

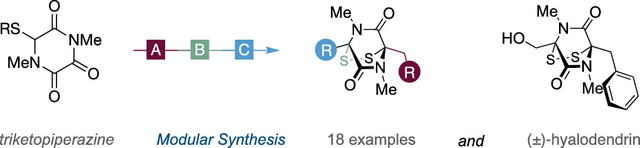
Abstract
Epidithiodiketopiperazines (ETPs) possess remarkably diverse biological activities and have attracted significant synthetic attention. The preparation of analogues is actively pursued; however, they are structurally challenging, and more direct and modular methods for their synthesis are desirable. To this end, the utility of a bifunctional triketopiperazine building block for the straightforward synthesis of ETPs is reported. A modular strategy consisting of enolate alkylation followed by site-selective nucleophile addition enables the concise synthesis of (±)-hyalodendrin and a range of analogues.
Graphical Abstract
Synthetic routes to epidithiodiketopiperazines (ETPs) have been intensely pursued. These fungal metabolites comprise 20 distinct families that are all characterized by a synthetically imposing disulfide-bridged diketopiperazine (DKP) (Figure 1a).1 The unusually stable transannular disulfide embedded within this heteroatom-rich motif 1 possesses a 0° CSSC dihedral angle, which demands a fully eclipsed arrangement of lone pairs on the adjacent S atoms and confers significant strain energy.2 This allows ETPs to engage in redox cycling to produce reactive oxygen species, ligate and eject Zn(II), or participate in rapid and reversible disulfide exchange reactions with the cysteine residues of proteins.3 The ETP core is solely responsible for the diversity of observed biological activities. For example, the enantiomers of hyalodendrin 2 are naturally occurring and have been isolated from different fungal sources, and they exhibit enantiomer-specific antimicrobial and antiviral/antibacterial activities.4 Similarly, the annulated ETPs dehydrogliotoxin 35 and glionitrin 46 constitute antipodal forms and differ only in arene decoration. The former exhibits antibacterial activity, whereas the latter is antibiotic/antitumor active. Chetomin 5, a rare heterodimeric indole containing two different ETP core units, has attracted significant interest as a chemotherapeutic agent.7 It is a potent in vitro and in vivo inhibitor of hypoxia inducible factor 1α (HIF-1α), a transcription factor that is essential to the growth of solid tumors.3g,8 Non-natural ETPs have also attracted significant attention.9 Overman and co-workers have developed NT-1721 (6), a candidate ETP with potent activity against acute myeloid leukemia.9a,b Finally, Matile and co-workers have employed the unique properties of ETPs for strain-promoted intracellular probe delivery (e.g., 7).10 This occurs with such extraordinary cellular uptake efficiencies that endosomal capture is avoided, leading to ETPs being harnessed as “unstoppable” transporters for thiol-mediated cellular uptake. Overall, it is clear that with such unique reactivity and properties ETPs will continue to serve as target compounds for therapeutic development and as design scaffolds for chemical biology applications. However, bespoke ETP synthesis continues to present significant synthetic challenges, which restrict their widespread potential to address biological problems. In order to remedy this, we herein report an experimentally straightforward, concise, and modular protocol for the preparation of ETPs.
Figure 1.
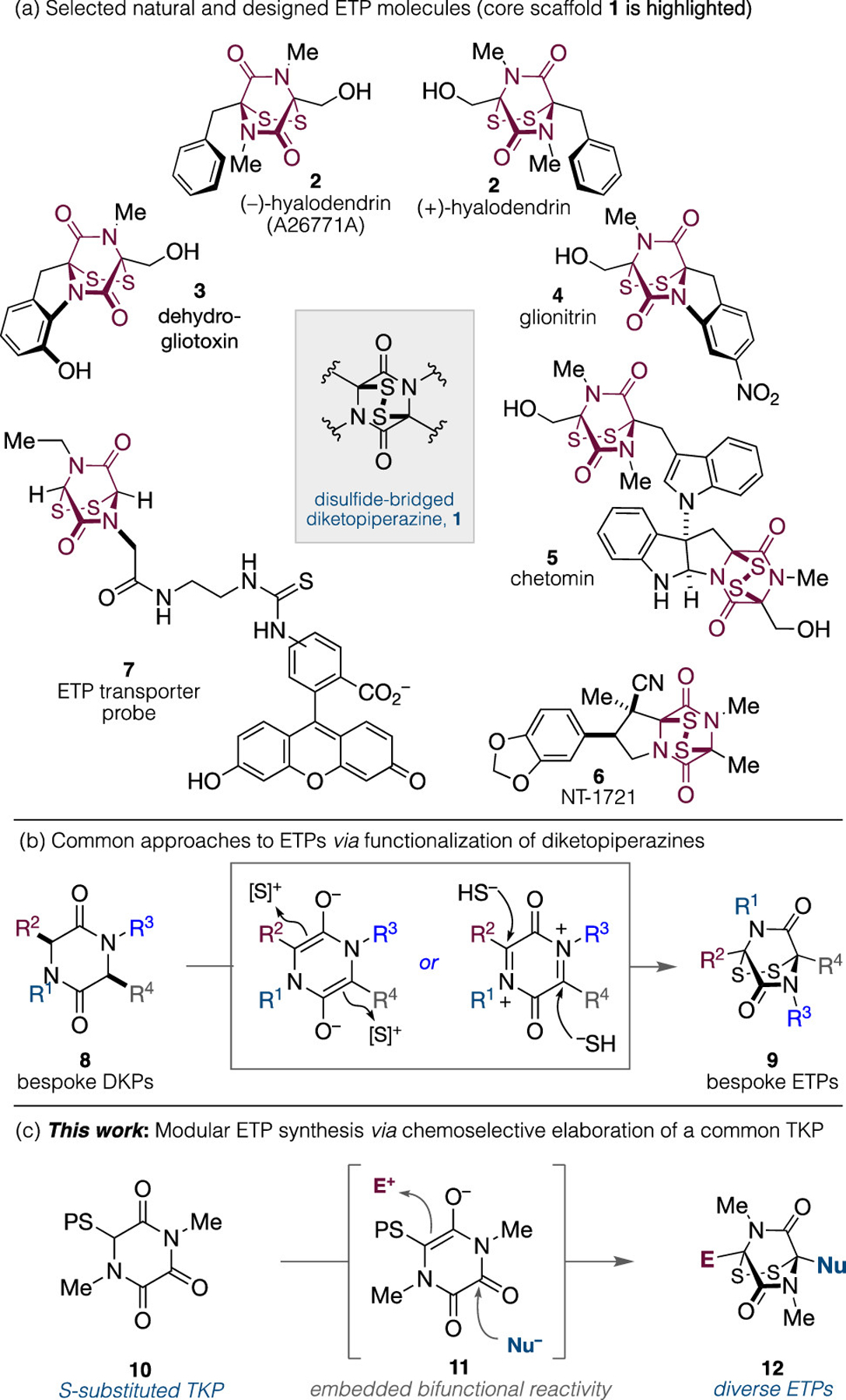
(a) Selected natural and non-natural ETPs. (b) ETP synthesis from DKP derived from parent amino acids. (c) Modular ETP synthesis which exploits the bifunctional reactivity of triketopiperazines (TKPs).
The synthesis of ETPs 9 typically involves the elaboration of preassembled diketopiperazines 8 via electrophilic (S+) or nucleophilic (S−) incorporation (Figure 1b).11 However, achieving site selectivity, functional group tolerance, and necessary syn stereoselectivity using such strategies can be challenging. Furthermore, the assembly of DKPs comprising different amino acids, as well as the preparation of bespoke amino acids themselves, can be challenging and requires significant synthetic investment.12 Triketopiperazines13 (TKPs) are rigid scaffolds that possess only one enolizable site. This removes the issue of site-selective enolization that complicates diketopiperazine (DKP) elaboration and should permit straightforward alkylation via the derived enolate 11 (Figure 1c). In addition, site-selective nucleophilic carbonyl addition is possible due dipole minimization of the 1,4-configured bis-amide motif, which confers disparate carbonyl electrophilicities to the vicinal dicarbonyl motif within the TKP (see 11).14 Tertiary alcohols resulting from nucleophile addition would serve as precursors to electrophilic N-acyliminium ions, which can be trapped by pendant sulfur nucleophiles to forge the challenging transannular disulfide.15 We expected that successful exploitation of this bifunctional reactivity within a common S-substituted TKP (10) would result in the straightforward preparation of both natural and non-natural N,N′-dimethyl ETPs (12). Herein, we report the successful realization of this goal in which a single TKP precursor can be elaborated via chemoselective functionalization events.
We began our investigations into the use of TKPs as useful synthetic intermediates by targeting the simplest of the ETPs, (±)-hyalodendrin 2 (Scheme 1).16 This naturally occurring phenylalanine–serine-derived ETP exhibits enantiomer-specific antimicrobial and antiviral/antibacterial activity. Beginning with N,N′-dimethyl triketopiperazine 13, which was readily prepared in two steps from sarcosine methyl ester,17 we sought to install a suitably protected thiol, which would later serve as an anchor for transannular disulfide assembly. Treatment of 13 with LiHMDS followed by trapping of the resulting enolate with Clive’s silyl ether protected sulfenating reagent18 efficiently provided S-substituted TKP 14 in excellent yield on 5 g scale. A second enolization with LiHMDS was then performed, and the resulting S-substituted enolate was trapped with benzyl bromide to provide the fully substituted carbon 15 that constitutes the phenylalanine subunit of hyalodendrin 2. Due to the limited solubility of the intermediate lithium enolate, the addition of DMPU to this second enolate alkylation was necessary to ensure consistently high yields. The next step involved installation of the crucial disulfide. Deprotection with a range of fluoride sources (TBAF, KHF2, HF, HF·pyr, CsF) was efficient; however, the instability of resulting thiol precluded efficient stepwise reaction with tritylsulfenyl chloride and resulted in mixtures of 16 and the corresponding desulfenylated product.19 Fortunately, this could be overcome by trapping the thiolate in situ; dropwise addition of TBAF to a solution of 15 and tritylsulfenyl chloride (TrSCl) in THF resulted in rapid and clean conversion to trityl-protected disulfide 16. Next, and in line with our synthetic design we required site selective addition of an appropriate nucleophile to the vicinal dicarbonyl moiety. After significant experimentation we established that treatment with methylmagnesium bromide gave tertiary alcohols 17 exclusively as an inconsequential 1:1 mixture of diastereomers. Organocopper and organozinc nucleophiles were ineffective whereas organolithium regents resulted a myriad of products presumably due to the highly oxophilicity of Li+. Thereafter, dehydration upon treatment of this mixture with a stoichiometric quantity of p-toluenesulfonic acid afforded a 3:1 mixture of both the desired dehydrated product 18 and the corresponding bridged disulfide product.20 However, employment of substoichiometric quantities of p-toluenesulfonic acid effectively suppressed disulfide formation and reliably provided the alkene 18 in 58% (over two steps). Upjohn dihydroxylation followed by treatment of the resulting diols 19 (dr 1:1) with BF3·OEt2 delivered (±)-hyalodendrin (2) in 70% yield via the putative N-acyliminium ion 20.21 In order to ensure efficient epidisulfide formation, purification of the intermediate diols 19 was necessary; direct exposure of the crude diols to BF3·OEt2 resulted in drastically lower conversion. Attempts to replace BF3·OEt2 with Hf(OTf)4,22a milder Lewis acid, were unsuccessful and resulted in no reaction.
Scheme 1. Synthesis of (±)-Hyalodendrin 2a.
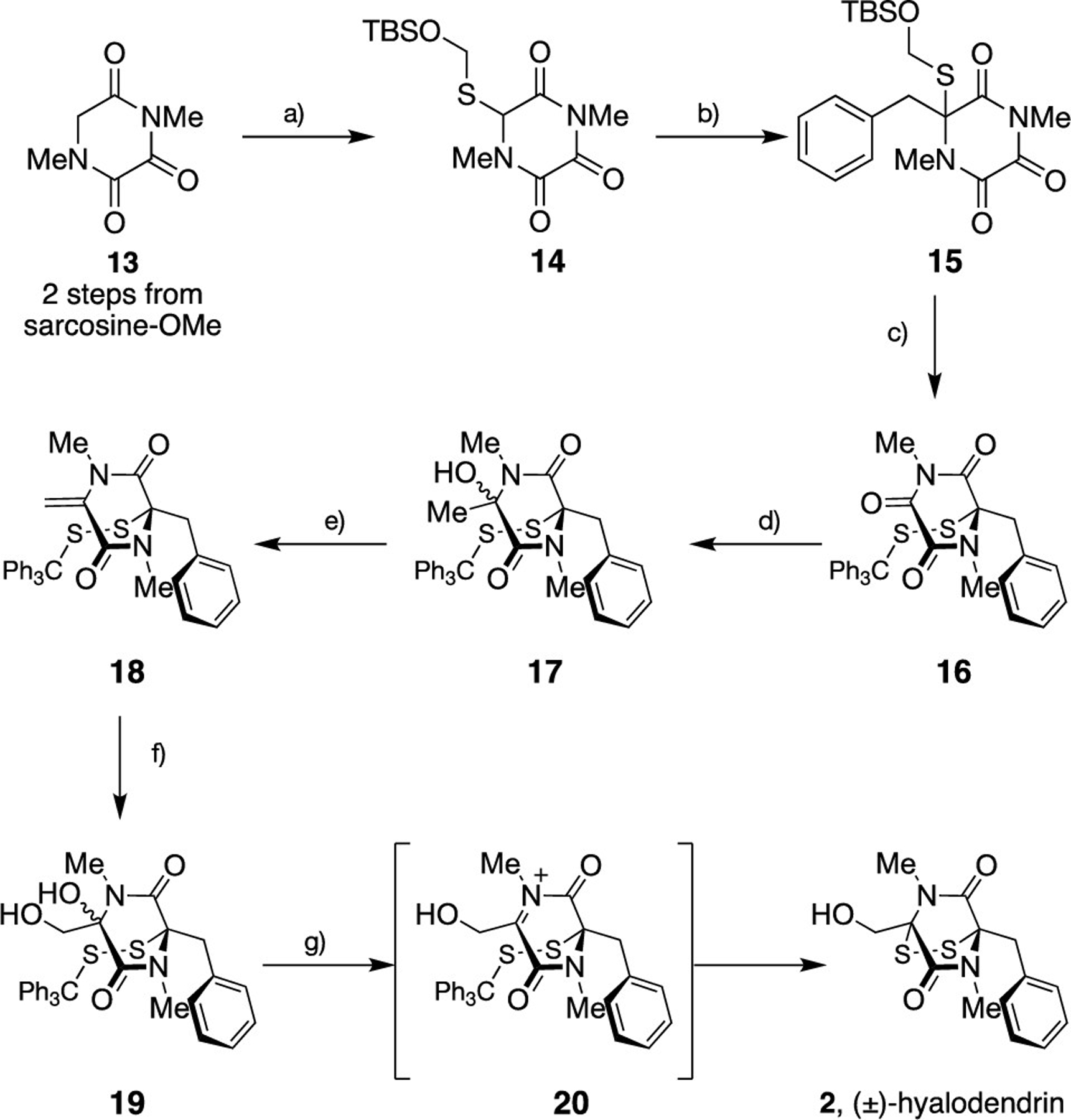
a Conditions: (a) LiHMDS, THF, −78 °C, S-[[(tert-butyldimethylsilyl)oxy]methyl] 4-methylbenzenesulfonothioate, 85%; (b) LiHMDS, THF, DMPU, 0 °C, BnBr, 84%; (c) TBAF, TrSCl, THF, 85%; (d) MeMgBr, THF, −78 °C; (e) PTSA, DCM, 58% (over two steps); (f) OsO4, NMO, acetone/H2O, 89%; (g) BF3·OEt2, DCM, −78 °C to rt, 70%.
Our synthesis of (±)-hyalodendrin 2 establishes the utility of bifunctional S-substituted triketopiperazine 14, and we next sought to exploit this in an operationally straightforward and modular synthesis of a focused library of ETP analogues of general structure 22 (Scheme 2). Starting from 14, diversification via a modular three-step alkylation/disulfide formation/Grignard addition–ring closure sequence provided a range of ETP analogues in useful yield on preparative scale. First, a range of electrophiles was explored for the initial alkylation of the lithium enolate derived from 14 (step A, substituents in red). Benzyl derivatives bearing substitution at the ortho, meta, and para positions (23–26) were well tolerated, with both electron deficient (24) and electron rich (25) and halogenated (23, 26) providing similar levels of efficiency. Disubstituted benzylic (28), π-extended naphthylic (27), and N-heterocyclic (29) electrophiles could also be employed, albeit in more modest yield.
Scheme 2.
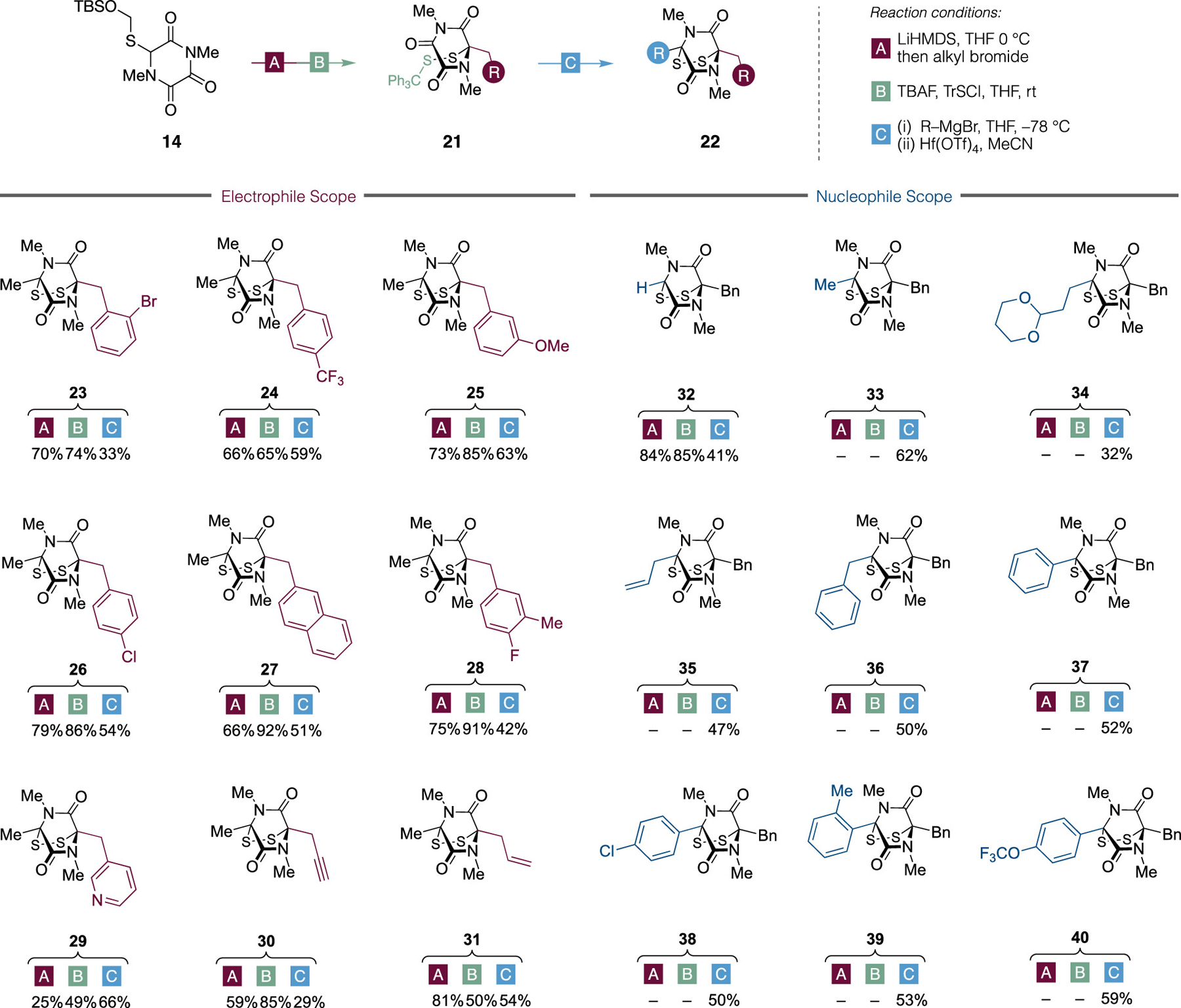
ETP Analogues Synthesized from Common TKP 14
Allylic and propargylic halides are also effective electrophiles (30 and 31) and provide potential handles for further functionalization within the context of broader SAR studies. These differentially alkylated TKPs were then converted to the corresponding ETPs via trityl disulfide formation, followed by site-selective nucleophilic addition of methylmagnesium bromide and hafnium(IV) triflate-mediated ring closure using a modification of Movassaghi’s protocol.22 The use of milder and more functional group tolerant Hf(OTf)4, where the number of equivalents of could be reduced from 10 to 1.5 equiv is noteworthy. During our hyalodendrin synthesis (cf. Scheme 2 19 → 2) this Lewis acid was ineffective for the conversion of 1,2-diols 19; however, it functions effectively here in the rapid and chemoselective activation of lone tertiary alcohols.
We next explored the nucleophile scope by treating benzylated and trityl-protected disulfide TKP 15 with a range of Grignard reagents, prior to epidisulfide formation. Both alkyl and aryl Grignard reagents performed well within the reaction and in each case the corresponding ETP was obtained with useful efficiency. Addition of i-butylmagnesium bromide to the benzyl-substituted TKP 15 resulted exclusively in hydride delivery and gave the phenylalanine–glycine containing ETP 3211f following ring closure, whereas the addition of methylmagnesium bromide afforded 33,11f the deoxygenated serine–alanine analogue of hyalodendrin, via the two-step sequence. Acetal-protected and allyl Grignard reagents provided ETPs 34 and 35, respectively, with functional handles for further manipulation. The nucleophilic addition of benzylmagnesium bromide was also successful and provided the C2-symmetric phenylalanine–phenylalanine-containing ETP 36.11c,d,f Finally, aromatic nucleophiles with various substitution patterns and functionality were also effective and gave ETPs 37–40 in good yield. These are particularly noteworthy as they would otherwise require independent syntheses of 2-arylglycines prior to sulfide-bridged DKP assembly.
In conclusion, we have demonstrated the utility of bifunctional S-containing triketopiperazine 14 as a common building block for the modular and flexible synthesis of epidithiodiketopiperazines. (±)-Hyalodendrin, a prototypical ETP target, was prepared from the parent sarcosine-derived TKP 13 in only seven steps and 22% overall yield. Using the same synthetic strategy, 14 could be elaborated via enolate alkylation with a range of previously inaccessible non-natural ETP analogues of general structure 22. Of particular significance is the fact that naturally occurring ETPs all derive from at least one aromatic amino acid; this strategy provides a straightforward means to divert from this as evidenced by the preparation of non-natural ETPs 30 and 31. Efforts to render the challenging enolate benzylation (14 to 15 or 21) enantioselective23,24 are ongoing and will be reported in due course.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Dung T. Do (Indiana University) for exploratory experimental efforts. We gratefully acknowledge Indiana University and the National Institutes of Health (R01GM121573) for generous financial support.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.9b01770.
Experimental procedures; characterization data; NMR spectra (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Welch TR; Williams RM Epidithiodioxopiperazines. Occurence, synthesis and biogenesis. Nat. Prod. Rep 2014, 31, 1376–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2). This has been observed via single-crystal analysis; see:; Overman LE; Sato T Construction of Epidithiodioxopiperazines by Directed Oxidation of Hydroxyproline-Derived Dioxopiperazines. Org. Lett 2007, 9, 5267–5270. [DOI] [PubMed] [Google Scholar]
- (3).(a) Munday R Studies on the mechanism of toxicity of the mycotoxin, sporidesmin. I. Generation of superoxide radical by sporidesmin. Chem.-Biol. Interact 1982, 41, 361–374. [DOI] [PubMed] [Google Scholar]; (b) Chai CLL; Waring P Redox sensitive epidithiodioxopiperazines in biological mechanisms of toxicity. Redox Rep. 2000, 5, 257–264. [DOI] [PubMed] [Google Scholar]; (c) Eichner RD; Waring P; Geue AM; Braithwaite AW; Mullbacher A Gliotoxin causes oxidative damage to plasmid and cellular DNA. J. Biol. Chem 1988, 263, 3772–3777. [PubMed] [Google Scholar]; (d) Mason JW; Kidd JG Effects of Gliotoxin and Other Sulfur-Containing Compounds on Tumor Cells in vitro. J. Immunol 1951, 66, 99–106. [PubMed] [Google Scholar]; (e) Hurne AM; Chai CLL; Waring P J. Biol. Chem 2000, 275, 25202–25206. [DOI] [PubMed] [Google Scholar]; (f) Mullbacher A; Waring P; Tiwari-Palni U; Eichner RD Structural relationship of epipolythiodioxopiperazines and their immunomodulating activity. Mol. Immunol 1986, 23, 231–235. [DOI] [PubMed] [Google Scholar]; (g) Cook KM; Hilton ST; Mecinovic J; Motherwell WB; Figg WD; Schofield CJ Epidithiodiketopiperazines Block the Interaction between Hypoxia-inducible Factor-1α (HIF-1α) and p300 by a Zinc Ejection Mechanism. J. Biol. Chem 2009, 284, 26831–26838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4). For the isolation of (+)-hyalodendrin, see:Strunz GM; Heissner CJ; Kakushima M; Stillwell MA Metabolites of Hyalodendron sp.: Bisdethiodi(methylthio)hyalodendrin. Can. J. Chem 1974, 52, 325–326. For the isolation of (−)-hyalodendrin (also known as A26771A), see:Michel KH; Chaney MO; Jones ND; Hoehn MM; Nagarajan R Epipolythiopiperazinedione Antibiotics from Penicillium Turbatum. J. Antibiot 1974, 27, 57–64.
- (5). For isolation, see:; (a) Lowe G; Taylor A; Vining LC Sporidesmins. Part VI. Isolation and structure of dehydrogliotoxin a metab olite of Penecillium terlikowskii. J. Chem. Soc. C 1966, 1799–1803. [DOI] [PubMed] [Google Scholar]; For syntheses, see:McMahon TC; Stanley S; Kazyanskaya E; Hung D; Wood JL A scaleable formal total synthesis of dehydrogliotoxin. Tetrahedron Lett. 2011, 52, 2262–2264.; (c) Kishi Y; Fukuyama T; Nakatsuka S Total synthesis of dehydrogliotoxin. J. Am. Chem. Soc 1973, 95, 6492–6493. [DOI] [PubMed] [Google Scholar]
- (6).Park HB; Kwon HC; Lee C-H; Yang HO Glionitrin A, an Antibiotic-Antitumor Metabolite Derived from Competitive Interaction between Abandoned Mine Microbes. J. Nat. Prod 2009, 72, 248–252. [DOI] [PubMed] [Google Scholar]
- (7).(a) Waksman SA; Bugie E Chaetomin, a New Antibiotic Substance Produced by Chaetominium cochliodes: I. Formation and Properties. J. Bacteriol 1944, 48, 527–530. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McInnes AG; Taylor A; Walter JA The Structure of Chetomin. J. Am. Chem. Soc 1976, 98, 6741. [DOI] [PubMed] [Google Scholar]; (c) Li G-Y; Li B-G; Yang T; Yan J-F; Liu G-Y; Zhang G-L Chaetocochins A–C, Epipolythiodioxopiperazines from Chaetonium cochliodes. J. Nat. Prod 2006, 69, 1374–1376. [DOI] [PubMed] [Google Scholar]; (d) Fujimoto H; Sumino M; Okuyama E; Ishibashi M Immunomodulatory Constituents from an Ascomycete, Chaetomium seminudum. J. Nat. Prod 2004, 67, 98–102. [DOI] [PubMed] [Google Scholar]
- (8).Kung AL; Zabludoff SD; France DS; Freedman SJ; Tanner EA; Viera A; Cornell-Kenyon S; Lee J; Wang B; Wang J; Memmert K; Naegeli HU; Petersen F; Eck MJ; Bair KW; Wood AW; Livingston DM Small Molecule Blockade of Transcriptional Coactivation of the Hypoxia-Inducible Factor Pathway. Cancer Cell 2004, 6, 33–43. [DOI] [PubMed] [Google Scholar]
- (9).(a) Kowolik CM; Lin M; Xie J; Overman LE; Horne DA NT1721, a Novel Epidithiodiketopiperazine, Exhibits Potent in vitro and in vivo Activity Against Acute Myeloid Leukemia. Oncotarget 2016, 7, 86186–86197. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Baumann M; Dieskau AP; Loertscher BM; Walton MC; Nam S; Kie J; Horne DA; Overman LE Tricyclic Analogues of Epidithiodioxopiperazine Alkaloids with Promising in vitro and in vivo Antitumor Activity. Chem. Sci 2015, 6, 4451–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Boyer N; Morrison KC; Kim J; Hergenrother PJ; Movassaghi M Synthesis and Anticancer Activity of Epipolythiodiketopiperazine Alkaloids. Chem. Sci 2013, 4, 1646–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Fujishiro S; Dodo K; Iwasa E; Teng E; Sohtome Y; Hamashima Y; Ito A; Yoshida M; Sodeoka M Epidithiodiketopiperazine as a Pharamcophore for Protein Lysine Methyltransferase G9a Inhibitors: Reducing Cyctotoxicity by Structural Simplification. Bioorg. Med. Chem. Lett 2013, 23, 733–736. [DOI] [PubMed] [Google Scholar]
- (10).(a) Zong L; Bartolami E; Abegg D; Adibekian A; Sakai N; Matile S Epidithiodiketopiperazines: Strain-Promoted Thiol-Mediated Cellular Uptake at the Highest Tension. ACS Cent. Sci 2017, 3, 449–453. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gasparini G; Sargsyan G; Bang E-K; Sakai N; Matile S Ring Tension Applied to Thiol-Mediated Cellular Uptake. Angew. Chem., Int. Ed 2015, 54, 7328–7331. [DOI] [PubMed] [Google Scholar]; (c) Chuard N; Gasparini G; Moreau D; Lörcher S; Palivan C; Meier W; Sakai N; Matile S Strain-Promoted Thiol-Mediated Cellular Uptake of Giant Substrates: Liposomes and Polymersomes. Angew. Chem., Int. Ed 2017, 56, 2947–2950. [DOI] [PubMed] [Google Scholar]
- (11). For selected examples, see:; (a) Kishi Y; Fukuyama T; Nakatsuka S New Method for the Synthesis of Epidithiodiketopiperazines. J. Am. Chem. Soc 1973, 95, 6490–6492. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC; Totokotsopoulos S; Giguère D; Sun Y-P; Sarlah D Total Synthesis of Epicoccin G. J. Am. Chem. Soc 2011, 133, 8150–8153. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nicolaou KC; Totokotsopoulos S; Giguère D; Sun Y-P; Sarlah D Synthesis and Biological Evaluation of Epidithio-, Eptetrathio-, and bis- (Methylthio)diketopiperazines: Synthetic Methodology, Enantioselective Total Synthesis of Epicoccin G 8,8′-epi-Rostratin B, Gliotoxin, Gliotoxin G, Emethallicin E, and Haematocin and Discovery of New Antiviral and Antimalarial Agents. J. Am. Chem. Soc 2012, 134, 17320–17332. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nicolaou KC; Giguère D; Totokotsopoulos S; Sun Y-P A Practical Sulfenylation of 2,5-Diketopiperazines. Angew. Chem., Int. Ed 2012, 51, 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Codelli JA; Puchlopek ALA; Reisman SE Enantioselective Total Synthesis of (−)-Acetylaranotin, a Dihydrooxepine Epidithiodiketopiperazine. J. Am. Chem. Soc 2012, 134, 1930–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Iwasa E; Hamashima Y; Fujishiro S; Higuchi E; Ito A; Yoshida M; Sodeoka M Total Synthesis of (+)-Chaetocin and its Analogues: Their Histone Methyltransferase G9a Inhibitory Activity. J. Am. Chem. Soc 2010, 132, 4078–4079. [DOI] [PubMed] [Google Scholar]; (g) Kim J; Ashenhurst JA; Movassaghi M Total Synthesis of (+)-11,11′-Dideoxyverticillin A. Science 2009, 324, 238–241. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Fukuyama T; Nakatsuka S; Kishi Y A New Synthesis of Epidithiapiperazinediones. Tetrahedron Lett. 1976, 17, 3393–3396. [Google Scholar]
- (12).Borthwick AD 2,5-Diketopiperazines: Synthesis, Reactions, Medicinal Chemistry, and Bioactive Natural Products. Chem. Rev 2012, 112, 3641–3716. [DOI] [PubMed] [Google Scholar]
- (13).(a) Cabanillas A; Davies CD; Male L; Simpkins NS Highly Enantioselective Access to Diketopiperazines via Cinchona Alkaloid Catalyzed Michael Additions. Chem. Sci 2015, 6, 1350–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Foster RW; Lenz EN; Simpkins NS; Stead D Organocatalytic Stereoconvergent Synthesis of α-CF3 Amides: Triketopiperazines and Their Hetereocyclic Metamorphosis. Chem. -Eur. J 2017, 23, 8810–8813. [DOI] [PubMed] [Google Scholar]
- (14).(a) Person D; Le Corre M Bull. Soc. Chim. Fr 1980, 5, 673–676. [Google Scholar]; (b) DeLorbe JE; Horne D; Jove R; Mennen SM; Nam S; Zhang F-L; Overman LE General Approach for Preparing Epidithiodioxopiperazines from Trioxopiperazine Precursors: Enantioselective Total Syntheses of (+)- and (−)-Gliocladine C, (+)-Leptosin D, (+)-T988C, (+)-Bionectin A, and (+)-Glitocladin A. J. Am. Chem. Soc 2013, 135, 4117–4128. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jabri SY; Overman LE Enantioselective Total Synthesis of Plectosphaeroic Acid B. J. Am. Chem. Soc 2013, 135, 4231–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Coste A; Kim J; Adams TC; Movassaghi M Concise total synthesis of (+)-bionectins A and C. Chem. Sci 2013, 4, 3191–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Takeuchi R; Shimokawa J; Fukuyama T Development of a route to chiral epidithiodioxopiperazine moieties and application to the asymmetric synthesis of (+)-hyalodendrin. Chem. Sci 2014, 5, 2003–2006. [Google Scholar]; See also refs 14b–d.
- (16). For previous syntheses, see:Szulc BR; Sil BC; Ruiz A; Hilton ST A Common Precurson Approach to Structurally Diverse Natural Products: The Synthesis of the Core Structure of (±)-Clausenamide and the Total Synthesis of (±)-Hyalodendrin. Eur. J. Org. Chem 2015, 2015, 7438–7442. Reference 15a..Fukuyama T; Nakatsuka S; Kishi Y Total synthesis of gliotoxin, dehydrogliotoxin and hyalodendrin. Tetrahedron 1981, 37, 2045–2078.Williams RM; Rastetter WH Syntheses of the fungal metabolites (±)-gliovictin and (±)-hyalodendrin. J. Org. Chem 1980, 45, 2625–2631.Strunz GM; Kakushima M Total synthesis of (±)-hyalodendrin. Experientia 1974, 30, 719–720.
- (17). Sarcosine-OMe ester was first converted to the corresponding N-methyl amide before reaction with oxalyldiimidazole according to Simpkins’ procedure (ref 13a). See the SI for details.
- (18).(a) Wang L; Clive DLJ [[(tert-Butyl)dimethylsilyl]oxy]-methyl Group for Sulfur Protection. Org. Lett 2011, 13, 1734–1737. [DOI] [PubMed] [Google Scholar]; (b) Dong S; Clive DLJ; Gao J-M [(tert-Butyldimethylsilyloxy])-methanethiol and [(tert-Butyldiphenylsilyloxy])methanethiol—nucle-ophilic protected H2S equivalents. Tetrahedron Lett. 2015, 56, 6857–6859. [Google Scholar]; (c) Wang L; Clive DLJ Synthetic studies related toMPC1001: formation of a model epidithiodiketopiperazine. Tetrahedron Lett. 2012, 53, 1504–1506. [Google Scholar]
- (19). See the SI for details.
- (20). See compound 33, Scheme 2..
- 21.Speckamp WN; Hiemstra H Intramolecular reactions of N-acyliminium intermediates. Tetrahedron 1985, 41, 4367–4416. [Google Scholar]
- (22). Hafnium(IV) triflate is a highly oxophilic but weakly thiophilic Lewis acid that has been used extensively in ETP synthesis. For an excellent discussion and lead references, see:; Kim J; Movassaghi M Biogenetically-Inspired Total Synthesis of Epidithiodiketopiperazines and Related Alkaloids. Acc. Chem. Res 2015, 48, 1159–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23). Despite significant efforts, we have been unable to render the transformation 14 to 15 enantioselective via phase-transfer-catalyzed benzylation. For a review of such reactions, see:; Shirakawa S; Maruoka K Recent Developments in Asymmetric Phase-Transfer Reactions. Angew. Chem., Int. Ed 2013, 52, 4312–4348. [DOI] [PubMed] [Google Scholar]
- (24). Similarly, and again despite significant efforts, we have been unable to render the transformation 14 to 15 enantioselective via Pd-catalyzed benzylation reactions. In contrast to Pd-catalyzed asymmetric allylic alkylation reactions of prochiral cyclic nucleophiles, there are few reports of the corresponding benzylic alkylations, and these are highly nucleophile specific; see:; (a) Trost BM; Czabaniuk LS Pd-Catalyzed Asymmetric Benzylation of Azlactones. Chem. -Eur. J 2013, 19, 15210–15218. [DOI] [PubMed] [Google Scholar]; (b) Trost BM; Czabaniuk LS Benzylic Phosphates and Electrophiles in the Palladium-Catalyzed Asymmetric Benzylation of Azlactones. J. Am. Chem. Soc 2012, 134, 5778–5781. [DOI] [PubMed] [Google Scholar]; (c) Trost BM; Czabaniuk LS Palladium-Catalyzed Asymmetric Benzylation of 3-Aryl Oxindoles. J. Am. Chem. Soc 2010, 132, 15534–15536. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.