
Extended Data Fig. 9. Mean CpG frequency per base correlates with methylation of motifs at accessible enhancer regions.
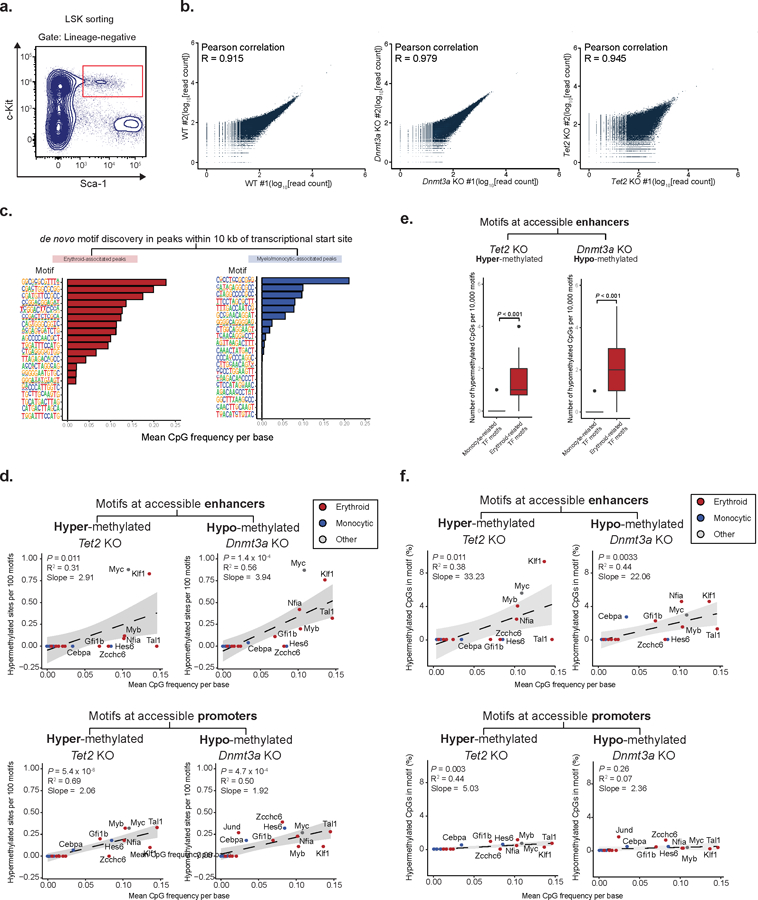
a) Gating for cell sorting for ATAC-Bseq experiments (LSK = lineage negative; Sca1 positive; c-Kit positive). b) Correlation between biological replicates for ATAC-Bseq experiments. Reads were downsampled to 30 x 106 reads per sample and the average read count per 10 kbp genomic windows was calculated (Pearson correlation). c) Examples of Homer output for de novo motif enrichment for either erythroid- or myelo-monocytic-associated accessible peaks within 10 kb of the closest transcriptional start site. d) Correlation between mean CpG frequency per base and the number of differentially (FDR<0.25, absolute methylation difference > 5%) hyper- or hypo-methylated CpGs between WT and Tet2 KO (n = 104,829 CpG sites) or Dnmt3a KO (250,353 CpG sites) respectively, per 100 motifs at accessible enhancers (upper panel) or accessible promoters (two-sided Students t-test; bottom panel; Spearman correlation). e) Number of hypermethylated CpGs per 10,000 motifs for erythroid- or monocyte-associated transcription factor motifs. 100 iterations of sampling without replacement were performed, sampling 10,000 motif sites each iteration, and measuring the number of differentially (FDR<0.25, absolute methylation difference > 5%) hypermethylated or hypomethylated sites captured in Tet2 KO (n = 2 mice) and Dnmt3a KO (n = 2 mice), respectively (two-sided Students t-test). f) Correlation between the percentage of hyper- or hypo-methylated CpGs between WT (n = 2 mice) and Tet2 KO (n = 2 mice) or Dnmt3a KO (n = 2 mice), respectively from total CpGs captured for each transcription factor DNA binding motif site and the mean CpG frequency per base, for motifs in accessible enhancers (middle panel) or accessible promoters (bottom panel; Spearman correlation; two-sided Students t-test).