
Figure 1: Transcription regulation induced by hypoxia.
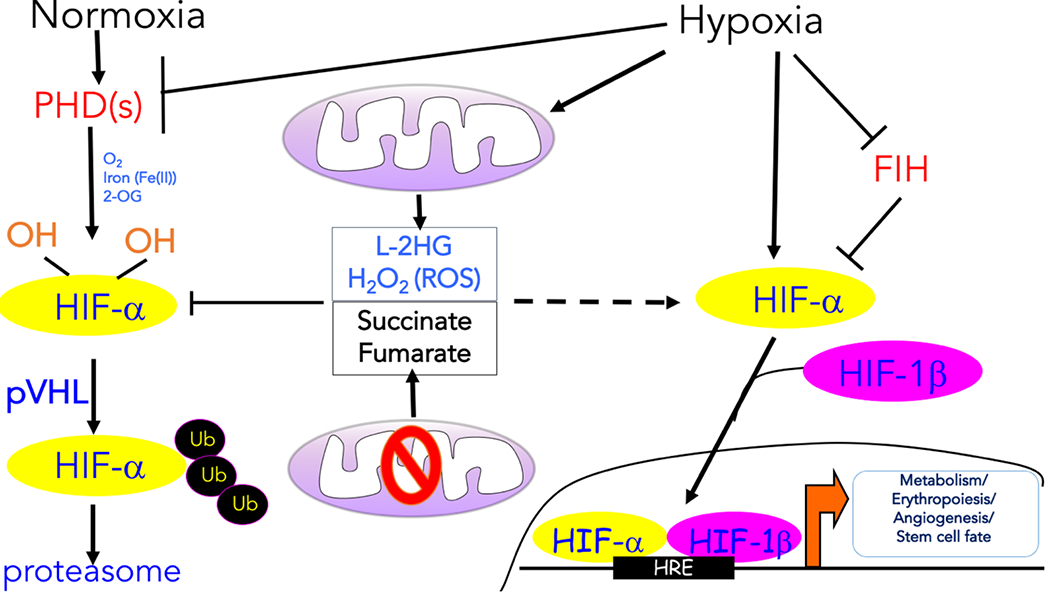
The oxygen-dependent interaction between the hypoxia inducible transcription factor α (HIF-α) subunits and the von Hippel–Lindau (VHL) tumour suppressor protein (pVHL) complex requires hydroxylation of two HIF-α proline residues by a family of α-ketoglutarate -dependent dioxygenases termed prolyl hydroxylases (PHDs), which requires oxygen (O2), iron (Fe2+), and α-ketoglutarate to function. Following hydroxylation, HIF-α subunits are polyubiquitylated by pVHL and targeted for proteasomal degradation. Hypoxia prevents the hydroxylation of the HIF-α protein subunits and their ubiquitin-mediated proteasomal degradation. As a result, the HIF-α protein subunits are allowed to dimerize with the HIF-1β protein subunits to form transcriptionally active complexes that bind to hypoxia response elements (HREs) to coordinate the induction of a large network of genes involved in metabolism, erythropoiesis, angiogenesis, and cell fate. Factor inhibiting HIF-1 (FIH-1) hydroxylates HIF-α subunits under normoxia to prevent recruitment of the transcription coactivators. Various mitochondrial products can also influence the hypoxic response. The production of reactive oxygen species (ROS) by mitochondrial complex III and L-2-hydroxyglutarate (L-2HG) under hypoxia can promote the stabilization of HIF-α protein levels. ROS likely inhibit PHDs by Cys oxidation, while L-2HG competes with α-ketoglutarate. Mutations in tricarboxylic acid (TCA) cycle components result in the accumulation of succinate and fumarate, which also inhibit PHD activity by competing with α-ketoglutarate, thereby causing an accumulation of the HIF-α protein even under normoxia.