
Figure 1.
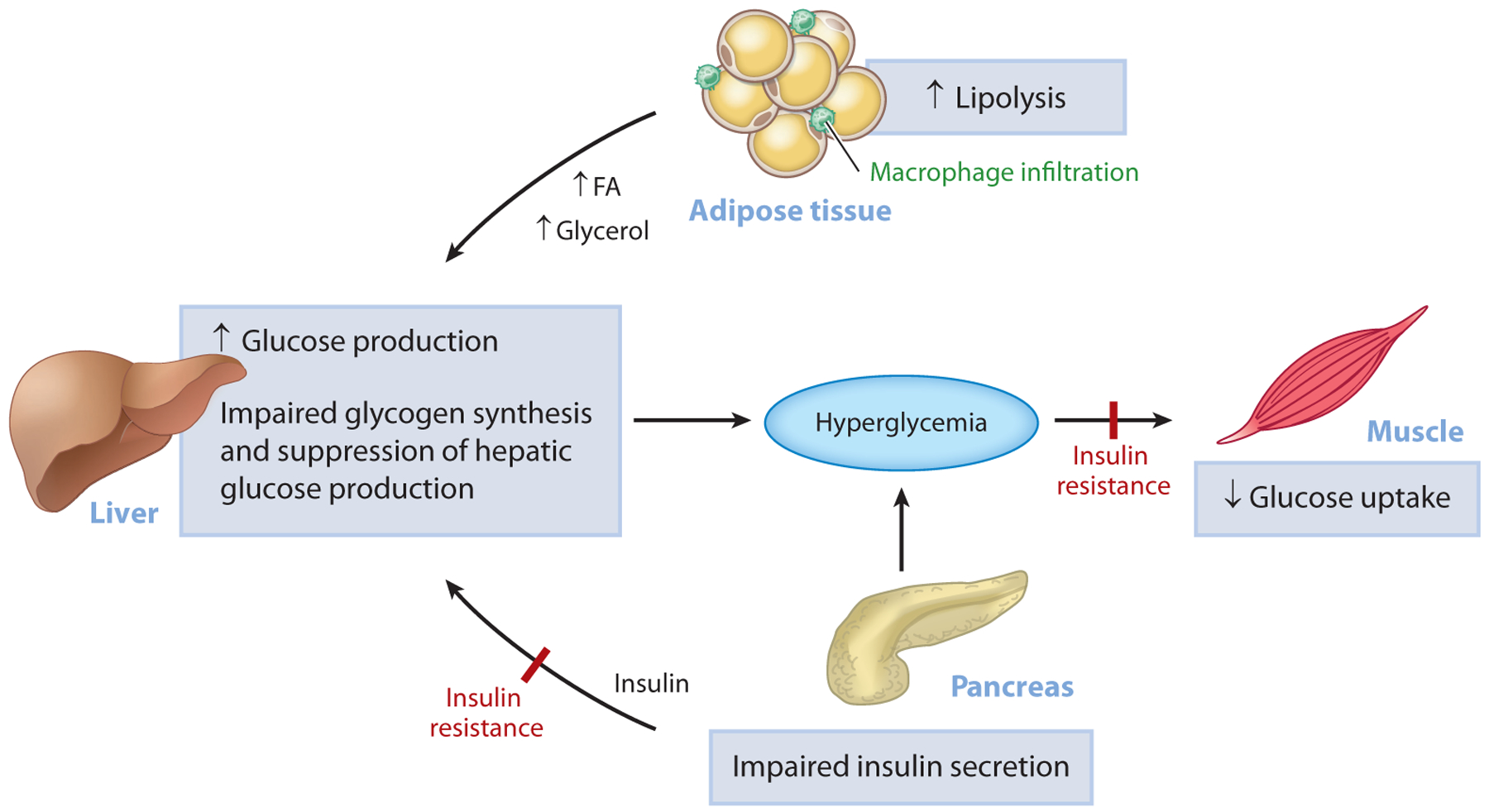
Pathogenesis of hyperglycemia in T2D. Uncontrolled hyperglycemia is a hallmark of T2D and is a major risk factor for long-term microvascular and macrovascular complications. While the progressive loss of pancreatic islet β-cell function is ultimately responsible for the progression from normoglycemia to hyperglycemia, insulin resistance predates β-cell dysfunction and plays a major role in the pathogenesis of T2D. In the muscle, insulin resistance is manifested as impaired glucose uptake following ingestion of a carbohydrate-rich meal and results in postprandial hyperglycemia. In the liver, insulin resistance is characterized by the inability of insulin to stimulate hepatic glycogen synthesis and suppress hepatic glucose production under postprandial conditions. In parallel, inappropriate increases in adipose tissue lipolysis (due to increases in inflammation) can drive hepatic gluconeogenesis through increases in hepatic acetyl-CoA content, allosteric activation of PC, and increased conversion of glycerol to glucose. Initially, the β-cell compensates for alterations in tissue insulin responsiveness by increasing insulin secretion; however, over time, this compensatory mechanism fails and β-cell mass declines, causing a further reduction in insulin secretion, worsening of hyperglycemia, and overt T2D. Abbreviations: FA, fatty acid; PC, pyruvate carboxylase; T2D, type 2 diabetes.