

Abstract
Objective
We evaluated ibrutinib, a once‐daily inhibitor of Bruton's tyrosine kinase, combined with bortezomib and dexamethasone in patients with relapsed or relapsed/refractory multiple myeloma who had received 1‐3 prior therapies.
Methods
This was a phase 2, single‐arm, open‐label, multicentre study (NCT02902965). The primary endpoint was progression‐free survival (PFS).
Results
Seventy‐six patients were enrolled; 74 received ≥1 dose of study treatment. After median follow‐up of 19.6 months, median PFS was 8.5 months (95% CI: 6.2‐10.8); median overall survival was not reached. Overall response rate was 57% (95% CI: 45‐68), and median duration of response was 9.5 months (95% CI: 6.9‐10.6). Grade 3/4 AEs occurred in 73% of patients and fatal AEs occurred in 15% of patients. Incidence of major haemorrhage was 5%; one patient died from cerebral haemorrhage. After an observed increased incidence of serious (42%) and fatal (11%) infections, enrolment was suspended to implement risk‐minimisation measures. The safety profile was otherwise consistent with known safety profiles of the individual drugs.
Conclusion
Ibrutinib combined with bortezomib and dexamethasone elicited clinical responses. However, efficacy assessments conducted at potential restart of enrolment indicated that the targeted PFS could not be reached with additional patient enrolment, and the study was terminated.
Keywords: bortezomib, dexamethasone, ibrutinib, multiple myeloma
Novelty statements.
What is the new aspect of your work?
This study evaluated the novel combination of ibrutinib, a once‐daily oral inhibitor of Bruton's tyrosine kinase, with bortezomib plus dexamethasone in patients with relapsed/refractory multiple myeloma. Bruton's tyrosine kinase is overexpressed on multiple myeloma cells and may contribute to development of drug resistance. Ibrutinib has demonstrated in vitro synergy with bortezomib and encouraging activity in combination with dexamethasone and carfilzomib plus dexamethasone in phase 1 and 2 studies in patients with relapsed/refractory multiple myeloma.
What is the central finding of your work?
Ibrutinib in combination with bortezomib and dexamethasone elicited clinical responses. However, the study was closed early after efficacy assessments indicated that the targeted median progression‐free survival of >12 months could not be reached with the enrolment of additional patients.
What is (or could be) the specific clinical relevance of your work?
The optimal treatment for multiple myeloma in the relapsed/refractory setting remains uncertain and an area of unmet clinical need. Despite strong rationale based on preclinical and early clinical studies, the combination of ibrutinib with bortezomib and dexamethasone did not demonstrate sufficient activity to warrant further study in patients with relapsed/refractory multiple myeloma. Clinical studies evaluating the safety and efficacy of novel combinations are critical to identifying more effective salvage treatment options for patients with multiple myeloma. The information gained from these studies of novel combinations, regardless of the outcome, can help to inform future studies seeking to address this unmet clinical need.
1. INTRODUCTION
Multiple myeloma (MM) is a malignancy of terminally differentiated plasma cells and is the second most common haematological malignancy after non‐Hodgkin's lymphoma.1, 2 Although novel drug combinations have improved outcomes for patients with MM, most patients will eventually relapse after initial treatment and the efficacy of salvage regimens declines with each subsequent relapse.1 Treatment choices in the relapsed setting are influenced by various factors, such as age, performance status, comorbidities, and/or efficacy and toxicity of previous treatments; the optimal treatment remains uncertain and an area of unmet clinical need.3 Current treatment strategies involve the use of multiple agents that target specific signalling pathways involved in tumour cell growth and survival.
Bruton's tyrosine kinase (BTK) is overexpressed on MM tumour cells relative to normal plasma cells and has been implicated in the growth and survival of MM tumour cells.4, 5 Overexpression of BTK in side population cells may contribute to the development of drug resistance in MM tumour cells.5 In particular, BTK‐overexpressing cells demonstrated increased activity of the ATP‐binding cassette (ABC) transporter efflux pump and increased expression of the ABCB1 transporter protein, thereby promoting resistance to bortezomib, doxorubicin, and etoposide.5 Inhibition of the drug efflux pump was able to restore sensitivity to bortezomib.5
Ibrutinib, a first‐in‐class, once‐daily oral inhibitor of BTK, is approved in the United States and European Union for the treatment of various B‐cell malignancies.6, 7 Ibrutinib has been shown to suppress tumour growth and improve MM‐induced bone lysis in a murine model4 and has demonstrated in vitro cytotoxicity and synergy with bortezomib and lenalidomide in malignant plasma cells from patients with MM.8 In patients with relapsed/refractory (R/R) MM, ibrutinib demonstrated encouraging activity in combination with dexamethasone9 and in combination with carfilzomib plus dexamethasone,10 warranting further investigation of ibrutinib with steroids and proteasome inhibitors. Bortezomib, in combination with dexamethasone, is currently approved for the treatment of R/RMM.11, 12
This study was designed to assess the efficacy, safety, and tolerability of the addition of ibrutinib to the established treatment regimen of bortezomib and dexamethasone in patients with relapsed or R/R MM.
2. METHODS
2.1. Study design and patients
PCYC‐1139 (ClinicalTrials.gov identifier, NCT02902965) was a non‐randomised, open‐label, international, multicentre, sponsor‐initiated, phase 2 study conducted at multiple sites in Europe. Eligible patients were aged ≥18 years with a diagnosis of active MM per International Myeloma Working Group (IMWG) criteria,13 had received one to three prior lines of therapy, and had demonstrated progressive disease (PD) since completion of their most recent treatment regimen. Patients could have received prior bortezomib treatment but must not have been refractory or non‐responsive to bortezomib treatment. All patients were required to have measurable disease, defined as at least one of the following: serum monoclonal protein (SPEP) ≥1 g/dL (for patients with immunoglobulin [Ig] A, IgD, IgE, or IgM MM SPEP ≥0.5 g/dL) or urine monoclonal protein ≥200 mg by 24‐hour urine electrophoresis. Additional inclusion criteria were Eastern Cooperative Oncology Group performance status ≤2 and adequate haematological (absolute neutrophil count ≥1.5 × 109/L; platelet count ≥75 × 109/L), hepatic, and renal function. Patients were excluded if they had primary refractory disease (defined as failure to achieve a minimal response or better with any therapy); had disease refractory (defined as progression on treatment or within 60 days of completion) or non‐responsive (defined as failure to achieve minimal response or better) to prior proteasome inhibitor therapy; had a history of plasma cell leukaemia, primary amyloidosis, or POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, skin abnormalities) syndrome within 12 months prior to first administration of study treatment; peripheral neuropathy grade ≥2 or grade 1 with pain at screening; had currently active or clinically significant cardiovascular disease (uncontrolled arrhythmia or class III or IV congestive heart failure as defined by the New York Heart Association Functional Classification), or a history of myocardial infarction, unstable angina, or acute coronary syndrome within 6 months prior to enrolment; had currently active, clinically significant hepatic impairment (mild impairment or worse by Child‐Pugh classification); or required treatment with strong CYP3A inhibitors.
After 74 patients had initiated study treatment, study enrolment was suspended because of an initially observed increase of serious and fatal infections. During the enrolment halt, enrolled patients continued to receive treatment with the immediate implementation of risk‐minimisation measures, which included modifications to the dexamethasone dosing regimen that effectively halved the dose of dexamethasone in the triple combination (described in “Treatments” section below), and strengthened guidance for infection prophylaxis. A Safety Review Committee was also established to evaluate the rate of new infections every 6 weeks following implementation of the risk‐minimisation measures and enrolment was approved to restart approximately 6 months after the enrolment halt as a result of these risk‐minimisation measures. However, an evaluation of the efficacy data performed at this time indicated that the primary endpoint was unlikely to be achieved. As a result, the study was terminated and enrolment was not restarted.
The study was conducted in accordance with International Conference on Harmonization guidelines for Good Clinical Practice and principles of the Declaration of Helsinki. The protocol and its amendments were approved by independent ethics committees of all participating institutions and all patients provided written informed consent.
2.2. Treatments
During cycles 1 to 8, patients received oral ibrutinib 840 mg once daily in combination with subcutaneous bortezomib 1.3 mg/m2 on days 1, 4, 8, and 11 and oral dexamethasone 20 mg on days 1, 2, 4, 5, 8, 9, 11, and 12 of each 21‐day cycle. With the implementation of risk‐minimisation measures, dosing of dexamethasone was limited to days 1, 4, 8, and 11. During cycles 9 to 12, patients received ibrutinib 840 mg once daily in combination with bortezomib 1.3 mg/m2 on days 1, 8, 22, and 29, and dexamethasone 20 mg on days 1, 2, 8, 9, 22, 23, 29, and 30 of each 42‐day cycle. With the implementation of risk‐minimisation measures, dosing of dexamethasone was limited to days 1, 8, 22, and 29. From cycle 13 onward, patients received ibrutinib 840 mg once daily in combination with dexamethasone 40 mg once weekly in each 28‐day cycle. For patients aged >75 years, adjustment of the dexamethasone dose to 10 mg was recommended for cycles 1 to 12 and adjustment to 20 mg was recommended for cycle 13 onward, at the discretion of the investigator. Treatment with ibrutinib plus dexamethasone was continued until PD, unacceptable toxicity, or other protocol‐specified reason for discontinuation.
2.3. Outcomes and assessments
Efficacy endpoints were assessed by investigators according to IMWG criteria.13 The primary efficacy endpoint was progression‐free survival (PFS), defined as the time from the first dose of study treatment to confirmed PD or death from any cause. Secondary endpoints were overall response rate (ORR), defined as partial response or better; PFS rate at landmark time points; duration of response, defined as the time from initial documentation of response to the first documented evidence of PD; overall survival (OS), defined as the time from the first dose of study treatment to death from any cause; time to progression, defined as the time from the first dose of study treatment until PD; and the safety and tolerability of ibrutinib in combination with bortezomib and dexamethasone. Pharmacokinetic characteristics of ibrutinib and bortezomib when given in combination with dexamethasone were assessed as an exploratory endpoint.
Response and progression were assessed per IMWG criteria13 every 3 weeks during cycles 2 to 12 and every 4 weeks thereafter. In case of suspected complete response, pertinent assessments were performed per the IMWG response assessment guidelines. Following confirmed PD, patients were contacted approximately every 12 weeks for long‐term survival follow‐up. Plasma samples for analysis of ibrutinib and bortezomib pharmacokinetics were collected predose and at 30 minutes; 1, 2, 4, and 6 hours postdose on day 1 of cycle 2; and predose on day 2 of cycle 2.
Safety assessments included adverse events (AEs), laboratory evaluations, physical examinations, and vital signs. Adverse events were monitored throughout treatment until 30 days after the last dose of study treatment. Adverse events were coded by preferred term using the Medical Dictionary for Regulatory Activities (MedDRA), version 21.1, and were graded according to National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03.
2.4. Statistical analysis
Efficacy and safety were evaluated in all patients who received at least one dose of study treatment. Time‐to‐event endpoints were estimated using the Kaplan‐Meier method. For PFS, two‐sided 95% confidence intervals (CIs) were calculated using the Brookmeyer‐Crowley method with the log‐log transformed Greenwood variance estimate to test the null hypothesis. For ORR, two‐sided 95% CIs were calculated using the exact binomial method. Pharmacokinetics were analysed by non‐compartmental methods using Phoenix WinNonlin (Certara USA Inc), and AEs were summarised descriptively.
A sample size of approximately 125 patients was required to achieve 80% power at a one‐sided 0.025 significance level to test the null hypothesis of median PFS ≤8 months versus ≥12 months under the alternative hypothesis, with the assumption that the PFS follows an exponential distribution. However, a review of the available efficacy data at the time of potential restart of enrolment indicated that the primary endpoint was unlikely to be achieved; therefore, hypothesis testing for PFS was not performed.
2.5. Data sharing statement
Requests for access to individual participant data from clinical studies conducted by Pharmacyclics LLC, an AbbVie Company, can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
3. RESULTS
3.1. Patients
A total of 76 patients were enrolled, and 74 patients received at least one dose of study treatment (Table 1). Median age was 67.5 years and 61% were aged ≥65 years (Table 2). All patients had received prior systemic therapy for MM; 61% of patients had received one line of therapy and 39% had received two or three lines of therapy. The most common prior systemic therapies (reported in ≥30% of patients) were dexamethasone (88%), bortezomib (81%), cyclophosphamide (65%), lenalidomide (39%), and melphalan (31%). Overall, 39% of all patients were refractory to the last line of therapy. At the time of final analysis, all patients had discontinued study treatment; the most common reasons for discontinuation were PD (50%), AEs (18%), and patient withdrawal (14%) (Table 1). The median follow‐up duration was 19.6 months (range, 0.2‐24.6).
Table 1.
Patient disposition
| Patients | |
|---|---|
| Enrolled, n | 76 |
| Did not receive treatment, n | 2 |
| Started treatment, n | 74 |
| Discontinued treatment, n (%) | 74 (100) |
| Progressive disease | 37 (50) |
| Adverse event | 13 (18) |
| Withdrawal by patient | 10 (14) |
| Death | 7 (9) |
| Physician decision | 4 (5) |
| Study termination by sponsor | 3 (4) |
Table 2.
Baseline demographic and disease characteristics
| Characteristics |
Patients N = 74 |
|---|---|
| Age | |
| Median, years (range) | 67.5 (37‐86) |
| ≥65 y, n (%) | 45 (61) |
| Male sex, n (%) | 35 (47) |
| White race, n (%) | 71 (96) |
| ECOG PS, n (%) | |
| 0 | 43 (58) |
| 1 | 27 (36) |
| 2 | 4 (5) |
| Median time since diagnosis, years (range) | 4.2 (0.4‐13.7) |
| ISS stage at baseline, n (%) | |
| I | 25 (34) |
| II | 29 (39) |
| III | 20 (27) |
| Measurable disease, n (%) | |
| UPEP only | 6 (8) |
| SFLC only | 0 |
| SPEP only | 42 (57) |
| SPEP and UPEP | 26 (35) |
| Immunoglobulin (Ig) heavy chain class, n (%) | |
| IgA | 21 (28) |
| IgE | 0 |
| IgG | 49 (66) |
| IgM | 1 (1) |
| None | 5 (7) |
| Median bone marrow plasma cell percentage (range) | 23 (2‐99) |
| Number of prior lines of therapy, n (%) | |
| 1 | 45 (61) |
| 2‐3 | 29 (39) |
| Previous transplantation, n (%) | 43 (58) |
| Prior systemic therapies, n (%) | |
| Dexamethasone | 65 (88) |
| Bortezomib | 60 (81) |
| Cyclophosphamide | 48 (65) |
| Lenalidomide | 29 (39) |
| Melphalan | 23 (31) |
| Refractory to last line of therapy, n (%) | 29 (39) |
Abbreviations: ANC, absolute neutrophil count; ECOG PS, Eastern Cooperative Oncology Group performance status; ISS, International Staging System; SFLC, serum‐free light‐chain assay; SPEP, serum monoclonal protein; UPEP, urine monoclonal protein.
3.2. Safety
The median duration of treatment was 5.7 months (range, 0.1‐23.7) for ibrutinib, 4.5 months (range, 0.0‐12.6) for bortezomib, and 5.6 months (range, 0.1‐20.1) for dexamethasone. The most common AEs of any grade were thrombocytopenia (61% of patients), diarrhoea (55%), anaemia (38%), asthenia (30%), fatigue (28%), peripheral oedema (28%), upper respiratory tract infection (26%), nausea (24%), peripheral sensory neuropathy (24%), cough (20%), and back pain (20%) (Table 3). Grade 3/4 AEs were reported in 73% of patients, and fatal AEs were reported in 15% of patients. The most common grade 3 or higher AEs (occurring in ≥10% of patients) were thrombocytopenia (34%), pneumonia (16%), anaemia (15%), and asthenia (14%). Serious AEs of any grade occurred in 47 patients (64%). The only serious AE occurring in ≥5% of patients was pneumonia, which occurred in 11 patients (15%); serious AEs of pneumonia were grade 3/4 in nine patients (12%), and grade 5 in two patients (3%).
Table 3.
Adverse events of any grade occurring in ≥15% of patients
| Adverse event, n (%) | Patients (N = 74) | ||
|---|---|---|---|
| Any Grade | Grade 3/4 | Grade 5 | |
| Any adverse event | 74 (100) | 54 (73) | 11 (15) |
| Thrombocytopenia | 45 (61) | 25 (34) | 0 |
| Diarrhoea | 41 (55) | 7 (9) | 0 |
| Anaemia | 28 (38) | 11 (15) | 0 |
| Asthenia | 22 (30) | 10 (14) | 0 |
| Fatigue | 21 (28) | 6 (8) | 0 |
| Oedema peripheral | 21 (28) | 0 | 0 |
| Upper respiratory tract infection | 19 (26) | 2 (3) | 0 |
| Nausea | 18 (24) | 1 (1) | 0 |
| Peripheral sensory neuropathy | 18 (24) | 1 (1) | 0 |
| Back pain | 15 (20) | 2 (3) | 0 |
| Cough | 15 (20) | 0 | 0 |
| Hypokalaemia | 13 (18) | 3 (4) | 0 |
| Pneumonia | 13 (18) | 10 (14) | 2 (3)a |
| Pyrexia | 13 (18) | 0 | 0 |
Does not include two additional grade 5 AEs of pneumonia reported as pneumonia escherichia and pneumonia bacterial (n = 1 each).
Infections were reported in 59 patients (80%) and were grade 3 or higher in 32 patients (43%); the most common infections of any grade (occurring in ≥10% of patients) were upper respiratory tract infection (26%), pneumonia (18%), bronchitis (15%), conjunctivitis (14%), nasopharyngitis (14%), and urinary tract infection (11%). Serious infections were reported in 31 patients (42%), with pneumonia being the most common serious infection (15%). Fatal infections were reported as AEs in eight patients (11%), coded as MedDRA‐preferred terms of sepsis (n = 3) and pseudomonal sepsis (n = 1) for a total of four fatal infections due to sepsis; and pneumonia (n = 2), pneumonia Escherichia (n = 1), and pneumonia bacterial (n = 1) for a total of four fatal infections due to pneumonia. Additional fatal AEs were cerebral haemorrhage, death (unknown cause), and sudden death (n = 1 each).
Bleeding events of any grade were reported in 23 patients (31%). Major haemorrhage was reported in four patients (5%); one event (cerebral haemorrhage) resulted in treatment discontinuation and was subsequently fatal. Atrial fibrillation occurred in seven patients (9%) and was grade 3/4 in three patients (4%). Adverse events leading to dose reduction of ibrutinib, bortezomib, and dexamethasone occurred in 38 (51%), 30 (41%), and 19 (26%) patients, respectively, and AEs leading to discontinuation of ibrutinib, bortezomib, and dexamethasone occurred in 18 (24%), 22 (30%), and 20 (27%) patients, respectively. The most common AEs leading to ibrutinib discontinuation were pneumonia (n = 3; 4%), atrial fibrillation, diarrhoea, and sepsis (n = 2 each; 3%). The most common AEs leading to bortezomib discontinuation were polyneuropathy (n = 3; 4%), atrial fibrillation, diarrhoea, peripheral sensorimotor neuropathy, pneumonia, and sepsis (n = 2 each; 3%). The most common AEs leading to discontinuation of dexamethasone were atrial fibrillation, diarrhoea, pneumonia, and sepsis (n = 2 each; 3%).
3.3. Efficacy
After a median follow‐up of 19.6 months, PFS events had occurred in 55 patients (74%). The median PFS was 8.5 months (95% CI: 6.2‐10.8) and estimated PFS at 20 months was 7% (Figure 1A).
Figure 1.
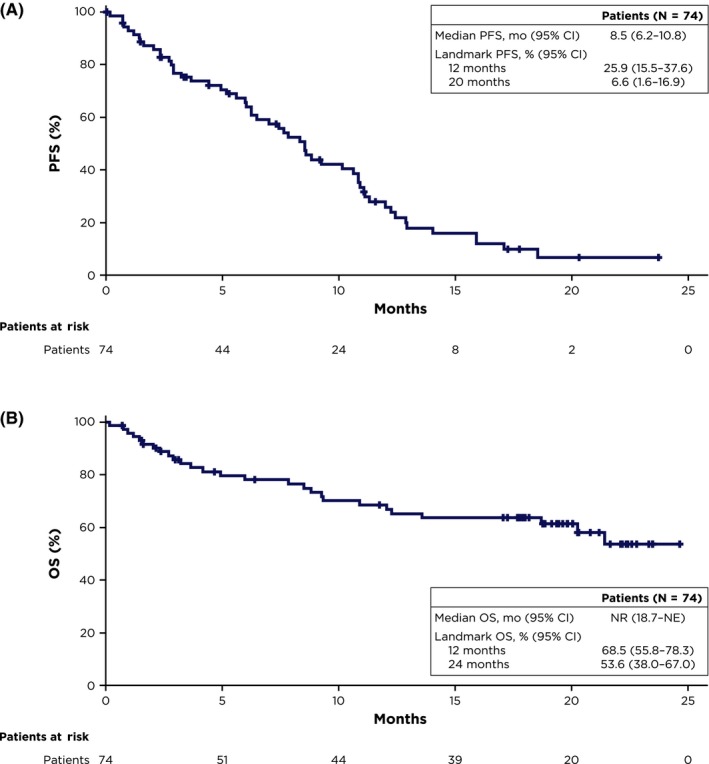
Kaplan‐Meier estimates of (A) PFS per investigator assessment and (B) OS. NE, not estimable; NR, not reached; OS, overall survival; PFS, progression‐free survival
The ORR was 57% (95% CI: 45‐68), including complete response in one patient, very good partial response in 13 patients, and partial response in 28 patients (Table 4). Among 42 patients who achieved partial response or better, the median duration of response was 9.5 months (95% CI: 6.9‐10.6) and the median time to progression was 10.6 months (95% CI: 7.8‐12.0). At the time of analysis, 27 patients (36%) had died and median OS was not reached (Figure 1B). Estimated OS at 24 months was 54%.
Table 4.
Best response per investigator assessment
|
Patients N = 74 |
|
|---|---|
| ORR, % (95% CI)a | 57 (45‐68) |
| Best response, n (%) | |
| sCR | 0 |
| CR | 1 (1) |
| VGPR | 13 (18) |
| PR | 28 (38) |
| MR | 11 (15) |
| SD | 13 (18) |
| PD | 2 (3) |
| Unconfirmed PD | 0 |
| Not evaluable | 1 (1) |
| Not done | 5 (7) |
Abbreviations: CI, confidence interval; CR, complete response; MR, minimal response; ORR, overall response rate; PD, progressive disease; PR, partial response; sCR, stringent CR; SD, stable disease; VGPR, very good partial response.
Confirmed sCR + CR + VGPR + PR.
3.4. Pharmacokinetics
Steady‐state plasma exposures of ibrutinib at the 840 mg dose level in this study were similar to those observed in patients with R/R MM receiving ibrutinib in combination with carfilzomib in the PCYC‐1119‐CA study.10 Steady‐state plasma exposures of bortezomib were consistent with those reported in the US prescribing information for bortezomib.14
4. DISCUSSION
This phase 2 study evaluated the efficacy and safety of ibrutinib in combination with bortezomib plus dexamethasone in patients with R/R MM who had received one to three previous lines of therapy. After 74 patients had received at least one dose of study treatment, study enrolment was suspended as a risk‐minimisation measure to address concerns over an increased incidence of serious and fatal infections compared with other studies of ibrutinib in R/R MM.9, 10 While the incidence of infection of any grade in this study (80%) was similar to rates reported with ibrutinib in other haematological malignancies (68%‐78%) over median follow‐up durations of 9.4 to 33.4 months, the incidence of grade ≥3 infections (43%) was higher than rates previously reported with ibrutinib (7%‐29%).15, 16, 17, 18, 19, 20 However, it should be noted that a higher dose of ibrutinib (840 mg once daily) was used in the current study compared with other haematological malignancies (420 mg or 560 mg once daily). The ibrutinib dose used in the current study was chosen based on clinical activity and a favourable safety profile observed at the 840 mg dose level of ibrutinib in combination with carfilzomib with or without dexamethasone and in combination with dexamethasone in two early phase studies in MM.9, 10 Plasma exposures to ibrutinib and bortezomib were consistent with those previously reported,10, 14 suggesting that drug‐drug interactions may not be contributing factors to the safety observations. Relative to other studies conducted in patients with MM, the incidence of grade ≥3 AEs observed with ibrutinib plus bortezomib and dexamethasone in the current study (88%) was similar to the incidence observed with ibrutinib plus carfilzomib with or without dexamethasone (86%),10 but higher than that observed with ibrutinib alone or in combination with dexamethasone without a proteasome inhibitor (57%).9
A review of the data after the implementation of risk‐minimisation measures showed a lower rate of serious infections and no fatal infections among patients already enrolled in the study. However, validation of the effectiveness of the risk‐minimisation measures was not possible as no new patients were enrolled. Apart from infections, the safety profile was consistent with the known safety profiles of the individual study drugs.6, 7, 11, 12 While safety profiles in general may vary depending on dosage, combination with other drugs, and by disease histology, incidences of AEs of interest with ibrutinib were largely consistent with previous reports across a range of haematological malignancies, including chronic lymphocytic leukaemia, Waldenström’s macroglobulinemia, and mantle cell lymphoma.15, 16, 17, 18, 19, 20 The median PFS of 8.5 months with ibrutinib plus bortezomib and dexamethasone was slightly longer than that observed in patients with R/R MM treated with placebo plus bortezomib and dexamethasone in the PANORAMA‐1 study (median 8.1 months).21 Such indirect comparisons should, however, be interpreted with caution given differences in study design. Clinical response was observed in 57% of patients and was generally durable, with a median response duration of 9.5 months. Overall survival data were not yet mature, with 64% of patients remaining alive after a median follow‐up duration of 19.6 months. Assessment of efficacy at the time of potential restart of enrolment indicated that the target median PFS for the alternative hypothesis (≥12 months) could not be reached by enrolling additional patients, and therefore, enrolment was not restarted. Because enrolment was suspended due to safety concerns before the target sample size had been reached, the hypothesis testing for the median PFS was not performed.
In summary, an initially increased incidence of serious infections was observed with ibrutinib in combination with bortezomib and dexamethasone, and in response, risk‐minimisation measures were introduced. Following the implementation of risk‐minimisation measures, very few serious and no fatal infections occurred, and regulatory authorities and ethics committees approved the restart of enrolment. Ibrutinib in combination with bortezomib and dexamethasone elicited clinical responses in patients with R/R MM. However, an efficacy assessment at the time of the potential restart of enrolment indicated that even with the pending enrolment of additional patients, the targeted endpoint of median PFS could not be reached and the study was terminated.
CONFLICT OF INTEREST
RH reports honoraria and consultancy/advisory role from Amgen, Celgene, Takeda, Bristol‐Myers Squibb, and Janssen, and research funding from Takeda, Novartis, Amgen, Janssen, Bristol‐Myers Squibb, and Celgene. LP reports nothing to disclose. MO reports honoraria from Takeda and Amgen; research funding from Janssen, Celgene, Takeda, Archigen Biotech, Roche, Merck Sharp & Dohme, Bayer, AbbVie, and Novartis; and travel, accommodations, or other expenses from Bristol‐Myers Squibb, Jazz, Roche, Sanofi, and Abdi Ibrahim. JMS reports travel, accommodations, or other expenses from Amgen Iberia. RGS reports honoraria from Amgen, Janssen, and Takeda; consultancy/advisory role for Bristol‐Myers Squibb and Takeda; research funding from Gilead, Incyte, Amgen, and Janssen; patents, royalties, or other intellectual property with Invivoscribe (EuroClonality/BIOMED‐2); and travel, accommodations, or other expenses from Janssen and Takeda. AA reports nothing to disclose. AO reports consultancy/advisory role for Celgene, Janssen, Amgen, and Takeda; and speakers bureau for Celgene and Amgen. NC reports nothing to disclose. AT, ED, and BH report employment with Pharmacyclics Switzerland GmbH, an AbbVie Company, and stock or other ownership in AbbVie. YL reports employment with Pharmacyclics LLC, an AbbVie Company, and stock or other ownership in AbbVie. EMB reports employment with Pharmacyclics Switzerland GmbH, an AbbVie Company. IS reports honoraria from and consultancy/advisory role with Celgene, Amgen, Janssen‐Cilag, Takeda, Bristol‐Myers Squibb, Novartis, and Sanofi; speakers bureau for Celgene, Amgen, Janssen‐Cilag, Takeda, Bristol‐Myers Squibb, and Novartis; and travel, accommodations, or other expenses from Celgene, Amgen, Janssen‐Cilag, and Bristol‐Myers Squibb.
AUTHOR CONTRIBUTIONS
RH designed the study in collaboration with the study sponsor. RH, LP, MO, JMS, RGS, AA, AO, NC, and IS collected data. YL, EMB, and ED confirmed the accuracy of the data, interpreted the data, and compiled it for analysis. YL performed statistical analyses. All authors had full access to the data, contributed to data interpretation, and vouch for accuracy of the data. All authors contributed to the manuscript preparation and approved the final version of the manuscript for submission.
ACKNOWLEDGEMENTS
We thank all the patients who participated in this study and their supportive families, as well as the investigators and clinical research staff from the study centres. We also thank Yvonne Pak, PhD, of Pharmacyclics LLC, an AbbVie Company, for her contributions to pharmacokinetic analyses and Melanie Sweetlove, MSc, for medical writing support, which was funded by Pharmacyclics LLC, an AbbVie Company.
Hajek R, Pour L, Ozcan M, et al. A phase 2 study of ibrutinib in combination with bortezomib and dexamethasone in patients with relapsed/refractory multiple myeloma. Eur J Haematol. 2020;104:435–442. 10.1111/ejh.13377
Funding information
This study was supported by Pharmacyclics Switzerland GmbH, an AbbVie Company.
REFERENCES
- 1. Kumar SK, Rajkumar V, Kyle RA, et al. Multiple myeloma. Nat Rev Dis Primers. 2017;3:17046. [DOI] [PubMed] [Google Scholar]
- 2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7‐30. [DOI] [PubMed] [Google Scholar]
- 3. Nijhof IS, van de Donk N, Zweegman S, Lokhorst HM. Current and new therapeutic strategies for relapsed and refractory multiple myeloma: an update. Drugs. 2018;78(1):19‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tai Y‐T, Chang BY, Kong S‐Y, et al. Bruton tyrosine kinase inhibition is a novel therapeutic strategy targeting tumor in the bone marrow microenvironment in multiple myeloma. Blood. 2012;120(9):1877‐1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang Y, Shi J, Gu Z, et al. Bruton tyrosine kinase is a therapeutic target in stem‐like cells from multiple myeloma. Cancer Res. 2015;75(3):594‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Janssen‐Cilag International NV . IMBRUVICA (ibrutinib) Summary of Product Characteristics 2018. Beerse, Belgium.
- 7. Pharmacyclics LLC . IMBRUVICA (ibrutinib) prescribing information 2019. Sunnyvale, CA.
- 8. Rushworth SA, Bowles KM, Barrera LN, Murray MY, Zaitseva L, MacEwan DJ. BTK inhibitor ibrutinib is cytotoxic to myeloma and potently enhances bortezomib and lenalidomide activities through NF‐kappaB. Cell Signal. 2013;25(1):106‐112. [DOI] [PubMed] [Google Scholar]
- 9. Richardson PG, Bensinger WI, Huff CA, et al. Ibrutinib alone or with dexamethasone for relapsed or relapsed and refractory multiple myeloma: phase 2 trial results. Br J Haematol. 2018;180(6):821‐830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chari A, Larson S, Holkova B, et al. Phase 1 trial of ibrutinib and carfilzomib combination therapy for relapsed or relapsed and refractory multiple myeloma. Leuk Lymphoma. 2018;59(11):2588‐2594. [DOI] [PubMed] [Google Scholar]
- 11. European Medicines Agency . Velcade: EPAR ‐ Product Information 2019; Amsterdam, The Netherlands. https://www.ema.europa.eu/en/documents/product-information/velcade-epar-product-information_en.pdf. Accessed July 28, 2019.
- 12. US Food and Drug Administration . VELCADE (bortezomib) for injection, for subcutaneous or intravenous use 2019. Silver Spring, Maryland, USA. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/021602s044lbl.pdf. Accessed July 28, 2019.
- 13. Rajkumar SV, Harousseau J‐L, Durie B, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood. 2011;117(18):4691‐4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Millennium Pharmaceuticals Inc . VELCADE (bortezomib) prescribing information 2019. Cambridge, MA.
- 15. Byrd JC, Brown JR, O'Brien S, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dimopoulos MA, Trotman J, Tedeschi A, et al. Ibrutinib for patients with rituximab‐refractory Waldenstrom's macroglobulinaemia (iNNOVATE): an open‐label substudy of an international, multicentre, phase 3 trial. Lancet Oncol. 2017;18(2):241‐250. [DOI] [PubMed] [Google Scholar]
- 17. Wang ML, Blum KA, Martin P, et al. Long‐term follow‐up of MCL patients treated with single‐agent ibrutinib: updated safety and efficacy results. Blood. 2015;126(6):739‐745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gopal AK, Schuster SJ, Fowler NH, et al. Ibrutinib as treatment for patients with relapsed/refractory follicular lymphoma: results from the open‐label, multicenter, phase II DAWN study. J Clin Oncol. 2018;36(23):2405‐2412. [DOI] [PubMed] [Google Scholar]
- 19. Chanan‐Khan A, Cramer P, Demirkan F, et al. Ibrutinib combined with bendamustine and rituximab compared with placebo, bendamustine, and rituximab for previously treated chronic lymphocytic leukaemia or small lymphocytic lymphoma (HELIOS): a randomised, double‐blind, phase 3 study. Lancet Oncol. 2016;17(2):200‐211. [DOI] [PubMed] [Google Scholar]
- 20. Shanafelt TD, Wang V, Kay NE, et al. Randomized phase III study of ibrutinib (PCI‐32765)‐based therapy vs. standard fludarabine, cyclophosphamide, and rituximab (FCR) chemoimmunotherapy in untreated younger patients with chronic lymphocytic leukemia (CLL): a trial of the ECOG‐ACRIN Cancer Research Group (E1912). Blood. 2018;132:LBA‐4. [Google Scholar]
- 21. San‐Miguel JF, Hungria VTM, Yoon S‐S, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double‐blind phase 3 trial. Lancet Oncol. 2014;15(11):1195‐1206. [DOI] [PubMed] [Google Scholar]