

Description
A 46-year-old African–American male patient with no significant medical history presented to the hospital with dyspnoea. He was in his usual state of health until 6 months ago when he experienced congestion, dry cough, exertional dyspnoea, fever, night sweats, decreased appetite and generalised fatigue in the setting of a sick contact (his spouse). He failed two courses of antibiotics in the outpatient setting.
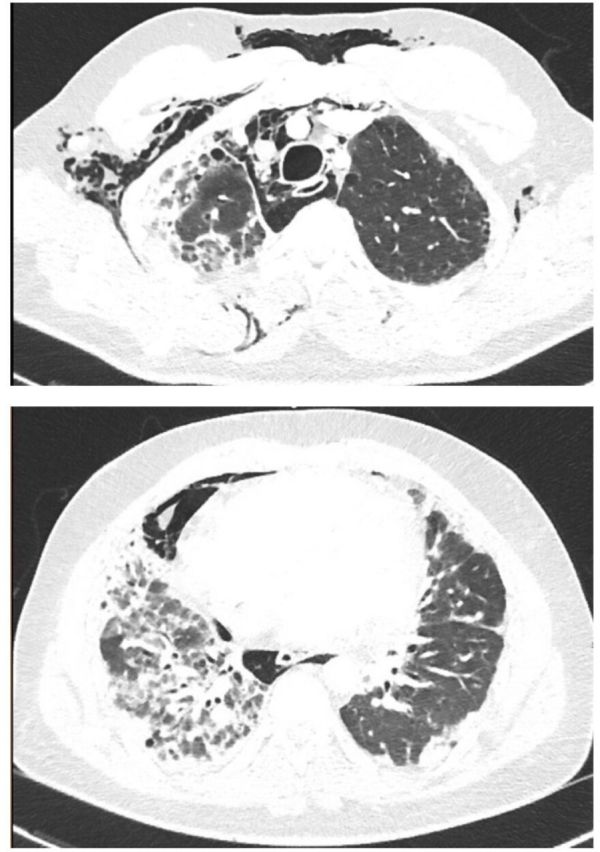
He was admitted to the hospital 3 months prior to presentation because he was unable to ambulate more than a few steps without feeling dyspneic. During that admission, he was diagnosed with cryptogenic organising pneumonia, after failing to improve with antibiotics and obtaining a CT chest consistent with this diagnosis (figure 1). He had a supraclavicular lymph node biopsy, which was non-diagnostic. He was ultimately started taking prednisone 60 mg with resolution of his cyclical fevers. There was a plan to a taper the prednisone by 10 mg each week, and to follow-up with rheumatology and pulmonology. He was sent home with supplemental oxygen.
Figure 1.

CT angiogram of the chest 3 months prior to the current admission, revealing diffuse, predominantly peripheral, patchy infiltrates.
He returned to the emergency department 3 months later, and he was febrile to 39.2°C, tachycardic to the 130 s–150 s, normotensive, tachypneic and hypoxic to the low 70 s on 3 L nasal cannula, thus prompting the initiation of high-flow nasal cannula. His arterial blood gas values were pH 7.5, Pco2 32 mm Hg and PO2 88 mm Hg. His ECG showed sinus tachycardia. Physical examination revealed bilateral inspiratory crackles, most appreciable at the basal third of the lung fields and normal strength on all four extremities. There was mild erythema of the palmar aspects of the distal fingertips (figure 2), as well as some distal digital hyperkeratosis with pinpoint pitting in the centre of the finger pad (figure 3). There is gross periungual erythema that is appreciable in greater detail on nail fold capillaroscopy. The most prominent findings can be seen on the fourth digit of the right hand, which exhibits several dilated capillary loops with one to two areas of dropout (figure 4). Laboratory findings revealed elevated C-reactive protein, erythrocyte sedimentation rate and high positive anti-melanoma differentiation-associated gene-5 (MDA-5) antibody with normal creatine kinase and aldolase level. A chest X-ray showed subcutaneous emphysema, R>L patchy infiltrates and no pneumothorax (figure 5). CT chest showed extensive pneumomediastinum and subcutaneous emphysema throughout the visualised mediastinum and increasing consolidations at the right middle lobe (figure 6). The patient received cefixime for 14 days, steroid treatment (1 mg/kg of methylprednisolone), tacrolimus and rituximab, but was still unable to wean off of high-flow nasal cannula. Currently, he is being evaluated for a lung transplant.
Figure 2.
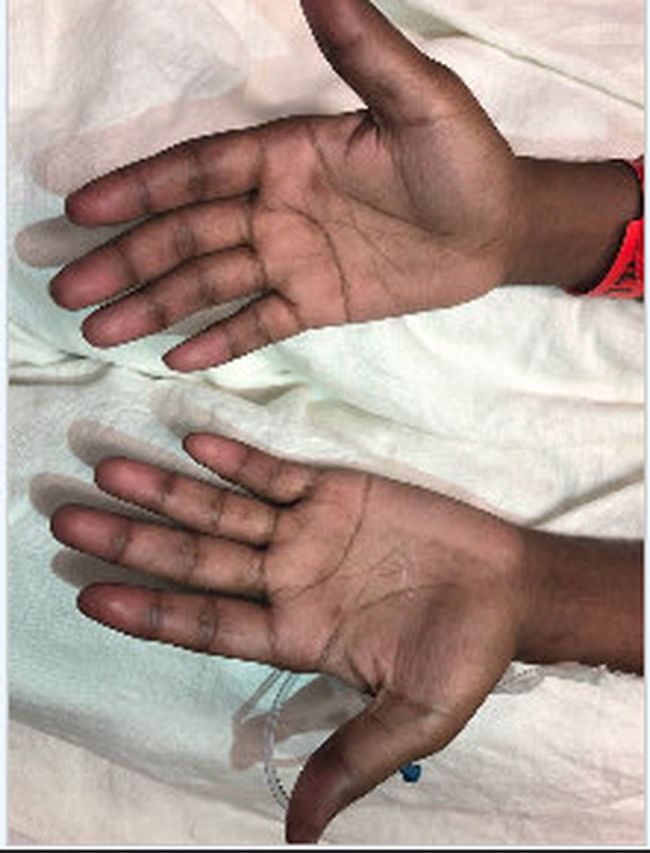
Mild erythema of the palmar aspects of the distal fingertips.
Figure 3.
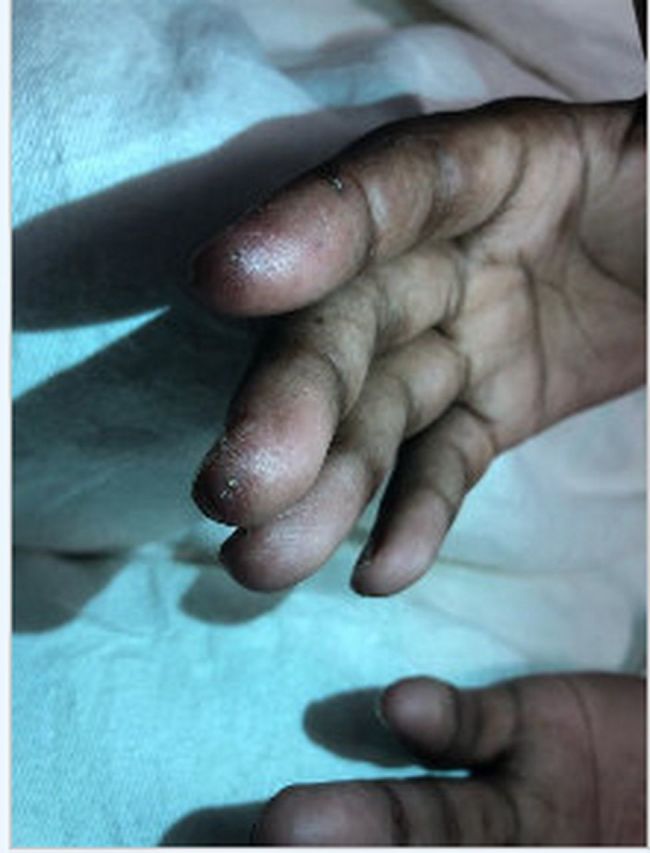
Distal digital hyperkeratosis with pinpoint pitting in the centre of the finger pad.
Figure 4.

Several dilated capillary loops with one to two areas of dropout.
Figure 5.

Subcutaneous emphysema and patchy infiltrates, right more than left.
Figure 6.
Extensive pneumomediastinum and subcutaneous emphysema throughout the visualised mediastinum, and consolidations of the right middle lobe.
Anti-MDA-5 antibody was first discovered in a Japanese population of amyopathic dermatomyositis patients in 2005. Autoantibodies were analysed from 298 patients with various connective tissue diseases, and anti-MDA-5 antibodies were detected in 8 of 42 patients with dermatomyositis. Those with anti-MDA-5 autoantibodies had significantly more rapidly progressive interstitial lung disease (ILD) when compared with patients without anti-MDA-5 autoantibodies.1
Another retrospective study found 10 out of 77 dermatomyositis patients were positive for the anti-MDA-5 antibody. This study found a characteristic cutaneous phenotype including skin ulceration, tender palmar papules or both. Hyperkeratosis of digital pulp, ulceration located on lateral nail folds, elbows, knees and Gottron papules were significantly associated with the disease. Patients with anti-MDA-5 antibodies also had an increased risk of hand swelling, arthritis/arthralgia and diffuse hair loss.2
Our patient did not present with classic signs and symptoms of dermatomyositis. He did not exhibit the typical skin findings of dermatomyositis except the periungual abnormalities. He also had normal muscle strength on examination with no evidence of myositis from the laboratory workup. Nonetheless, we still treated him as MDA-5 dermatomyositis because he had the typical skin findings of MDA-5 dermatomyositis with a positive anti-MDA-5 antibody with progressive ILD.2 Patients with anti-MDA-5 antibody typically present without evidence of myositis, this disease was previously referred to as anti-clinically amyopathic dermatomyositis-140 autoantibodies. He might develop typical cutaneous findings of dermatomyositis during the disease process in the future, hence, the absence of these manifestations does not rule out this diagnosis. There are not currently any guidelines or randomised trials in treating MDA-5 dermatomyositis. In our case, we used methylprednisolone, tacrolimus and rituximab. We did not give a pulse dose steroid as the patient initially responded well with the 1 mg/kg of methylprednisolone, we were able to wean him down from the high-flow nasal cannula to nasal cannula. Unfortunately, his condition regressed after the initial improvement, which prompted the initiation of rituximab, tacrolimus and eventually evaluation for a lung transplant.
Learning points.
Cases positive for anti-melanoma differentiation-associated gene-5 (MDA-5) antibodies have been emerging since the autoantigen was introduced in 2005. This antibody is usually found in amyopathic dermatomyositis (DM) patients, who have little evidence of myositis.
The anti-MDA-5 antibody is associated with progressive interstitial lung disease in DM patients.
It is important for clinicians to increase awareness of this disease. Skin manifestations (palmar nodules, skin ulcer and classic skin findings in DM) are the keys to diagnosis and possible early treatment and prognostication.
Footnotes
Contributors: AF, KF and SL wrote the primary manuscript and was revised by AH.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Patient consent for publication: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Sato S, Hirakata M, Kuwana M, et al. . Autoantibodies to a 140-kD polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum 2005;52:1571–6. 10.1002/art.21023 [DOI] [PubMed] [Google Scholar]
- 2.Fiorentino D, Chung L, Zwerner J, et al. . The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol 2011;65:25–34. 10.1016/j.jaad.2010.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]