
FIGURE 2.
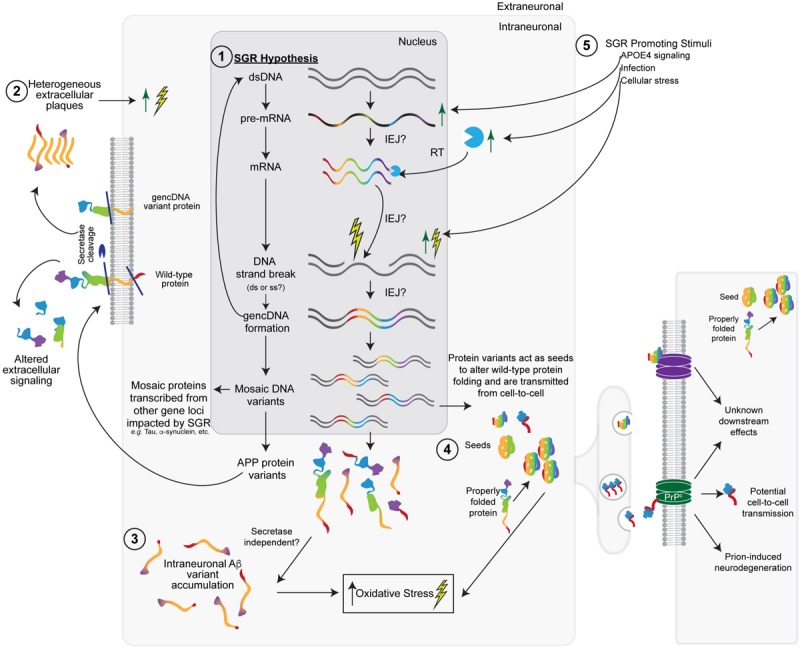
The somatic gene recombination (SGR) hypothesis for SAD. (1) Dysregulated SGR of APP through the insertion of reverse-transcribed mRNA leads to mosaic genomic APP variants that result in variant proteins with a number of downstream effects. Other gene loci may also be impacted by SGR. (2) In keeping with the Aβ-hypothesis, some variants would be transported to the cellular membrane, where wild-type, SNV-containing, and gencDNA variant proteins may or may not be cleaved by the traditional secretase pathways to produce heterogeneous extracellular plaques and altered extracellular signaling pathways. (3) Intracellular variants may also accumulate without the need for secretase cleavage. The accumulation of intraneuronal Aβ variants likely increases cellular (oxidative) stress, leading to an increase in DNA strand breaks, the insertion of gencDNAs, and the production of variants, creating a feed-forward loop that promotes a disease. (4) Variant proteins may also act as “seeds,” which alter the conformation of wild-type APP or other gencDNA variant proteins to create more aggregates. These may be propagated from cell-to-cell and cause prion-like transfer and neurodegeneration through the possible involvement of prion protein receptors (PrP). (5) Various stimuli (e.g., APOE4, infection, and cellular stress) could promote SGR via actions at multiple steps, including increased APP transcription.