
Figure 5.
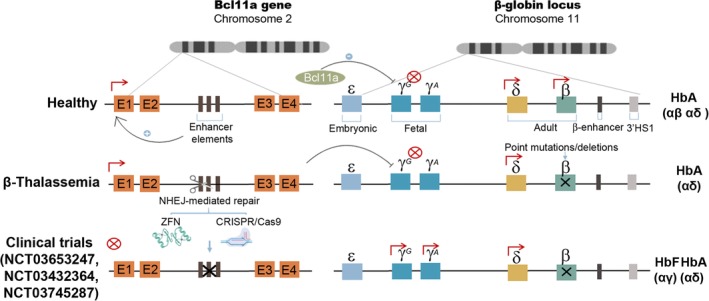
NHEJ GE strategies for treating β‐thalassemic and sickle cell disease (SCD) patients. Schematic representation of the β‐globin cluster in healthy individuals (top drawing). Only adult globins, δ‐globin and β‐globin, are expressed in healthy adult individuals. The Bcl11a gene is expressed thanks to the erythroid‐specific enhancer and blocks fetal globin (γG and γA) expression. In β‐thalassemia and SCD patients (middle drawing), mutations in the β‐globin gene abrogate its normal expression, preventing the generation of the predominant adult hemoglobin form (αβ). Three different clinical trials are currently on‐going to investigate the feasibility of eliminating the erythroid‐specific enhancer of the Bcl11a gene on HSPCs (Bottom drawing) using ZFNs (NCT03432364 and NCT03745287) or CRISPR/Cas9 (NCT03653247). By disrupting Bcl11a gene expression in erythroid cells, the fetal γ‐globin will be expressed in adults forming fetal hemoglobin (αγ) which should restore normal function