL1/IL1RL2, STAT4/NAB1, SLC19A3/CCL20, PLCL2, CD80, TIMMDC1/TMEM39A, IL12A/IL12A‐AS1/IQCJ/SCHIP1, DGKQ, NFKB1/MANBA, IL7R/CAPSL, NUDT12/C5orf30, IL12B, OLIG3/TNFAIP3, ELMO1, IRF5/TNPO3, RPS6KA4, DDX6/CXCR5, TNFRSF1A, SH2B3, TNFSF11, ATXN2/BRAP, DLEU1/BCMS, RAD51B,TNFAIP3, IL16 EXOC3L4, RMI2/CLEC16A/SOCS1, IRF8/FOXF1, ZPBP2/GSDMB/IKZF3, MAPT, TYK2, SPIB, and SYNGR1/PDGFB/RPL3. In addition, Asian‐specific susceptibility regions for PBC (CD58, CD28/CTLA4, IL21‐AS1, TNFSF15/TNFSF8, POU2AF1, IL16, IL21R, PRKCB, CSNK2A2/CCDC113, and ARID3A) were identified in Japanese and Chinese populations.( 13 , 14 , 15 , 16 ) These results indicated that T‐cell activation pathways that produce interferon‐gamma (IFNG) (i.e., interleukin [IL]‐12 production in antigen‐presenting cells and subsequent IFNG production in T cells by IL‐12 and tumor necrosis factor [TNF] superfamily [TNFSF] 15 stimulation) and B‐cell differentiation and maturation pathways involving various transcription factors, which are encoded by SPIB, POU2AF1, IKZF3, and ARID3A, play important roles in the development of PBC.( 17 , 18 , 19 ) In addition, gene‐expression profiling microarray analysis using liver tissue samples or peripheral blood mononuclear cells indicated that many genes implicated in intrahepatic inflammation, fibrosis, and regeneration are up‐regulated in PBC.( 20 , 21 , 22 , 23 ) However, messenger RNA (mRNA) expression profile data are very limited in scope. Comprehensive analysis of GWAS and mRNA expression profiles has not been performed yet. Here we identified IFNG and CD40 ligand (CD40L) as the central upstream regulators in the pathogenesis of PBC using computational analysis combining GWAS and mRNA microarray data in the Japanese population. In addition, we identified several potential therapeutic downstream targets, including B‐cell maturation antigen (BCMA) (also known as tumor necrosis factor receptor superfamily 17 [TNFRSF17]) and a B‐cell‐activating factor (BAFF) (also known as TNFSF13B) belonging to the TNF family receptor (BAFFR) in B cells, by combining our mRNA expression profile data set of livers with PBC and a data set of cell type–specific mRNA expression in normal livers published in the literature.( 24 )
Materials and Methods
Subjects
DNA samples for GWASs were collected from 3,690 Japanese individuals (1,920 patients with PBC and 1,770 healthy controls) as described.( 13 , 14 , 16 ) The diagnosis of PBC was performed according to the diagnostic criteria by the American Association for the Study of Liver Diseases.( 2 ) The demographics and clinical features of these 1,920 patients are found in Supporting Table S1.Written informed consent was obtained from all participants. The protocol of this study was approved by the Committee on Research Ethics and Genetically Modified Organisms of the Graduate School of Medicine, University of Tokyo, Tokyo, Japan, and the ethics committees of the National Hospital Organization and all other participating institutions. For the mRNA microarray analysis, liver needle biopsy specimens were collected from 36 patients with PBC (mean age, 61.3 years; female, 83.3%; undergoing ursodeoxycholic acid treatment, 22.2%). Normal liver tissue was collected within 30 minutes after liver resection from 5 patients with metastatic liver cancer (mean age, 74.0 years; female, 80.0%).
Genotyping, Imputation, and Association Analysis
Genotyping was performed using the Japonica V1 array (Toshiba, Tokyo, Japan) and the Axiom Genome‐Wide ASI 1 Array (Affymetrix Japan, Tokyo, Japan). Genotype calling was performed with the apt‐probeset‐genotype program in Affymetrix Power Tools version 1.18.2 (Thermo Fisher Scientific, Waltham, MA). Sample quality control was conducted according to the manufacturer’s recommendations (dish quality control >0.82 and sample call rate >97%). Clustering of each single‐nucleotide polymorphism (SNP) was evaluated by the ps‐classification function in the SNPolisher package version 1.5.2 (Thermo Fisher Scientific). SNPs that were determined to be “recommended” by the Ps classification function were used for downstream analyses. SNPs that satisfied the following criteria were used for genotype imputation: call rate >99.0%, Hardy‐Weinberg Equilibrium P > 0.0001, and minor allele frequency >0.5%. Prephasing was conducted with EAGLE version 2.3.2( 25 ) with default settings. Genotype imputation was conducted with IMPUTE4 version 1.0( 26 ) using a phased reference panel of 2,049 Japanese individuals from a prospective general population cohort study performed by the Tohoku Medical Megabank Organization, Sendai, Japan.( 27 , 28 ) These procedures were conducted using default settings. Cryptic relatives were excluded using PRIMUS( 29 ) with default settings. In addition, principal component analysis was performed using East Asian samples from the International 1000 Genome Project (104 Japanese in Tokyo, 103 Han Chinese in Beijing, 93 Southern Han Chinese, 91 Chinese Dai in Xishuangbanna, and 99 Kinh in Ho Chi Minh City samples) in addition to the case and control samples. Based on the Smirnov‐Grubbs test with a Bonferroni corrected P < 0.05, principal component analysis identified outliers for exclusion.
Association analysis was performed with PLINK version 1.9 for each data set (i.e., 2,897 Applied Scientific Instrumentation array data and 1,148 Japonica array data). The following options were used for PLINK: call rate >97.0%, Hardy‐Weinberg Equilibrium P > 0.000001, minor allele frequency >0.1%, and logistic regression modeling. The meta‐analysis option in PLINK was used after excluding duplicates between the two data sets. The fixed‐effects meta‐analysis P value was used.
Gene Allocation
After association analysis, SNPs with P < 0.00001 were extracted. Genes were allocated to those SNPs only when they were located within genes; 261 genes were allocated to at least 1 of the 6,760 SNPs with P < 0.00001. The refGene.txt.gz file downloaded from the University of California, Santa Cruz (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/), was converted to a BED file as follows:
cat refGene.txt | awk ‐v OFS='\t' ‐v FS='\t' '{print $3,$5,$6,$13,1,$4}'
Bedtools was used to allocate genes as follows( 30 ):
bedtools intersect ‐a SNP_bed (conversion from.assoc after association analysis) ‐b reference_bed (conversion from refGene.txt.gz) –wao
Quantitation of mRNA Expression in Liver Tissues
Liver tissue samples were preserved in RNA stabilization reagent (RNAlater; QIAGEN, Hilden, Germany), and total RNA was extracted with the RNeasy Mini Kit (QIAGEN). From each liver tissue sample, 50 ng of total RNA was labeled using the Agilent Low‐Input Quick Amp Labeling Kit and applied for hybridization, washing, and scanning according to the manufacturer’s protocol (Agilent, Santa Clara, CA). Quantitative DNA microarray data were obtained using Agilent Feature Extraction software. Data normalization for all probes excluding long intergenic noncoding RNA (lincRNA) was performed using the quantile method. Adjusted P values to correct for multiple testing were calculated using limma (http://www.bioconductor.org/).( 31 )
Analysis of the Microarray Data
We applied the quantile normalization method from the limma package in R to 41 samples (PBC: 36; normal: 5) to normalize the data on expression excluding lincRNAs. Statistical analysis was also performed with limma.( 31 ) We extracted the probes where at least one sample had a present (P) flag. The fold change and the P value for PBC versus normal samples were calculated for each probe. The 3,172 genes with a ratio >2 and P < 0.05 were extracted.
These 3,172 genes were uploaded to ingenuity pathway analysis (IPA) and mapped to 2,525 genes. We input these data into GeneSpring. If genes have duplicates, GeneSpring calculates their average expression. Through this method, the expression matrix of the 2,522 genes was completed.
Of the 2,522 genes, those whose expression levels were altered as the result of liver resection (e.g., ischemia due to ligation of the hepatic artery and portal vein before resection of the liver with metastatic cancer) were excluded with reference to publicly available data (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE28619, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE48452) in Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/). In fact, five normal biopsy samples in each data set (GSM709348, GSM709349, GSM709350, GSM709351, and GSM709352 from http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE28619; GSM1178970, GSM1178971, GSM1178972, GSM1178973, and GSM1178974 from http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE48452) were normalized with our 36 PBC samples. Fold changes in these publicly available data sets and our data set of 2,522 significant genes were compared. A gene was excluded if it met either of the following conditions: (1) The direction of the fold change in the data set of our samples and the data set including public samples was different; or (2) expression change occurred in the data set of our samples and not in the data set of public samples. After these steps, 1,574 genes were extracted. Hierarchical clustering with a Euclidean distance metric and a Ward linkage rule was performed with these 1,574 genes, both on the genes and on the samples, using GeneSpring version 14.9.1.
Pathway and Upstream Analysis
IPA was used to identify the signaling networks involved in the 261 GWAS genes allocated to significant SNPs and in the 1,574 significant microarray genes (Fig. 1). Signaling networks were estimated from each gene set. Each signaling network was assigned a Benjamini‐Hochberg (BH) P value reflecting the probability of this network being generated at random. BH–P values are corrected P values based on the BH method to account for multiple testing. Upstream regulators were estimated using Fisher’s exact test with P < 0.05.
Fig. 1.
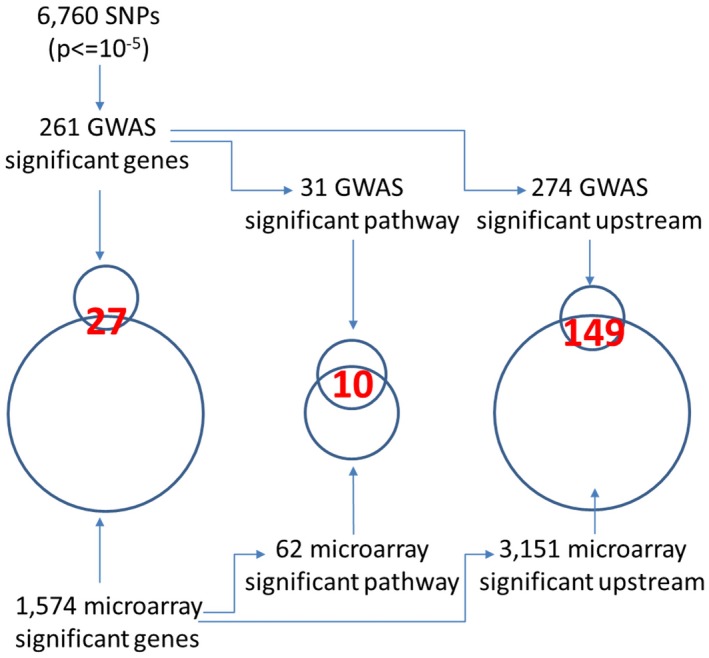
Relationships between GWAS and microarray data. (left) Intersection of genes from GWAS and microarray analysis. (center) Intersection of pathways based on genes identified through GWAS and microarray analyses. (right) Intersection of upstream regulators based on genes identified through GWAS and microarray analyses.
Identification of Cell Type–Specific Gene Expression in the Liver
There were 361 cell type–specific genes listed in a previous study that were categorized into 20 cell types.( 24 ) Because those categories contained duplicates, we merged the duplicated categories into nine categories: B cell, T cell, natural killer (NK)‐like cell, Kupffer cell, cholangiocyte, endothelial cell, erythroid, hepatocyte, and stellate cell. We categorized genes identified in the microarray analysis into these nine cell types.
Results
Integrated Analysis of GWAS and Microarray Data
As summarized in Fig. 1, there were 6,760 SNPs with P < 0.00001 identified after GWAS analysis (Supporting Table S2). These SNPs were allocated to 261 genes when an SNP was located within a gene (Supporting Table S3). Next, we focused on the intersection between the 261 genes from GWAS analysis and 1,574 significant genes (Supporting Table S4) from the microarray analysis. The number of the genes in the intersection was 27 (Table 1), indicating that approximately 10% of genes identified through GWASs were functionally operative in the liver. In addition, 24 of 27 genes in the intersection were located on chromosome 6, including HLA genes (HLA‐DQA1, HLA‐DQB2, HLA‐DPA1, HLA‐DPB1, HLA‐DMB, and HLA‐DPB2), whereas only four genes were located in non‐HLA regions. These were PRKCB on chromosome 16, UBE2D3 on chromosome 4, KCNMA1 on chromosome 10, and SLC17A4 on chromosome 6.
Table 1.
Genes in the Intersection
| Gene | Chromosome | Position | rs ID | P Value | Fold Change |
|---|---|---|---|---|---|
| HLA‐DQA1 | 6 | 32605917 | rs118073417 | 5.91E‐23 | 2.95 |
| TNXB | 6 | 32070239 | rs117481809 | 3.07E‐22 | −2.67 |
| CYP21A2 | 6 | 32006722 | rs11757034 | 7.56E‐22 | −4.24 |
| HLA‐DQB2 | 6 | 32727550 | rs117548357 | 1.02E‐21 | 2.11 |
| PSMB9 | 6 | 32822186 | rs4148878 | 4.29E‐21 | 2.36 |
| HLA‐DPA1 | 6 | 33047432 | rs2856822 | 4.48E‐21 | 2.75 |
| HLA‐DPB1 | 6 | 33047432 | rs2856822 | 4.48E‐21 | 2.17 |
| LY6G5B | 6 | 31639845 | rs11758242 | 5.05E‐20 | −3.63 |
| HLA‐DMB | 6 | 32903086 | rs112252170 | 8.38E‐20 | 2.12 |
| POU5F1 | 6 | 31133682 | rs200370996 | 1.72E‐19 | −2.21 |
| HCP5 | 6 | 31432006 | rs2263318 | 4.82E‐19 | 3.98 |
| PPP1R10 | 6 | 30568448 | rs7774485 | 5.84E‐18 | −2.09 |
| PPP1R18 | 6 | 30650596 | rs199834657 | 1.25E‐17 | −2.58 |
| TRIM15 | 6 | 30138936 | rs118105893 | 3.05E‐17 | −5.17 |
| HLA‐DPB2 | 6 | 33096380 | rs28361077 | 1.28E‐16 | 2.71 |
| COL11A2 | 6 | 33137689 | rs9277929 | 8.86E‐16 | −2.23 |
| UBD | 6 | 29527621 | rs362535 | 3.15E‐15 | 3.05 |
| ZKSCAN4 | 6 | 28226171 | rs10708573 | 1.05E‐14 | 2.56 |
| ZKSCAN3 | 6 | 28321836 | rs79144650 | 2.07E‐14 | 2.61 |
| IP6K3 | 6 | 33705528 | rs12193658 | 5.17E‐14 | −2.78 |
| SLC17A4 | 6 | 25779727 | rs3734525 | 1.37E‐12 | 2.14 |
| PRR3 | 6 | 30529475 | rs4713337 | 3.02E‐09 | −2.24 |
| HLA‐F | 6 | 29691713 | rs2076182 | 2.73E‐08 | 2.24 |
| PRKCB | 16 | 23888840 | rs7404928 | 4.74E‐08 | 2.32 |
| UBE2D3 | 4 | 103732866 | rs223413 | 5.83E‐08 | −3.08 |
| HLA‐J | 6 | 29973925 | rs4313034 | 9.24E‐08 | 3.38 |
| KCNMA1 | 10 | 78682672 | rs11001933 | 8.56E‐06 | −2.64 |
Note: These 27 genes are found in the intersection between the 261 significant genes in the GWAS analysis and the 1,574 significant genes in the microarray analysis. The lowest P value for SNPs located within a gene is shown for each annotated gene. Fold‐change values for mRNA expression in PBC versus healthy controls are also shown.
Abbreviations: COL11A2, Collagen Type XI Alpha 2 Chain; CYP21A2, Cytochrome P450 Family 21 Subfamily A Member 2; HCP5, HLA Complex P5; HLA‐DMB, HLA Class II Histocompatibility Antigen, DM Beta Chain; HLA‐DPA1, HLA Class II Histocompatibility Antigen, DQ Alpha 1 Chain; HLA‐DPB1, HLA Class II Histocompatibility Antigen, DP(W4) Beta Chain; HLA‐DPB2, HLA Class II Histocompatibility Antigen, DQ Beta 2 Chain; HLA‐DQA1, HLA Class II Histocompatibility Antigen, DQ Alpha 1 Chain; HLA‐DQB2, HLA Class II Histocompatibility Antigen, DQ Beta 2 Chain; HLA‐F, HLA Class I Histocompatibility Antigen, Alpha Chain F; HLA‐J, Major Histocompatibility Complex, Class I, J (Pseudogene); IP6K3, Inositol Hexakisphosphate Kinase 3; KCNMA1, Potassium Calcium‐Activated Channel Subfamily M Alpha 1; LY6G5B, Lymphocyte Antigen 6 Family Member G5B; POU5F1, POU Class 5 Homeobox 1; PPP1R10, Protein Phosphatase 1 Regulatory Subunit 10; PPP1R18, Protein Phosphatase 1 Regulatory Subunit 18; PRKCB, Protein Kinase C Beta; PRR3, Proline Rich 3; PSMB9, Proteasome 20S Subunit Beta 9; SLC17A4, Solute Carrier Family 17 Member 4; TNXB, Tenascin XB; TRIM15, Tripartite Motif Containing 15; UBD, Ubiquitin D; UBE2D3, Ubiquitin Conjugating Enzyme E2 D3; ZKSCAN3, Zinc Finger With KRAB And SCAN Domains 3; ZKSCAN4, Zinc Finger With KRAB And SCAN Domains 4.
IPA identified 31 significant operative pathways based on the GWAS analysis of 261 genes (Supporting Table S5) and 62 based on the microarray analysis of 1,574 genes (Supporting Table S6). There were 10 pathways in the intersection (Table 2), indicating that at least 32% of the pathways identified through GWASs were functionally operative in the liver. Notably, all of these pathways were related to the immune system, such as the antigen presentation pathway, T helper 1 (Th1) pathway, T helper 2 (Th2) pathway, and communication between innate and adaptive immune cells.
Table 2.
Pathways in the Intersection
| Common 10 Pathways | GWAS Rank | Array Rank | GWAS P Value | Array P Value |
|---|---|---|---|---|
| Antigen presentation pathway | 1 | 22 | 7.94E‐29 | 0.002188 |
| Th1 pathway | 6 | 2 | 7.94E‐12 | 1.00E‐09 |
| Th2 pathway | 8 | 14 | 3.16E‐11 | 0.000398 |
| Type 1 diabetes mellitus signaling | 9 | 34 | 7.94E‐11 | 0.009333 |
| Th1 and Th2 activation pathway | 11 | 1 | 5.37E‐10 | 5.01E‐11 |
| Crosstalk between dendritic cells and NK cells | 12 | 29 | 9.12E‐10 | 0.007943 |
| T helper cell differentiation | 14 | 17 | 2.95E‐08 | 0.001259 |
| Altered T‐cell and B‐cell signaling in rheumatoid arthritis | 17 | 18 | 1.95E‐07 | 0.001259 |
| Communication between innate and adaptive immune cells | 23 | 13 | 3.63E‐05 | 0.000316 |
| Neuroinflammation signaling pathway | 24 | 6 | 8.91E‐05 | 2.95E‐06 |
Note: These 10 pathways are found in the intersection between the 31 significant pathways based on the GWAS analysis and the 62 significant pathways based on the microarray analysis.
Upstream Regulators Associated With Disease Susceptibility
As summarized in Fig. 1, there were 274 significant operative upstream regulators based on the GWAS analysis of 261 genes (Supporting Table S7) and 3,151 based on the microarray analysis of 1,574 genes (Supporting Table S8). There were 149 upstream regulators in the intersection (Supporting Table S9), indicating that approximately 54% of upstream regulators identified through GWASs were functionally operative in the liver. Of these upstream regulators, mRNA expression of 11 (encoded by IFNG, CD40L, B2M, OSM, CRP, IRF1, PRDM1, SPIB, STAT3, and IL‐6) was significantly increased (fold change >2) or decreased (fold change < 2) in livers with PBC compared with normal livers (Supporting Table S9). Among these significant upstream regulators, fold change for IL‐6, oncostatin M, C‐reactive protein, and signal transducer and activator of transcription 3 (STAT3) were −42.56, −12.694, −6.75, and − 2.18, respectively. Because the IL‐6–STAT3 signaling pathway is markedly activated in the resected liver,( 32 ) it was considered that significant alteration in the levels of these mRNA was due to the effect of the procedure of sample collection. Therefore, we removed four upstream regulators related to the IL‐6 signaling pathway (IL‐6, oncostatin M, C‐reactive protein, and STAT3). We only presented seven molecules (IFNG, CD40L, beta‐2‐microglobulin, IFN regulatory factor 1, PR domain zinc finger protein 1, transcription factor Spi‐B, and Fas ligand) as being potential significant upstream regulators identified through both GWAS and microarray analyses (Table 3).
Table 3.
Upstream Regulators in the Intersection
| Upstream Regulator | GWAS Rank | Array Rank | GWAS P Value | Array P Value | Fold Change | Molecule Type |
|---|---|---|---|---|---|---|
| IFNG | 6 | 9 | 1.62E‐08 | 2.52E‐26 | 5.76 | Cytokine |
| CD40L | 12 | 33 | 1.39E‐06 | 2.62E‐19 | 3.77 | Cytokine |
| B2M | 30 | 2,090 | 0.000163 | 0.0101 | 2.35 | Transmembrane receptor |
| IRF1 | 48 | 217 | 0.00118 | 2.69E‐09 | −2.24 | Transcription regulator |
| PRDM1 | 75 | 1,159 | 0.00591 | 0.000589 | −2.29 | Transcription regulator |
| SPIB | 83 | 317 | 0.00877 | 7.35E‐08 | 2.47 | Transcription regulator |
| FASLG | 239 | 772 | 0.0444 | 5.74E‐05 | 2.27 | Cytokine |
Note: These seven upstream regulators are found in the intersection between 274 significant upstream regulators based on the GWAS analysis and 3,151 significant upstream regulators based on the microarray analysis.
Abbreviations: B2M, beta‐2‐microglobulin; FASLG, Fas ligand; IRF1, interferon regulatory factor 1; PRDM1, PR domain zinc finger protein 1; SPIB, transcription factor Spi‐B.
Hierarchical Clustering of Gene Expression in Livers With PBC
To investigate the biological significance of genes expressed in livers with PBC, we performed hierarchical clustering of gene expression using data on 1,574 significant genes identified through the microarray analysis in 36 patients with PBC and 5 healthy controls (Fig. 2A). The 36 patients with PBC were clearly divided into two clusters (PBC group 1 and PBC group 2) based on their gene‐expression pattern. To investigate phenotypic difference between the two groups, we compared the levels of several serum markers. We found that serum levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), and alkaline phosphatase (ALP), markers of disease activity in PBC, were significantly higher (P = 0.024, 0.018, and 0.041, respectively) in PBC group 2 than in PBC group 1 (Fig. 2B). A heatmap revealed some genes or gene clusters with higher expression in PBC group 2 than in PBC group 1; therefore, we performed t tests to identify genes that were differentially expressed between these two groups. There were 183 genes whose expression was significantly higher in PBC group 2 than in PBC group 1, and 12 genes whose expression was significantly lower in PBC group 2 than in PBC group 1 (Supporting Table S10).
Fig. 2.
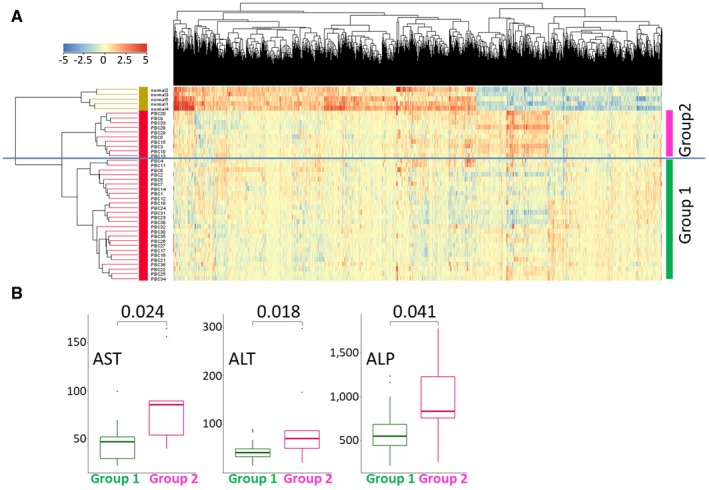
(A) Hierarchical clustering of 1,574 significant genes from the microarray analysis in 36 patients with PBC and 5 controls. The samples were divided into three groups: PBC group 1, PBC group 2, and normal. (B) Distribution of serum markers of PBC activity (AST, ALT, and ALP) in patients with PBC. Mean AST, ALT, and ALP values were all significantly higher in PBC group 2 than in PBC group 1. P values based on the Wilcoxon test are shown.
Upstream Regulators Associated With Disease Activity
Next, we performed upstream analysis with the 195 significant genes in livers with PBC. There were an estimated 2,813 upstream regulators (Supporting Table S11) in which the expression of 40 upstream regulators was significantly different between PBC group 1 and PBC group 2 (Table 4). Of these 40 upstream regulators significantly associated with PBC disease activity, IFNG and CD40L were the most and second‐most significant upstream regulators, respectively, followed by various cytokines, chemokines, and their transmembrane receptors (Table 4). Five significant upstream regulators for PBC disease susceptibility (IFNG, CD40L, PR domain zinc finger protein 1, Fas ligand, and transcription factor Spi‐B) (Table 3) were also identified as upstream regulators for disease activity (Table 4).
Table 4.
Upstream Regulators Associated With Disease Activity
| Upstream Regulator | P Value | Fold Change (Group 2 vs. Group 1) | Molecular Type |
|---|---|---|---|
| IFNG | 2.27E‐18 | 3.33 | Cytokine |
| CD40L | 6.50E‐18 | 2.43 | Cytokine |
| IL‐6 | 7.04E‐17 | 3.13 | Cytokine |
| CCL2 | 1.06E‐11 | 3.05 | Cytokine |
| CCL11 | 5.88E‐11 | 3.01 | Cytokine |
| CCR2 | 3.04E‐10 | 2.79 | G protein–coupled receptor |
| TLR7 | 5.99E‐10 | 2.33 | Transmembrane receptor |
| CCR5 | 2.23E‐09 | 2.46 | G protein–coupled receptor |
| PRDM1 | 0.0000001 | 2.58 | Transcription regulator |
| TLR8 | 0.000000102 | 2.08 | Transmembrane receptor |
| CCL5 | 0.000000308 | 2.29 | Cytokine |
| FASLG | 0.000000471 | 2.34 | Cytokine |
| PLAUR | 0.00000107 | 2.38 | Transmembrane receptor |
| CXCL10 | 0.00000198 | 4.32 | Cytokine |
| CXCL3 | 0.0000143 | 2.23 | Cytokine |
| SPIB | 0.0000834 | 3.57 | Transcription regulator |
| CYP27B1 | 0.000195 | 2.18 | Enzyme |
| CCL19 | 0.000208 | 4.03 | Cytokine |
| GZMA | 0.000216 | 2.43 | Peptidase |
| CDK1 | 0.000454 | 2.44 | Kinase |
| ICOS | 0.000688 | 3.42 | Transmembrane receptor |
| SH2D1A | 0.000726 | 2.49 | Other |
| LTB | 0.000955 | 2.71 | Cytokine |
| TLR10 | 0.00106 | 2.64 | Transmembrane receptor |
| CCL18 | 0.00148 | 4.22 | Cytokine |
| LTF | 0.00154 | 2.27 | Peptidase |
| CD48 | 0.00251 | 2.24 | Other |
| CXCL8 | 0.00513 | 6.51 | Cytokine |
| SOCS3 | 0.00686 | 2.41 | Phosphatase |
| CD96 | 0.00853 | 2.34 | Other |
| CENPF | 0.00853 | 2.10 | Other |
| CXCR6 | 0.00853 | 2.70 | G protein–coupled receptor |
| EGR2 | 0.00991 | 3.42 | Transcription regulator |
| CXCR3 | 0.015 | 3.62 | G protein–coupled receptor |
| LEF1 | 0.0174 | 3.01 | Transcription regulator |
| CXCL11 | 0.0254 | 3.91 | Cytokine |
| CD72 | 0.0254 | 2.48 | Transmembrane receptor |
| CXCL9 | 0.0337 | 4.27 | Cytokine |
| MMP2 | 0.0378 | 2.14 | Peptidase |
| SLAMF6 | 0.042 | 2.89 | Transmembrane receptor |
Note: These 40 upstream regulators are from the 195 genes with a significant difference in expression between PBC group 1 and PBC group 2. Fold change values in PBC group 2 versus PBC group 1 are also shown.
Abbreviations: CCL11, C‐C motif chemokine ligand 11; CCL18, C‐C motif chemokine ligand 18; CCL19, C‐C motif chemokine ligand 19; CCL2, C‐C motif chemokine ligand 2; CCL5, C‐C motif chemokine ligand 5; CCR2, C‐C motif chemokine receptor 2; CCR5, C‐C motif chemokine receptor 5; CD48, CD48 antigen; CD72, CD72 antigen; CD96, CD96 antigen; CDK1, cyclin‐dependent kinase 1; CENPF, centromere protein F; CXCL3, C‐X‐C motif chemokine ligand 3; CXCL8, C‐X‐C motif chemokine ligand 8; CXCL9, C‐X‐C motif chemokine ligand 9; CXCL10, C‐X‐C motif chemokine ligand 10; CXCL11, C‐X‐C motif chemokine ligand 11; CXCR3, C‐X‐C motif chemokine receptor 3; CXCR6, C‐X‐C motif chemokine receptor 6; CYP27B1, cytochrome P450 family 27 subfamily B member 1; EGR2, E3 SUMO‐protein ligase EGR2; GZMA, granzyme A; ICOS, inducible T‐cell co‐stimulator; IL‐6, interleukin‐6; LEF1, lymphoid enhancer‐binding factor 1; LTB, lymphotoxin‐beta; LTF, lactotransferrin; MMP2, matrix metallopeptidase 2; PLAUR, plasminogen activator urokinase receptor; PRDM1, PR domain zinc finger protein 1; SH2D1A, SH2 domain‐containing protein 1A; SLAMF6, SLAM family member 6; SOCS3, suppressor of cytokine signaling 3; TLR7, toll‐like receptor 7; TLR8, toll‐like receptor 8; TLR10, toll‐like receptor 10.
Identification and Categorization of Cell Type–Specific Genes
A recently reported list of 361 cell type–specific genes expressed in the liver( 24 ) was used to categorize the 1,574 significant genes identified through microarray analysis in this study. From these 361 cell type–specific genes, we extracted 62 genes whose expression was significantly different (fold change > 2) in PBC as compared with healthy controls. Of these 62 cell type–specific genes, 37 genes had higher expression in PBC than in healthy controls, whereas 25 genes had lower expression in PBC than in healthy controls (Supporting Table S12A). Among the 37 up‐regulated genes, 36 genes were categorized as part of cell populations related to the immune system (12 genes in B cells, 13 genes in T cells, 8 genes in NK‐like cells, 7 genes in Kupffer cells, with 4 genes categorized in two categories), whereas only one gene was categorized as related to hepatocytes (Fig. 3A; Supporting Table S12A). In contrast, most of the 25 down‐regulated genes were categorized as part of cell populations that are not related to the immune system (5 genes in endothelial cells, 4 genes in cholangiocytes, 3 genes in erythroid, 10 genes in hepatocytes, 1 gene in stellate cells, with 1 gene categorized in two categories) (Fig. 3A; Supporting Table S12A). In addition, there were 21 genes whose expression was higher in PBC group 2 than in PBC group 1. There was one gene whose expression was higher in PBC group 1 than in PBC group 2 (Fig. 3B; Supporting Table S12B). All of these 21 up‐regulated genes were categorized as part of cell populations related to the immune system (9 genes in B cells, 11 genes in T cells, 3 genes in NK‐like cells), but none were categorized as related to Kupffer cells (Fig. 3B; Supporting Table S12B). One down‐regulated mRNA, encoded by CLDN4, was categorized as related to cholangiocytes.
Fig. 3.
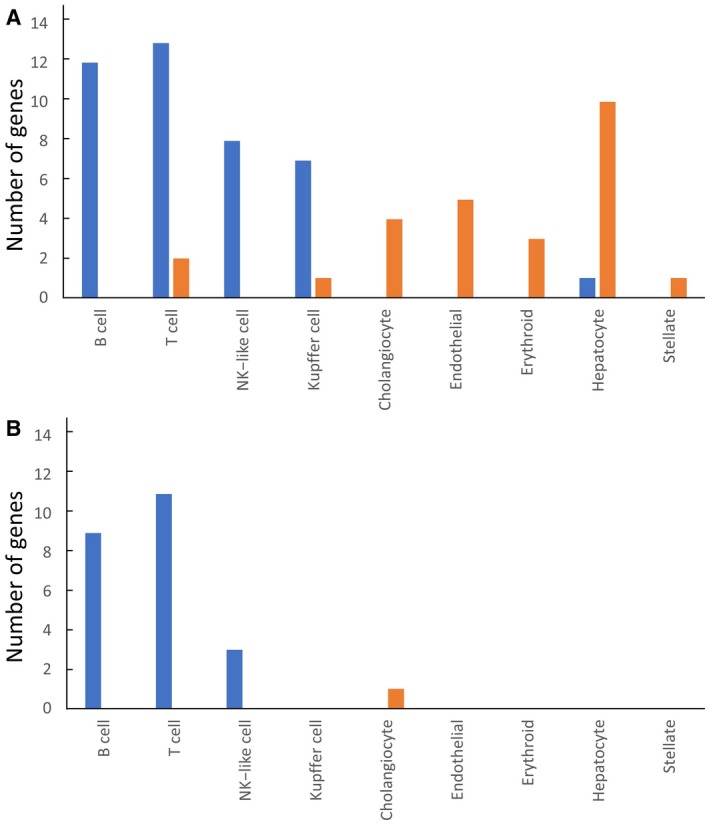
(A) Number of significant genes identified in the microarray analysis, which were categorized into nine distinct cell types in the liver. With reference to 361 cell type–specific genes in the liver,( 24 ) 62 cell type–specific genes were identified from the 1,574 significant genes in the microarray analysis and categorized into nine distinct cell types in the liver. The number of genes in each cell type is shown. Blue and orange bars indicate up‐regulated and down‐regulated genes, respectively, in patients with PBC versus healthy controls. (B) Number of categorized genes with significant differences in expression between PBC group 1 and PBC group 2. With reference to 361 cell type–specific genes in the liver,( 24 ) 22 cell type–specific genes with significant differences in expression between PBC group 1 and PBC group 2 were identified and categorized into nine distinct cell types in the liver. Blue and orange bars indicate up‐regulated and down‐regulated genes, respectively, in PBC group 2 versus PBC group 1.
Downstream Molecules in the IFNG and CD40L Pathway
Because IFNG was predicted as the most significant upstream regulator in both GWAS and microarray analyses, we identified 10 downstream molecules of IFNG in 62 cell type–specific genes (Table 5). Increased mRNA expression of C‐C motif chemokine ligand 5 in T cells, X‐C motif chemokine ligand 1 in NK‐like cells, and lymphotoxin‐beta and HLA‐DQA1 in B cells was involved in both PBC development and disease activity (Table 5). In contrast, complement C1q subcomponent subunit A, complement C1q subcomponent subunit B, and macrophage scavenger receptor 1 in Kupffer cells were involved in the development of PBC but not in disease activity (Table 5). In addition, because CD40L was predicted as the second‐most significant upstream regulator, we similarly identified seven downstream molecules of CD40L in 62 cell type–specific genes (Table 6). C‐C motif chemokine ligand 5 in T cells, lymphotoxin‐beta, HLA‐DQA1, and TNFRSF17 (also known as BCMA) in B cells were involved in both PBC development and disease activity. TNFRSF13C (also known as BAFFR) in B cells was involved in the development of PBC (Table 6). Among these potential molecular targets listed in Tables 5 and 6, several drugs, including a monoclonal antibody to BAFFR (e.g., VAY736), have been under clinical trials according to the Global Online Biomarker Database (https://www.gobiomdbplus.com/).
Table 5.
Downstream Molecules of IFNG
| Cell Type | Downstream Molecule | Fold Change (PBC vs. Control) | Fold Change (Group 2 vs. Group 1) | Chromosome | GWAS P Value | Drugs (Target Disease) |
|---|---|---|---|---|---|---|
| NK‐like cell | XCL1 | 2.97 | 2.04 | 1 | . | . |
| B cell | HLA‐DQA1 | 2.95 | 2.30 | 6 | 5.91E‐23 | Insulin (type 2 diabetes mellitus) |
| Kupffer cell | C1QA | 2.27 | 1 | . | Bevacizumab (colorectal cancer) | |
| B cell | LTB | 2.17 | 2.71 | 6 | . | . |
| T cell | CCL5 | 2.14 | 2.29 | 17 | . | . |
| Kupffer cell | C1QB | 2.11 | 1 | . | Bevacizumab (colorectal cancer) | |
| Kupffer cell | MSR1 | 2.01 | 8 | . | . | |
| Endothelial | HSPG2 | −2.33 | 1 | . | Palifermin (head and neck cancer) | |
| T cell | JUNB | −7.44 | 19 | . | . | |
| Hepatocyte | SERPINA1 | −18.45 | 14 | . | . |
Note: These 10 downstream molecules of IFNG are identified from 62 cell type–specific genes in PBC liver. The 62 cell type–specific genes were extracted from our microarray data of PBC liver biopsy samples with reference to a previously reported 361 cell type–specific genes in the liver.( 24 ) The GWAS P‐value column indicates the lowest P value of SNPs located within a gene. For drug information, the Global Online Biomarker Database was used (http://www.gobiomdb.com).
Abbreviations: C1QA, complement C1q subcomponent subunit A; C1QB, complement C1q subcomponent subunit B; CCL5, C‐C motif chemokine ligand 5; HLA‐DQA1, HLA class II histocompatibility antigen, DQ alpha 1 chain; HSPG2, heparan sulfate proteoglycan 2; JUNB, transcription factor Jun‐B; LTB, lymphotoxin‐beta; MSR1, macrophage scavenger receptor 1; SERPINA1, serpin family A member 1; XCL1, X‐C motif chemokine ligand 1.
Table 6.
Downstream Molecules of CD40L
| Cell Type | Downstream Molecule | Fold Change (PBC vs. Control) | Fold Change (Group 2 vs. Group 1) | Chromosome | GWAS P Value | Drugs (Target Disease) |
|---|---|---|---|---|---|---|
| B cell | TNFRSF17 | 5.49 | 5.11 | 16 | . | . |
| B cell | HLA‐DQA1 | 2.95 | 2.30 | 6 | 5.91E‐23 | Insulin (type 2 diabetes mellitus) |
| B cell | LTB | 2.17 | 2.71 | 6 | . | . |
| T cell | CCL5 | 2.14 | 2.29 | 17 | . | . |
| Hepatocyte | TM7SF2 | 2.06 | 11 | . | . | |
| B cell | TNFRSF13C | 2.05 | 22 | . | VAY736 (Sicca syndrome) | |
| Erythroid | YBX3 | −2.64 | 12 | . | . |
Note: These seven molecules downstream of CD40L are identified from 62 cell type–specific genes in PBC liver. The 62 cell type–specific genes were extracted from our microarray data of PBC liver biopsy samples with reference to a previously reported 361 cell type–specific genes in the liver.( 24 ) The GWAS P‐value column indicates the lowest P value of SNPs located within a gene. For drug information, the Global Online Biomarker Database was used (http://www.gobiomdb.com).
Abbreviations: CCL5, C‐C motif chemokine ligand 5; HLA‐DQA1, HLA class II histocompatibility antigen, DQ alpha 1 chain; LTB, lymphotoxin‐beta; TM7SF2, delta(14)‐sterol reductase TM7SF2; YBX3, Y‐Box Binding Protein 3.
Discussion
GWASs have been used to identify causal variants, disease pathways, and upstream or downstream regulators in various complex diseases.( 33 ) However, functional characterization of mechanisms at GWAS loci is a multifaceted challenge. The post‐GWAS approach is currently not satisfactory for identifying critical molecular targets in these complex diseases. One reason for this limitation may be insufficient information on the transcriptome of the affected organs or cell types used for integrated analysis with GWASs.
In this study, we first performed a GWAS analysis of PBC in a large Japanese population and a transcriptome analysis of liver specimens with PBC. We then objectively combined the results of the GWAS and transcriptome analyses, focusing on the overlap or intersection. We predicted that IFNG and CD40L were the central regulators in both PBC development and disease activity based on computational analysis with IPA (Table 3). In addition, we could identify genes expressed in distinct cell populations (B cells, T cells, NK‐like cells, and Kupffer cells) that are involved in PBC development, disease activity, or both by combining our transcriptome data of livers with PBC with data on single‐cell RNA sequences in the human liver from the literature.( 24 )
IFNG is a cytokine critical for various immune responses. It is produced predominantly by NK and NK T cells as a part of the innate immune system and by CD4T and CD8T cells as a part of the adaptive immune system.( 34 ) Aberrant IFNG expression is associated with a number of autoinflammatory and autoimmune diseases.( 35 ) In PBC, a recent GWAS indicated the importance of IL‐12 to IFNG pathways in the development of PBC.( 5 , 12 , 13 ) Correspondingly, IFNG has been implicated in the pathogenesis of PBC in both human and mice models.( 36 , 37 , 38 , 39 ) IFNG protein expression was up‐regulated in infiltrating mononuclear cells and small bile ducts in livers with PBC.( 36 , 37 ) Chronic expression of IFNG leads to murine autoimmune cholangitis with a female predominance that mimics human PBC, manifested by portal inflammation, liver granulomas, elevated bile salt and serum immunoglobulin M (IgM) levels, and production of antimitochondrial antibodies in IFNG‐3′‐untranslated region adenylate‐rich element‐deleted mice.( 38 ) The interplay of type I and type II interferons is also implicated as a cause of human PBC( 40 ) and in a murine model of autoimmune cholangitis.( 39 ) This study strongly supports the importance of IFNG as a key cytokine in PBC development and disease activity. Hierarchical clustering of gene expression in livers with PBC in conjunction with data on single‐cell RNA sequences in the human liver identified several cell type–specific genes in the downstream of IFNG (Table 5), thus providing a rationale for the development of anti‐IFNG pathway therapies in a cell type–specific manner.( 41 )
CD40L is a TNFRSF member predominantly expressed on activated CD4T cells and plays an important role to transduce accelerated IFNG signaling from T‐cell side to B‐cell side, resulting in the maintenance of a vicious cycle of autoimmunity (i.e., the interaction of CD40L with CD40 on B cells plays a critical role in B‐cell activation and differentiation, germinal center formation, and production of class‐switched antibodies).( 42 , 43 ) In addition, the interaction of CD40L on activated CD4T cells with CD40 on antigen‐presenting cells induces IL‐12 production in antigen‐presenting cells, which leads to IFNG production by activated CD4T cells. The interaction of CD40L with CD40 expressed on epithelial cells induces the production of inflammatory mediators, such as TNF, IL‐1b, IL‐6, IL‐8, monocyte chemoattractant protein 1, and matrix metallopeptidase, and also up‐regulates adhesion molecules, such as intracellular adhesion molecule 1, on epithelial cells.( 42 ) CD40L expression is elevated in patients with various autoimmune diseases, including rheumatoid arthritis, systemic lupus erythematosus, and Sjogren’s syndrome, indicating the critical role of the CD40L–CD40 pathway in the pathogenesis of autoimmune diseases.( 42 )
In this study, combined GWAS and transcriptome analysis predicted that CD40L is the second‐most significant upstream regulator in PBC development and disease activity. Several reports have already indicated the involvement of the CD40L–CD40 signaling pathway in the pathogenesis of PBC. First, CD40L is up‐regulated in the cells infiltrating the portal tracts and biliary epithelial cells of the small bile ducts in livers with PBC.( 44 ) Second, IgM levels are inversely correlated with CD40L promoter methylation in patients with PBC.( 45 ) Third, co‐culture of human liver macrophages and cholangiocytes leads to CD40‐dependent apoptosis and cytokine secretion.( 46 ) Fourth, anti–CD‐40L monoclonal antibodies delay the progression of murine autoimmune cholangitis.( 47 ) In fact, several molecules targeting the CD40L–CD40 pathway have been generated and evaluated in clinical settings in some human autoimmune diseases. The results appear to be promising, except for safety concerns due to reports of thromboembolic events.( 42 ) The present study might provide a rationale for the development of anti–CD40L‐CD40 pathway therapy in PBC.
It was noteworthy that molecules involved in B‐cell development and survival, such as the BAFF (also known as TNFSF13B), a proliferation‐inducing ligand (APRIL) system, were identified as the potential therapeutic targets on pathways regulated by CD40L (Table 6). We previously reported that serum levels of BAFF are significantly higher in patients with PBC who have advanced interface hepatitis, indicating the involvement of BAFF–BAFFR (also known as TNFRSF13C) and/or APRIL–BCMA (also known as TNFRSF17) signaling in the development of PBC.( 48 ) Belimumab, a monoclonal antibody to BAFF that blocks BAFF–BAFFR signaling, has been shown to be effective for systemic lupus erythematosus and approved by the U.S. Food and Drug Administration and European Medicines Agency for human use.( 49 ) VAY736, a monoclonal antibody to BAFFR, has been shown to be effective for primary Sjogren’s syndrome and is now under clinical trial (Table 6). Blockade of APRIL–BCMA signaling by a monoclonal antibody to APRIL was shown to be effective in murine model of systemic lupus erythematosus and immunoglobulin A nephropathy.( 50 ) Altogether, these findings may indicate the importance of further detailed investigation of the BAFF/APRIL system as a potential therapeutic target for PBC.
It was also noteworthy that the genes categorized to B cells, T cells, and NK‐like cells are involved in both PBC disease susceptibility and disease activity, whereas the genes in Kupffer cells are involved only in disease susceptibility (Fig. 3; Supporting Table S12). These results may indicate the distinct role of Kupffer cells in the pathogenesis of PBC as compared with B cells, T cells, and NK‐like cells. In fact, the number of Kupffer cells is markedly increased in livers with PBC, regardless of disease activity (data not shown). Further studies are needed to dissect the role of each gene expression in cell type–specific manner.
In conclusion, this study introduced an objective systematic approach combining GWAS and transcriptome analysis in PBC. It predicted the central role of IFNG and CD40L in both PBC disease susceptibility and disease activity. In addition, potential therapeutic targets, including BCMA and BAFFR, were identified in pathways regulated by IFNG and CD40L in a cell type–specific manner. Although the database of IPA and the single‐cell RNA sequence data of the liver used in the present study are still not satisfactory enough to clarify the whole picture of the disease‐pathways and therapeutic targets, our approach could provide a rationale for selecting potential therapeutic targets among thousands of molecules identified by GWAS and microarray analyses in various complex diseases, including PBC.
Supporting information
Table S1‐S12
Acknowledgment
We thank all the patients and volunteers who enrolled in the study and all clinical staffs who participated in collection of samples (serum, DNA, and liver biopsy samples) and clinical information and obtaining informed consents from PBC patients. We also thank Ms. Mayumi Ishii and Takayo Tsuchiura (National Center for Global Health and Medicine) and Ms. Natsumi Baba, Ms. Yoshimi Shigemori, and Ms. Tomoko Suzuki (University of Tokyo) for their technical and administrative assistance.
Potential conflict of interest: Nothing to report.
Financial Support: The Japan Agency for Medical Research and Development, a Grant‐in‐Aid for Clinical Research from the National Hospital Organization, the Uehara Memorial Foundation, Platforms Program for Promotion of Genome Medicine (16 km 0405205h0101), a grant from the Research Program of Intractable Disease provided by the Ministry of Health, Labor, and Welfare of Japan, Japan Society for the Promotion of Science (15K19314, 17K15924, 15K19357, 17K09449, and 23591), and the Takeda Science Foundation.
References
Author names in bold designate shared co‐first authorship.
- 1. Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med 2005;353:1261‐127 3. [DOI] [PubMed] [Google Scholar]
- 2. Lindor KD, Bowlus CL, Boyer J, Levy C, Mayo M. Primary biliary cholangitis: 2018 practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2019;69:394‐419. [DOI] [PubMed] [Google Scholar]
- 3. Corpechot C, Chazouilleres O, Rousseau A, Le Gruyer A, Habersetzer F, Mathurin P, et al. A placebo‐controlled trial of bezafibrate in primary biliary cholangitis. N Engl J Med 2018;378:2171‐2181. [DOI] [PubMed] [Google Scholar]
- 4. Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, et al. A placebo‐controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med 2016;375:631‐643. [DOI] [PubMed] [Google Scholar]
- 5. Hirschfield GM, Liu X, Xu C, Lu Y, Xie G, Lu Y, et al. Primary biliary cholangitis associated with HLA, IL12A, and IL12RB2 variants. N Engl J Med 2009;360:2544‐2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hirschfield GM, Liu X, Han Y, Gorlov IP, Lu Y, Xu C, et al. Variants at IRF5‐TNPO3, 17q12‐21 and MMEL1 are associated with primary biliary cholangitis. Nat Genet 2010;42:655‐657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu X, Invernizzi P, Lu Y, Kosoy R, Lu Y, Bianchi I, et al. Genome‐wide meta‐analyses identify three loci associated with primary biliary cholangitis. Nat Genet 2010;42:658‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mells GF, Floyd JA, Morley KI, Cordell HJ, Franklin CS, Shin SY, et al. Genome‐wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet 2011;43:329‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hirschfield GM, Xie G, Lu E, Sun Y, Juran BD, Chellappa V, et al. Association of primary biliary cholangitis with variants in the CLEC16A, SOCS1, SPIB and SIAE immunomodulatory genes. Genes Immun 2012;13:328‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu JZ, Almarri MA, Gaffney DJ, Mells GF, Jostins L, Cordell HJ, et al. Dense fine‐mapping study identifies new susceptibility loci for primary biliary cholangitis. Nat Genet 2012;44:1137‐1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Juran BD, Hirschfield GM, Invernizzi P, Atkinson EJ, Li Y, Xie G, et al. Immunochip analyses identify a novel risk locus for primary biliary cirrhosis at 13q14, multiple independent associations at four established risk loci and epistasis between 1p31 and 7q32 risk variants. Hum Mol Genet 2012;21:5209‐5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cordell HJ, Han Y, Mells GF, Li Y, Hirschfield GM, Greene CS, et al. International genome‐wide meta‐analysis identifies new primary biliary cholangitis risk loci and targetable pathogenic pathways. Nat Commun 2015;6:8019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakamura M, Nishida N, Kawashima M, Aiba Y, Tanaka A, Yasunami M, et al. Genome‐wide association study identified TNFSF15 and POU2AF1 as susceptibility locus for primary biliary cholangitis in the Japanese population. Am J Hum Genet 2012;91:721‐728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kawashima M, Hitomi Y, Aiba Y, Nishida N, Kojima K, Kawai Y, et al. Genome‐wide association study identified PRKCB as a genetic susceptibility locus for primary biliary cholangitis in a Japanese population. Hum Mol Genet 2017;26:650‐659. [DOI] [PubMed] [Google Scholar]
- 15. Qiu F, Tang R, Zuo X, Shi X, Wei Y, Zheng X, et al. A genome‐wide association study identifies six novel risk loci for primary biliary cholangitis. Nat Commun 2017;8:14828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hitomi Y, Ueno K, Kawai Y, Nishida N, Kojima K, Kawashima M, et al. POGLUT1, the putative effector gene driven by rs2293370 in primary biliary cholangitis susceptibility locus chromosome 3q13.33. Sci Rep 2019;14:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Webb GJ, Hirschfield GM. Using GWAS to identify genetic predisposition in hepatic autoimmunity. J Autoimmun 2016;66:26‐39. [DOI] [PubMed] [Google Scholar]
- 18. Trivedi PJ, Hirschfield GM. The immunogenetics of autoimmune cholestasis. Clin Liver Dis 2016;20:15‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liaskou E, Hirschfield GM. Genetic association studies and the risk factors for developing the “Immuno‐bile‐logic” disease primary biliary cholangitis. Hepatology 2018;67:1620‐1622. [DOI] [PubMed] [Google Scholar]
- 20. Luo Z, Jegga AG, Bezerra JA. Gene‐disease associations identify a connectome with molecular pathways in human cholangiopathies. Hepatology 2018;67:21‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shackel NA, McGuinness PH, Abbott CA, Gorrell MD, McCaughan GW. Identification of novel molecules and pathogenic pathways in primary biliary cirrhosis: cDNA array analysis of intrahepatic differential gene expression. Gut 2001;49:565‐576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Honda M, Kawai H, Shirota H, Yamashita T, Kaneko S. Differential gene expression profiles in stage 1 primary biliary cirrhosis. Am J Gastroenterol 2005;100:2019‐2030. [DOI] [PubMed] [Google Scholar]
- 23. Chen L, Borozan I, Milkiewicz P, Sun J, Meng X, Coltescu C, et al. Gene expression profiling of early primary biliary cirrhosis: possible insights into the mechanism of action of ursodeoxycholic acid. Liver Int 2008;28:997‐1010. [DOI] [PubMed] [Google Scholar]
- 24. MacParland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun 2018;9:4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Loh PR, Danecek P, Palamara PF, Fuchsberger C, A Reshef Y, K Finucane H, et al. Reference‐based phasing using the Haplotype Reference Consortium panel. Nat Genet 2016;48:1443‐1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. Genome‐wide genetic data on ~500,000 UK Biobank participants. bioRxiv 2017;166298. [Google Scholar]
- 27. Nagasaki M, Yasuda J, Katsuoka F, Nariai N, Kojima K, Kawai Y, et al. Rare variant discovery by deep whole‐genome sequencing of 1,070 Japanese individuals. Nat Commun 2015;6:8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yamaguchi‐Kabata Y, Nariai N, Kawai Y, Sato Y, Kojima K, Tateno M, et al. iJGVD: an integrative Japanese genome variation database based on whole‐genome sequencing. Hum Genome Var 2015;2:15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Staples J, Qiao D, Cho MH, Silverman EK, Nickerson DA, University of Washington Center for Mendelian Genomics , et al. PRIMUS: rapid reconstruction of pedigrees from genome‐wide estimates of identity by descent. Am J Hum Genet 2014;95:553‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease‐associated variation in regulatory DNA. Science 2012;337:1190‐1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schmidt‐Arras D, Rose‐John S. IL‐6 pathway in the liver: from physiopathology to therapy. J Hepatol 2016;64:1403‐1415. [DOI] [PubMed] [Google Scholar]
- 33. Cannon ME, Mohlke KL. Deciphering the emerging complexities of molecular mechanisms at GWAS loci. Am J Hum Genet 2018;103:637‐653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon‐g: an overview of signals, mechanisms and functions. J Leukoc Biol 2004;75:163‐189. [DOI] [PubMed] [Google Scholar]
- 35. Hu X, Ivashkiv LB. Cross‐regulation of signaling pathways by interferon‐gamma: implications for immune responses and autoimmune diseases. Immunity 2009;31:539‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Harada K, Van de Water J, Leung PS, Coppel RL, Ansari A, Nakanuma Y, et al. In situ nucleic acid hybridization of cytokines in primary biliary cirrhosis: predominance of the Th1 subset. Hepatology 1997;25:791‐796. [DOI] [PubMed] [Google Scholar]
- 37. Yang C‐Y, Ma X, Tsuneyama K, Huang S, Takahashi T, Chalasam NP, et al. IL‐12/Th1 and IL23/Th17 biliary microenvironment in primary biliary cirrhosis: implications for therapy. Hepatology 2014;59:1944‐1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bae HR, Leung PSC, Tsuneyama K, Valencia JC, Hodge DL, Kim S, et al. Chronic expression of interferon‐gamma leads to murine autoimmune cholangitis with a female predominance. Hepatology 2016;64:1189‐1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bae HR, Hodge DL, Yang G‐X, Leung PSC, Chodisetti SB, Valencia JC, et al. The interplay of type I and type II interferons in murine autoimmune cholangitis as a basis for sex‐biased autoimmunity. Hepatology 2018;67:1408‐1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Takii Y, Nakamura M, Ito M, Yokoyama T, Komori A, Shimizu‐Yoshida Y, et al. Enhanced expression of type 1 interferon and toll‐like receptor 3 in primary biliary cirrhosis. Lab Invest 2005;85:908‐920. [DOI] [PubMed] [Google Scholar]
- 41. Singhania A, Graham CM, Gabrysova L, Moreira‐Teixeira L, Stavropoulos E, Pit JM, et al. Transcriptional profiling unveils type 1 and 2 interferon networks in blood and tissues across diseases. Nat Commun 2019;10:2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Karnell JL, Rieder SA, Ettinger R, Kolbeck R. Targetting the CD40‐CD40L pathway in autoimmune diseases: humoral immunity and beyond. Adv Drug Deliv Rev 2019;15:92‐103. [DOI] [PubMed] [Google Scholar]
- 43. Wang L, Sun Y, Zhang Z, Jia Y, Zou Z, Ding J, et al. CXCR5+ CD4+ T follicular helper cells participate in the pathogenesis of primary biliary cirrhosis. Hepatology 2015;61:627‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Afford SC, Ahmed‐Choudhury J, Randhawa S, Russell C, Youster J, Crosby HA, et al. CD40 activation‐induced, FAS‐dependent apoptosis and NF‐kappa B/AP‐1 signaling in human intrahepatic epithelial cells. FASEB J 2001;15:2345‐2354. [DOI] [PubMed] [Google Scholar]
- 45. Ileo A, Liao J, Invernizzi P, Zhao M, Bernuzzi F, Ma L, et al. Immunoglobulin M levels inversely correlate with CD40 ligand promoter methylation in patients with primary biliary cholangitis. Hepatology 2012;55:153‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Alabraba EB, Lai V, Boon L, Wigmore SJ, Adams DH, Afford SC. Culture of human liver macrophages and cholangiocytes leads to CD40‐dependent apoptosis and cytokine secretion. Hepatology 2008;47:552‐562. [DOI] [PubMed] [Google Scholar]
- 47. Tanaka H, Yang GX, Iwakoshi N, Knechtie K, Tsuneyama K, Leung O, et al. Anti‐CD40L monoclonal antibody delays the progression of murine autoimmune cholangitis. Clin Exp Immunol 2013;174:364‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Migita K, Ilyassova B, Kovzel EF, Nersesov A, Abiru S, Maeda Y, et al. Serum BAFF and APRIL levels in patients with PBC. Clin Immunol 2010;134:217‐225. [DOI] [PubMed] [Google Scholar]
- 49. Smulski CR, Eibel H. BAFF and BAFF‐receptor in B cell selection and survival. Front Immunol 2018;9:2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Myette JR, Kano T, Suzuki H, Sloan SE, Szretter KJ, Ramakrishnan B, et al. A proliferation inducing ligand (APRIL) targeted antibody is a safe and effective treatment of murine IgA nephropathy. Kidney Int 2019;96:104‐116. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐S12