

Abstract
Standard-of-care treatment for haemophilia A or B is to maintain adequate coagulation factor levels through clotting factor administration. The current study aimed to evaluate annualised bleeding rates (ABR) and treatment adherence for haemophilia A or B patients receiving standard half-life (SHL) vs. extended half-life (EHL) factor replacement products. We analysed data from the Adelphi Disease-Specific Programmes, a health record–based survey of United States and European haematologists. Analysis included 651 males with moderate-to-severe haemophilia A or B (the United States, n = 132; Europe, n = 519). The haemophilia A analysis included 501 patients (SHL, n = 435; EHL, n = 66). In the combined United States/European population, mean (SD) ABR was 1.7 (1.69) for the SHL group and 1.8 (2.00) for the EHL group. A total of 72% of patients receiving SHL factor VIII and 75% of patients receiving EHL factor VIII in the combined population were fully adherent (no doses missed of the last 10 doses), as reported by physicians. The haemophilia B analysis included 150 patients (SHL, n = 114; EHL, n = 36). The mean (SD) ABR in the combined population was 2.1 (2.16) for patients receiving SHL factor IX (FIX) and 1.4 (1.48) for patients receiving EHL FIX. The percentage of fully adherent patients (physician-reported) was similar in both treatment groups (SHL FIX, 68%; EHL FIX, 73%). In this preliminary real-world survey in a relatively small sample of patients, measures of ABR and adherence between SHL and EHL products were evaluated. Additional real-world research on prescribing patterns, SHL vs. EHL effectiveness, and adherence is warranted.
Keywords: blood coagulation disorders, blood coagulation factor, factor IX deficiency, factor VIII deficiency, healthcare survey, medication adherence
Introduction
Haemophilia A and B are X-linked inherited diseases in which mutation of the gene for factor VIII (FVIII) and factor IX (FIX), respectively, results in coagulation factor deficiencies. The severity of haemophilia symptoms is related to coagulation factor levels, and patients with moderate [1–5 international unit (IU)/dl] to severe (<1 IU/dl) haemophilia experience spontaneous bleeding episodes and prolonged, sometimes life-threatening, bleeding events with minor trauma [1]. The standard of care for the treatment and prevention of bleeding events and their complications is to maintain adequate coagulation factor levels through periodic intravenous clotting factor concentration administration [1]. Patients may treat prophylactically or on demand, although prophylaxis is the standard of care for those with a severe bleeding phenotype [1,2].
Recombinant factor replacement products with half-lives of approximately 8–12 h for FVIII and 18–40 h for FIX are referred to as standard half-life (SHL) products [3,4]. In contrast, extended half-life (EHL) factor replacement products have comparably longer half-lives; for FVIII, EHL products have an approximately 1.5 times longer half-life than SHL FVIII products, and for FIX, the half-life is extended by up to five times compared with SHL FIX products [3]. Factor products with longer half-lives may allow for a longer time between injections, with the potential to substantially reduce the burden of treatment [5,6]. Both SHL and EHL FVIII and FIX products are available in the United States and Europe.
While it may seem that reducing the frequency of injections with EHL replacement products could improve patient adherence and, by extension, promote lower bleeding event rates compared with SHL products [7–10], limited data are available in the published literature to support these expectations. The objective of this exploratory analysis of real-world data from a patient health record–based survey was to evaluate the reported annualised bleeding rate (ABR) and patient adherence to treatment (based on number of missed doses reported) in patients with moderate to severe haemophilia A or B receiving SHL vs. EHL FVIII or FIX replacement products.
Methods
The Adelphi Disease-Specific Programmes (DSP; Adelphi Real World, Bollington, Cheshire, UK) is an aggregated, multinational data source of patient-level real-world evidence on specific chronic diseases. The data are collected under research guidelines from physician interviews, physician workload questionnaires, detailed patient record forms completed by physicians, and self-report questionnaires completed by patients and by their caregivers [11]. The current analysis was performed using Adelphi DSP validated methodology in compliance with the European Pharmaceutical Market Research Association and the US Health Insurance Portability and Accountability Act of 1996.
The haemophilia DSP database includes medical data from 1667 male patients with haemophilia A or B, excluding acquired haemophilia A, collected from physicians (haematologists and haematologists-oncologists) and haemophilia patients in the United States and five European countries (France, Germany, Italy, Spain, and the United Kingdom). In total, 205 physicians were recruited for participation in the haemophilia DSP (the United States, n = 75; France, n = 25; Germany, n = 20; Italy, n = 35; Spain, n = 20; the United Kingdom, n = 30) and asked to complete patient record forms for the next eight consecutive patients with haemophilia A or B (with or without inhibitors) who met the eligibility criteria. The patient record form is a detailed electronic record completed prospectively (in about 20–25 min) by the physician for each patient presenting with haemophilia. Details collected include demographics (such as age, sex, and race/ethnicity), diagnosis, severity of the condition, specific symptoms, treatment history, and medication adherence. Patients were then asked to complete a self-reported record (adult caregiver completion on behalf of paediatric patients), independent of physician input. Physician responses were included in the database, regardless of whether or not the patient completed the patient record.
Data included in this analysis were collected from May 2017 to November 2017. Deidentified data from the haemophilia DSP database were used to retrospectively identify male patients with moderate-to-severe haemophilia A or B who were using an SHL or EHL factor replacement product at the time of data collection. Table 1 lists all products included in this analysis.
Table 1.
Standard and extended half-life factor replacement products for haemophilia A and B included in this analysis
| Standard half-life products | Extended half-life products |
| Haemophilia A (factor VIII) | |
| Xyntha/ReFacto AF (moroctocog alfa; Pfizer) | Adynovate (rurioctocog alfa pegol; Shire) |
| Advate (octocog alfa; Shire) | Eloctate/Elocta (efmoroctocog alfa; Bioverativ Therapeutics/SOBI) |
| Helixate FS/Helixate NexGen (octocog alfa; CSL Behring) | |
| Kogenate FS/Kogenate (octocog alfa; Bayer HealthCare) | |
| Kovaltry (octocog alfa; Bayer HealthCare) | |
| Recombinate (octocog alfa; Shire) | |
| Nuwiq (simoctocog alfa; Octapharma) | |
| NovoEight (turoctocog alfa; Novo Nordisk) | |
| Haemophilia B (factor IX) | |
| Betafact (nonacog alfa; LFB Biomedicaments) | Alprolix (eftrenonacog alfa; Bioverativ Therapeutics) |
| Immunine VH (human coagulation FIX, vapour heated; Shire) | Idelvion (albutrepenonacog alfa; CSL Behring) |
| Octanine F (human coagulation FIX; Octapharma) | |
| Haemonine (human coagulation FIX; Biotest Pharma) | |
| Berinin P (human coagulation FIX; CSL Behring) | |
| BeneFIX (nonacog alfa; Pfizer) | |
| Rixubis (nonacog gamma; Shire) | |
FIX, factor IX.
Baseline clinical characteristics collected included physician-reported severity of haemophilia and inhibitor status (never had inhibitors vs. had inhibitors in the past). Key outcome measures were the ABR, calculated as the number of bleeding events reported by patients in the previous 12 months, and the physician-reported adherence to the last 10 doses of factor replacement product, based on their clinical interactions with patients and their knowledge of patient usage characteristics. For adherence to the last 10 doses, physicians were asked on the patient record form, ‘Of the last 10 doses of factor infusion the patient should have taken, how many do you think they have missed?’ A missed dose was defined as the failure to administer a scheduled dose in a prophylaxis protocol or the failure to administer one of the requisite repeated injections over the course of treatment for a bleeding episode. Patients were considered fully adherent if they had taken all of the last 10 doses prescribed. Treatment information collected on the patient record form, as completed by the physicians, included the total dose of replacement factor per week and per dose, and the frequency of replacement factor administration per week.
Statistical analysis
Patients with missing data were excluded from the analysis of the missing variable only; patients were included in any analysis for which they had evaluable data. Descriptive statistics (mean, SD, median, and range) were calculated for all measures; the study was not designed with formal sample size calculations and was not powered to detect statistical differences. Outcomes were analysed for patients in the United States only, for those in Europe only, and for the combined populations (the United States and Europe). Data for haemophilia A and B were analysed separately.
Results
A sample of 651 patients with haemophilia A or B met the inclusion criteria (the United States, n = 132; Europe, n = 519). In both the United States and European populations, age, weight, and BMI were similar between the SHL and EHL groups within the haemophilia A and B groups (Tables 2 and 3). In patients with haemophilia A (combined population), physicians reported that joint bleeds, nosebleeds, and bruises were most commonly reported (affecting at least 30% of patients in either treatment group) in the previous 12 months; similar percentages of patients experienced joint bleeds (SHL, 44%; EHL, 48%) and nosebleeds (SHL, 38%; EHL, 36%), but more patients who received FVIII EHL experienced bruises (SHL, 26%; EHL, 40%). Similarly, bleeding events common among patients with haemophilia B (combined population) in the last 12 months were joint bleeds (SHL, 46%; EHL, 17%), nosebleeds (SHL, 33%; EHL, 13%), and bruises (SHL, 37%; EHL, 43%).
Table 2.
Demographics and clinical and treatment characteristics for patients with haemophilia A receiving standard half-life vs. extended half-life factor VIII replacement products in the United States and Europe
| The United States, N = 110 | Europe, N = 391 | Combined, N = 501 | ||||
| Characteristic | SHL group, n = 74 | EHL group, n = 36 | SHL group, n = 361 | EHL group, n = 30 | SHL group, n = 435 | EHL group, n = 66 |
| Age, mean (SD) (years) | 27.5 (13.9) | 24.0 (12.2) | 29.9 (15.9) | 27.2 (13.5) | 29.5 (15.6) | 25.5 (12.8) |
| Weight, mean (SD) (kg) | 74.3 (20.5) | 70.9 (19.8) | 68.0 (15.0) | 65.4 (12.2) | 68.0 (15.0) | 65.4 (12.2) |
| BMI, mean (SD) (kg/m2) | 25.0 (5.0) | 24.3 (4.1) | 23.4 (3.3) | 22.4 (2.9) | 23.7 (3.7) | 23.5 (3.7) |
| Race/ethnicity, n (%)a | ||||||
| White | 54 (73.0) | 31 (86.1) | 337 (93.4) | 27 (90.0) | 391 (89.9) | 58 (87.9) |
| Black/African American | 11 (14.9) | 3 (8.3) | 0 | 0 | 11 (2.5) | 3 (4.5) |
| Hispanic/Latino | 4 (5.4) | 1 (2.8) | 2 (0.6) | 1 (3.3) | 6 (1.4) | 2 (3.0) |
| Otherb | 5 (6.8) | 1 (2.8) | 22 (6.1) | 2 (6.7) | 27 (6.2) | 3 (4.5) |
| Haemophilia severity, n (%) | ||||||
| Moderate | 50 (68) | 18 (50) | 180 (50) | 21 (70) | 230 (53) | 39 (59) |
| Severe | 24 (32) | 18 (50) | 181 (50) | 9 (30) | 205 (47) | 27 (41) |
| Inhibitor status, n (%) | ||||||
| Never had inhibitors | 69 (93) | 32 (89) | 319 (88) | 26 (87) | 388 (89) | 58 (88) |
| Had inhibitors in the pastc | 5 (7) | 4 (11) | 42 (12) | 4 (13) | 47 (11) | 8 (12) |
| Treatment | ||||||
| Frequency of dose per week, mean (SD) | 2.9 (1.97) | 2.0 (0.52) | 3.0 (1.12) | 1.9 (0.98) | 3.0 (1.28) | 2.0 (0.73) |
| Factor per dose, mean (SD) (IU/kg) | 38.5 (13.9) | 49.5 (9.1) | 34.8 (14.7) | 53.0 (28.0) | 35.4 (14.6) | 50.8 (18.4) |
| Total dose per week, mean (SD) (IU/kg) | 106.2 (51.74) | 101.29 (37.97) | 102.8 (48.98) | 71.5 (25.29) | 103.3 (49.33) | 91.8 (36.93) |
EHL, extended half-life; SHL, standard half-life.
aTotal percentage may not equal 100% because of rounding.
bOther includes Native American, Afro-Caribbean, Asian-Indian subcontinent, Asian-other, Chinese, Middle Eastern, mixed race, and unknown.
cNo patient had inhibitors at baseline.
Table 3.
Demographics and clinical and treatment characteristics for patients with haemophilia B receiving standard half-life vs. extended half-life factor IX replacement products in the United States and Europe
| Characteristic | The United States, N = 22 | Europe, N = 128 | Combined, N = 150 | |||
| SHL group, n = 10 | EHL group, n = 12 | SHL group, n = 104 | EHL group, n = 24 | SHL group, n = 114 | EHL group, n = 36 | |
| Age, mean (SD) (years) | 21.5 (11.2) | 18.5 (8.1) | 28.8 (14.1) | 29.1 (12.9) | 28.1 (14.0) | 25.6 (12.4) |
| Weight, mean (SD) (kg) | 71.8 (33.2) | 64.2 (25.9) | 69.9 (20.1) | 64.8 (18.5) | 69.9 (20.1) | 64.8 (18.5) |
| BMI, mean (SD) (kg/m2) | 26.6 (7.0) | 21.9 (5.4) | 24.1 (5.9) | 23.1 (3.3) | 24.4 (6.0) | 22.7 (4.1) |
| Race/ethnicity, n (%)a | ||||||
| White | 6 (60.0) | 6 (50.0) | 91 (87.5) | 23 (95.8) | 97 (85.1) | 29 (80.6) |
| Black/African American | 3 (30.0) | 2 (16.7) | 0 | 0 | 3 (2.6) | 2 (5.6) |
| Hispanic/Latino | 0 | 4 (33.3) | 1 (1.0) | 0 | 1 (0.9) | 4 (11.1) |
| Otherb | 1 (10.0) | 0 | 12 (11.5) | 1 (4.2) | 13 (11.4) | 1 (2.8) |
| Haemophilia severity, n (%) | ||||||
| Moderate | 3 (30) | 1 (8) | 61 (59) | 17 (71) | 64 (56) | 18 (50) |
| Severe | 7 (70) | 11 (92) | 43 (41) | 7 (29) | 50 (44) | 18 (50) |
| Inhibitor status, n (%) | ||||||
| Never had inhibitors | 10 (100) | 10 (83) | 95 (91) | 24 (100) | 105 (92) | 34 (94) |
| Had inhibitors in the pastc | 0 | 2 (17) | 9 (9) | 0 | 9 (8) | 2 (6) |
| Treatment | ||||||
| Frequency of dose per week, mean (SD) | 2.4 (0.90) | 0.9 (0.36) | 2.1 (0.72) | 1.2 (0.95) | 2.1 (0.74) | 1.1 (0.80) |
| Factor per dose, mean (SD) (IU/kg) | 50.0 (9.40) | 43.8 (10.30) | 41.7 (14.0) | 53.9 (26.2) | 42.5 (13.8) | 50.2 (22.1) |
| Total dose per week, mean (SD) (IU/kg) | 120.0 (32.1) | 39.8 (17.14) | 87.2 (40.49) | 53.7 (27.09) | 90.8 (40.76) | 48.1 (24.28) |
EHL, extended half-life; SHL, standard half-life.
aTotal percentage may not equal 100% because of rounding.
bOther includes Afro-Caribbean, Asian-Indian subcontinent, other Asian, Middle Eastern, and unknown.
cNo patient had inhibitors at baseline.
Haemophilia A
A total of 501 patients were included in the haemophilia A analysis, with 110 from the United States (SHL, n = 74; EHL, n = 36) and 391 from Europe (SHL, n = 361; EHL, n = 30). Patients with haemophilia A ranged in age from 1 to 95 years, with a median age of 25 years. None of the patients had inhibitors at baseline, and approximately 90% (n = 446) never had inhibitors in the past. The demographics and clinical characteristics of SHL and EHL FVIII groups in both the United States and Europe are presented in Table 2. Of the 501 patients included in the analysis, 333 (66%) were on prophylactic rFVIII treatment. Treatment patterns (frequency of dosing per week, factor per dose, and total dose per week) are shown for the United States and European populations by treatment group in Table 2. While the total dose per week was similar between the SHL and EHL FVIII groups in the United States, the SHL FVIII total dose per week was numerically higher than the EHL FVIII dose in the European and combined populations.
In the combined US and Europe population, the mean (SD) ABR was 1.7 (1.69) for patients receiving SHL FVIII and 1.8 (2.00) for those receiving EHL FVIII, with a median of 1.0 for both groups (Fig. 1a). In the combined population of patients with bleeding event data, 92 of 388 (24%) patients treated with SHL FVIII and 15 of 57 (26%) patients treated with EHL FVIII reported having no bleeding events during the previous 12 months. The mean ABR was generally higher in patients from Europe than in patients from the United States. The median ABR was 1 for both treatment groups in the United States (range: SHL, 0–10; EHL, 0–8), and 2 for both treatment groups in Europe (range: SHL, 0–8; EHL, 0–9).
Fig. 1.
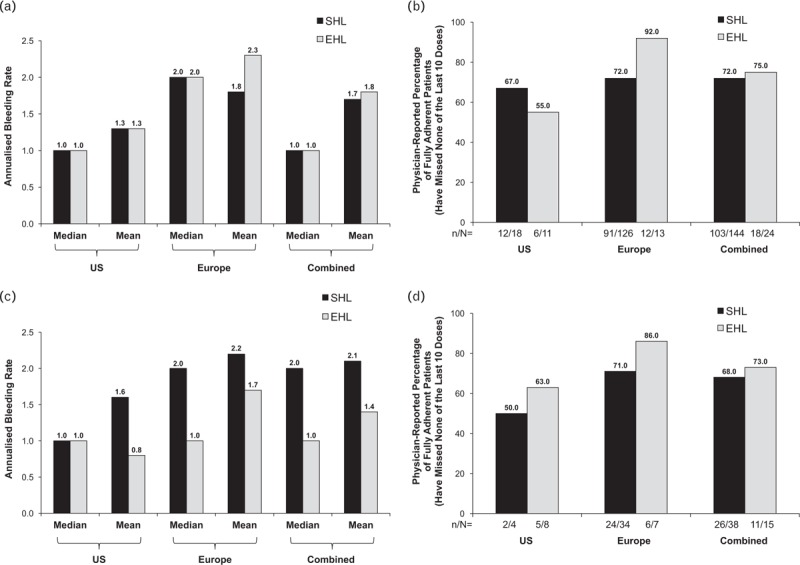
Annualised bleeding rates and adherence with standard half-life vs. extended half-life factor replacement products. (a) The annualised bleeding rate in patients with haemophilia A receiving standard half-life vs. extended half-life factor VIII replacement products in the United States, Europe, and combined populations. (b) The percentage of patients with haemophilia A receiving standard half-life vs. extended half-life factor VIII replacement products in the United States, Europe, and combined populations who were fully adherent to their last 10 doses of factor replacement (physician-reported). (c) The annualised bleeding rate in patients with haemophilia B receiving standard half-life vs. extended half-life factor IX replacement products in the United States, Europe, and combined populations. (d) The percentage of patients with haemophilia B receiving standard half-life vs. extended half-life factor IX replacement products in the United States, Europe, and combined populations who were fully adherent to their last 10 doses of factor replacement (physician-reported). Panel (a) United States: standard half-life, n = 62 and extended half-life, n = 30; Europe: standard half-life, n = 326 and extended half-life, n = 27. Panels (b) and (d): n/N: n, number of patients with no missed doses in the last 10 doses; N, patients with physician-reported dose data. Panel (c) United States: standard half-life, n = 9 and extended half-life, n = 11; Europe: standard half-life, n = 99 and extended half-life, n = 23. EHL, extended half-life; SHL, standard half-life.
In patients with dosing data, 103 of 144 (72%) patients treated with SHL FVIII and 18 of 24 (75%) patients treated with EHL FVIII were fully adherent to their last 10 doses of factor (i.e., had no physician-reported missed doses of the last 10 doses the patient should have taken) in the combined population (Fig. 1b). The mean (SD) number of doses missed was 0.7 (1.85) for patients receiving the SHL FVIII product and 1.0 (2.40) for patients receiving the EHL FVIII product in the combined population. The mean number of missed doses was numerically higher in the EHL than the SHL FVIII group in the United States (1.7 vs. 0.4), whereas in Europe, the opposite was observed (EHL FVIII, 0.4; SHL FVIII, 0.8). The median number of missed doses was 0 for all groups in the United States (range: SHL, 0–2; EHL, 0–10), European (range: SHL, 0–10; EHL, 0–5), and combined populations.
Haemophilia B
A total of 150 patients, 22 from the United States (SHL, n = 10; EHL, n = 12) and 128 from Europe (SHL, n = 104; EHL, n = 24), were included in the haemophilia B analysis. The demographics and clinical characteristics of the SHL FIX and EHL FIX groups in the United States, Europe, and combined populations are shown in Table 3. Among patients with haemophilia B, ages ranged from 4 to 71 years (median, 25 years). The majority of patients never had inhibitors (n = 139; 93%); of those who had, none had inhibitors at baseline. Of the 150 patients included in the analysis, 101 (67%) were on prophylactic rFIX treatment. Dosing information is listed by treatment group for the United States and European populations in Table 3. The total dose per week was numerically higher for the SHL than the EHL FIX products in both the United States and Europe.
In the United States, European, and combined populations, the mean (SD) ABR for the SHL FIX group and the EHL FIX group were as follows for the United States: SHL, 1.6 (1.88); EHL, 0.8 (0.75); Europe: SHL, 2.2 (2.18); EHL, 1.7 (1.66); and patients in the two populations combined: SHL, 2.1 (2.16); EHL, 1.4 (1.48) (Fig. 1c). In the combined population of patients with bleeding data, 21 of 108 (19%) patients treated with SHL FIX and 11 of 34 (32%) patients treated with EHL FIX reported having no bleeding events during the previous 12 months. The mean ABR was numerically greater in patients in Europe and in the combined population than in patients in the United States. The median ABR was 1 for both treatment groups in the United States (range: SHL, 0–6; EHL, 0–2), and was 2 for the SHL FIX group (range, 0–11) and 1 for the EHL FIX group (range, 0–5) in Europe.
In a subanalysis of patients in the EHL FIX group, 21 were taking eftrenonacog alfa (the United States, six; Europe, 15) and 15 were taking albutrepenonacog alfa (the United States, six; Europe, nine). Of these 36 patients, ABR data were available for 34 patients, 19 in the eftrenonacog alfa group and 15 in the albutrepenon-acog alfa group. In the combined United States and European population, the mean ABR was 1.4 and the median ABR was 1 (range, 0–5) in both the eftrenonacog alfa and albutrepenonacog alfa groups.
The percentage of patients who were fully adherent, as reported by their physicians, was similar between treatment groups in the combined population, with 26 of 38 (68%) patients treated with SHL FIX and 11 of 15 (73%) patients treated with EHL FIX having missed none of their last 10 doses of factor (Fig. 1d). The mean number of missed doses was comparable in the SHL and EHL FIX groups in the United States [SHL, 0.8 (0.96) vs. EHL 0.5 (0.76)] and in Europe [SHL, 0.5 (1.16) vs. EHL, 0.1 (0.38)]. The median number of doses missed of the last 10 doses the patient should have taken was 0.5 for the SHL FIX group in the United States (range: 0–2) and 0 for all other groups (range: the United States EHL, 0–2; Europe SHL, 0–6; EHL, 0–1).
Discussion
In this exploratory analysis of real-world data from a patient health record–based survey, ABR and adherence data from haemophilia A and B patients receiving SHL vs. EHL FVIII or FIX replacement products were collected. The small sample size of patients included in this analysis precluded inferences from being made about differences between groups. The EHL factor replacement products have been shown in clinical trials to be safe and effective for prophylaxis and for treatment of episodic bleeding events in patients with severe haemophilia A or B [12,13]; however, no head-to-head trials comparing the efficacy of EHL vs. SHL factor replacement products have been reported [14]. One analysis based on administrative pharmaceutical claims data, presented at the 2018 Annual Meeting of the Academy of Managed Care Pharmacy, reported no difference in the occurrence of bleeding events in haemophilia patients who switched from SHL to EHL factor replacement products, with the caveat that medical claims data do not necessarily identify all bleeding events [15]. Likewise, in a retrospective chart review from a single US-based centre, ABR following the switch from an SHL to EHL product was numerically lower (i.e., preswitch ABR of 2.3 and 2.5 improved to 1.3 and 0.82 for haemophilia A and B patients, respectively), but statistical testing was not performed [10].
A reduced ABR in patients receiving EHL vs. SHL FVIII or FIX replacement products might be anticipated if use of the EHL product provided better control of trough plasma factor levels in patients receiving prophylactic treatment, and/or encouraged a switch from episodic to prophylactic treatment of haemophilia A or B [5,6]. Prophylactic treatment substantially improves ABR relative to treatment of bleeding episodes [12]; consequently, the National Hemophilia Foundation recommends that prophylaxis be considered optimal therapy for individuals with severe haemophilia A or B [2]. Based on this historical perspective, it is hypothesized that patients with greater disease severity may benefit from switching to an EHL product but, at the time of this study, no evidence-based guidelines on the use of EHL products were available [16,17]. Reducing the burden of treatment may also encourage patients to switch from episodic to prophylactic treatment [6]. It would be expected that longer half-life factor replacement products may potentially reduce the burden of treatment by allowing greater spacing between injections while maintaining patients’ trough factor levels of at least 1 IU/dl [5,6].
Several discussions of the potential benefits of a longer half-life factor replacement option have raised the possibility that reducing the burden of treatment may improve patient adherence [7,8,18]. Because poor adherence to prophylactic treatment is associated with an increased frequency of bleeding events [19], changes to treatment that increase patient adherence to a prophylactic regimen should improve haemophilia patient outcomes [19,20]. Likewise, failure to adhere to the recommended dose and frequency of factor infusions in patients’ episodic treatment regimens may affect efficacy and long-term treatment outcomes [21]. To our knowledge, however, no direct evidence that adherence to treatment is improved with the use of EHL factor replacement products has been reported in the literature. The current analysis used an indirect method to estimate treatment adherence for haemophilia A or B patients receiving SHL vs. EHL products. Based on physician impression, the percentage of patients who took all of their last 10 doses of a factor replacement product was comparable between the SHL and EHL treatments. To the best of our knowledge, there are no validated measures of physician-reported patient adherence; however, when patient-reported adherence data are scarce, physician-reported adherence data may be a valuable proxy. Additional studies that include patient-reported adherence outcomes in patients receiving prophylactic SHL vs. EHL factor replacement products will be critical to understanding whether EHL products can, in fact, improve adherence.
Differences in the total IU dose administered for SHL vs. EHL replacement factors is of particular interest given that clotting factor has been estimated to account for more than 90% of direct medical costs and 80% of total cost of illness in individuals with haemophilia A [22]. In line with published pharmacoeconomic and health claims database analyses of SHL vs. EHL factor replacement products, the total IU dose administered in the current analysis was numerically lower in the EHL than SHL factor group for most comparisons (haemophilia A in Europe and haemophilia B in both the United States and Europe) [15,23,24]. At the same time, the price per unit is higher for EHL than SHL products [24]. While one European retrospective claims database analysis found lower costs associated with EHL vs. SHL FVIII replacement products for both prophylactic treatment of haemophilia A and for treating bleeding episodes [25], US-based analyses have instead shown an increase in expenditures after a switch from an SHL to EHL FIX [24] or FVIII and FIX [15] replacement products, even when the total IUs dispensed were reduced after the switch. Conclusions regarding the pharmacoeconomics of replacement product switches are unlikely to be definitive given that costs per IU are variable across countries and regions [26].
The data collection and analysis in this study are subject to several important limitations. The analyses were retrospective in nature and were thus limited by the information available from the physician and patient data available in the DSP. Measures of treatment adherence and bleeding rates were based on reporting of past events by physicians and patients and may have been affected by recall bias, a problem inherent in this method [9]. Selection bias was introduced, as the results were based on the responses of physicians who chose to participate. The study design lacked random selection, which may have influenced the results. Further, the data were cross-sectional and cannot be used to demonstrate cause and effect. Given that haemophilia is a rare disease, the sample size for some groups was small; in particular, both the FVIII and FIX EHL groups had fewer patients relative to the respective SHL groups. In addition, the data were not adjusted for treatment regimens, changes in regimen based on clinical presentation of the patients, or the reason for switching from SHL to EHL therapy. It is possible that the EHL group represents patients with a prior history of compliance issues when using a prophylactic SHL regimen or patients who have more severe disease. In a recent retrospective study that included 13 patients switching from an SHL to EHL product, patient reasons for switching were varied [27]. Additional research that provides further longitudinal data on the same cohort of patients who switch from SHL to EHL therapies or provides insight into patient preferences for treatment would be informative.
Patient adherence to replacement factor therapy is complicated and likely depends on a number of factors. For example, patients taking a suboptimal therapy may take all of their doses to maintain a bleeding event-free or low-bleed status, while those on a fully optimized product may miss doses, knowing that they will still maintain a low ABR. The clinical implications of a single missed dose with a once-weekly regimen vs. a regimen with multiple infusions per week should be considered when interpreting these results. In future studies, assessing other haemophilia outcomes, such as measures of joint health or pharmacokinetics, may be important to determine the full implications of maintaining adherence.
In summary, this analysis of patients with moderate-to-severe haemophilia in the United States and Europe evaluated ABR and treatment adherence between SHL and EHL FVIII or FIX replacement products. This analysis was based on a limited sample, and further analyses of larger samples, incorporating essential clinical characteristics, should be explored to confirm these preliminary findings. When making clinical decisions regarding factor replacement, clinicians should consider these data in the context of individual patients, their treatment history, and their clinical presentation.
Acknowledgements
The authors thank Charlotte Johnston of Adelphi Real World for her contributions to data collection and analysis. Data collection was undertaken by Adelphi Real World as part of an independent survey titled the Adelphi Haemophilia Disease-Specific Programmes, sponsored by multiple pharmaceutical companies, including Pfizer. Pfizer did not influence the original survey through contributions to either the design of the questionnaires or data collection.
Author contributions: A.C., D.S., B.J.T., J.M., J.d.C., and J.A. contributed to the study design. J.M. and J.d.C. participated in the collection and assembly of data. All authors had full access to the data, and all authors contributed to the drafting, critical review, and revision of the article, with the support of medical writers provided by Pfizer Inc. All authors granted approval of the final article for submission.
Conflicts of interest
The current study was sponsored by Pfizer Inc. A.C., D.S., P.F.F., B.J.T., E.R., S.H., and J.A. are employees of Pfizer and may own stock or options in the company. A.M.P. was an employee of Pfizer Inc at the time of this research. J.M and J.d.C are employees of Adelphi Real World. The study described here using data from the Adelphi Haemophilia Disease-Specific Programmes was funded by Pfizer Inc. Medical writing and editorial support were provided by Kathleen Dorries, PhD, and Jessica D. Herr, PharmD, of Peloton Advantage, LLC, an OPEN Health company, and funded by Pfizer Inc.
Footnotes
Current affiliation: Endpoints and Evidence, LLC, Surf City, North Carolina, USA.
References
- 1.Srivastava A, Brewer AK, Mauser-Bunschoten EP, Key NS, Kitchen S, Llinas A, et al. Guidelines for the management of hemophilia. Haemophilia 2013; 19:e1–e47. [DOI] [PubMed] [Google Scholar]
- 2. MASAC recommendation concerning prophylaxis (regular administration of clotting factor concentrate to prevent bleeding). MASAC Document #241 (replaces #179). National Hemophilia Foundation; New York, NY. Available at: https://www.hemophilia.org/sites/default/files/document/files/241Prophylaxis.pdf. [Accessed 23 January 2020]. [Google Scholar]
- 3.Peyvandi F, Garagiola I, Young G. The past and future of haemophilia: diagnosis, treatments, and its complications. Lancet 2016; 388:187–197. [DOI] [PubMed] [Google Scholar]
- 4.Hua B, Wu R, Sun F, Luo B, Alvey C, Labadie R, et al. Confirmation of longer FIX activity half-life with prolonged sample collection after single doses of nonacog alfa in patients with haemophilia B. Thromb Haemost 2017; 117:1052–1057. [DOI] [PubMed] [Google Scholar]
- 5.Shapiro AD, Ragni MV, Kulkarni R, Oldenberg J, Srivastava A, Quon DV, et al. Recombinant factor VIII Fc fusion protein: extended-interval dosing maintains low bleeding rates and correlates with von Willebrand factor levels. J Thromb Haemost 2014; 12:1788–1800. [DOI] [PubMed] [Google Scholar]
- 6.Mancuso ME, Santagostino E. Outcome of clinical trials with new extended half-life FVIII/IX concentrates. J Clin Med 2017; 6:E39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carcao M. Changing paradigm of prophylaxis with longer acting factor concentrates. Haemophilia 2014; 20: Suppl 4: 99–105. [DOI] [PubMed] [Google Scholar]
- 8.von Mackensen S, Kalnins W, Krucker J, Weiss J, Miesbach W, Albisetti M, et al. Haemophilia patients’ unmet needs and their expectations of the new extended half-life factor concentrates. Haemophilia 2017; 23:566–574. [DOI] [PubMed] [Google Scholar]
- 9.Thornburg CD, Duncan NA. Treatment adherence in hemophilia. Patient Prefer Adherence 2017; 11:1677–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang C, Young G. Clinical use of recombinant factor VIII Fc and recombinant factor IX Fc in patients with haemophilia A and B. Haemophilia 2018; 24:414–419. [DOI] [PubMed] [Google Scholar]
- 11.Anderson P, Benford M, Harris N, Karavali M, Piercy J. Real-world physician and patient behaviour across countries: disease-specific programmes – a means to understand. Curr Med Res Opin 2008; 24:3063–3072. [DOI] [PubMed] [Google Scholar]
- 12.Powell JS, Pasi KJ, Ragni MV, Ozelo MC, Valentino LA, Mahlangu JN, et al. Phase 3 study of recombinant factor IX Fc fusion protein in hemophilia B. N Engl J Med 2013; 369:2313–2323. [DOI] [PubMed] [Google Scholar]
- 13.Mahlangu J, Powell JS, Ragni MV, Chowdary P, Josephson NC, Pabinger I, et al. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood 2014; 123:317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Young G, Mahlangu JN. Extended half-life clotting factor concentrates: results from published clinical trials. Haemophilia 2016; 22: Suppl 5: 25–30. [DOI] [PubMed] [Google Scholar]
- 15.Bowen K, Gleason P. Incremental cost of switching to extended half-life coagulation factor products to treat hemophilia among 15 million commercially insured members. J Manag Care Spec Pharm 2018; 24: Suppl: S33–S34. [Abstract D4]. [Google Scholar]
- 16.Dunn A. The long and short of it: using the new factor products. Hematology Am Soc Hematol Educ Program 2015; 2015:26–32. [DOI] [PubMed] [Google Scholar]
- 17.Lambert T, Benson G, Dolan G, Hermans C, Jimenez-Yuste V, Ljung R, et al. Practical aspects of extended half-life products for the treatment of haemophilia. Ther Adv Hematol 2018; 9:295–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miguelino MG, Powell JS. Clinical utility and patient perspectives on the use of extended half-life rFIXFc in the management of hemophilia B. Patient Prefer Adherence 2014; 8:1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krishnan S, Vietri J, Furlan R, Duncan N. Adherence to prophylaxis is associated with better outcomes in moderate and severe haemophilia: results of a patient survey. Haemophilia 2015; 21:64–70. [DOI] [PubMed] [Google Scholar]
- 20.Iorio A, Krishnan S, Myren KJ, Lethagen S, McCormick N, Yermakov S, et al. Indirect comparisons of efficacy and weekly factor consumption during continuous prophylaxis with recombinant factor VIII Fc fusion protein and conventional recombinant factor VIII products. Haemophilia 2017; 23:408–416. [DOI] [PubMed] [Google Scholar]
- 21.Duncan NA, Kronenberger WG, Roberson CP, Shapiro AD. VERITAS-PRN: a new measure of adherence to episodic treatment regimens in haemophilia. Haemophilia 2010; 16:47–53. [DOI] [PubMed] [Google Scholar]
- 22.Zhou ZY, Koerper MA, Johnson KA, Riske B, Baker JR, Ullman M, et al. Burden of illness: direct and indirect costs among persons with hemophilia A in the United States. J Med Econ 2015; 18:457–465. [DOI] [PubMed] [Google Scholar]
- 23.Bjorkman S. A commentary on the differences in pharmacokinetics between recombinant and plasma-derived factor IX and their implications for dosing. Haemophilia 2011; 17:179–184. [DOI] [PubMed] [Google Scholar]
- 24.Tortella BJ, Alvir J, McDonald M, Spurden D, Fogarty PF, Chhabra A, et al. Real-world analysis of dispensed IUs of coagulation factor IX and resultant expenditures in hemophilia B patients receiving standard half-life versus extended half-life products and those switching from standard half-life to extended half-life products. J Manag Care Spec Pharm 2018; 24:643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henry N, Jovanovic J, Schlueter M, Kritikou P, Wilson K, Myren KJ. Cost-utility analysis of life-long prophylaxis with recombinant factor VIIIFc vs recombinant factor VIII for the management of severe hemophilia A in Sweden. J Med Econ 2018; 21:318–325. [DOI] [PubMed] [Google Scholar]
- 26.Thorat T, Neumann PJ, Chambers JD. Hemophilia burden of disease: a systematic review of the cost-utility literature for hemophilia. J Manag Care Spec Pharm 2018; 24:632–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dunn AL, Ahuja SP, Mullins ES. Real-world experience with use of antihemophilic factor (recombinant), PEGylated for prophylaxis in severe haemophilia A. Haemophilia 2018; 24:e84–e92. [DOI] [PubMed] [Google Scholar]