

Abstract
Intrinsically disordered proteins (IDPs) constitute a significant fraction of eukaryotic proteomes. High-resolution characterization of IDP conformational ensembles can help elucidate their roles in a wide range of biological processes but remains challenging both experimentally and computationally. Here, we present a generic algorithm to improve the accuracy of coarse-grained IDP models using a diverse set of experimental measurements. It combines maximum entropy optimization and least squares regression to systematically adjust model parameters and improve the agreement between simulation and experiment. We successfully applied the algorithm to derive a transferable force field, which we term as MOFF, for de novo prediction of IDP structures. Statistical analysis of force field parameters reveals features of amino acid interactions not captured by potentials designed to work well for folded proteins. We anticipate its combination of efficiency and accuracy will make MOFF useful for studying the phase separation of IDPs, which drives the formation of various biological compartments.
Graphical Abstract
Introduction
The classical structure-function paradigm suggests that proteins must fold into unique three-dimensional conformations to perform their functions within the cell.1,2 It has helped establish conceptual frameworks that are instrumental in understanding crucial biological features such as the enzymatic activity3 and protein evolution.4 However, recent evidence suggests that 30 to 40% of eukaryotic proteomes contain disordered regions that do not fold into defined tertiary structures.5 These so-called intrinsically disordered proteins (IDPs), instead, function with an ensemble of different conformations. They can promote liquid-liquid phase separation via multivalent interactions and are critical for the formation of cellular compartments such as stress granules, P granules, super-enhancers, and heterochromatin.6–10 It has been argued that disorderness and multivalent interactions are indeed evolutionarily advantageous for performing increasingly more complicated tasks in higher-order organism.11 An appreciation of this emerging disorder-function relationship for IDPs requires a detailed characterization of their structures and the set of physicochemical interactions that dictate their organization.12–18
Though significant progress has been made, high-resolution structural characterization remains challenging for IDPs. In particular, traditional experimental techniques that found great success with folded proteins, such as X-Ray scattering and Cryo-electron microscopy, face difficulty in resolving the fuzzy conformation of IDPs.19–21 The two popular techniques that are often used for studying IDPs are small-angle X-ray scattering (SAXS) and Förster resonance energy transfer (FRET).22–25 Both are low-resolution approaches that fall short at providing detailed atomic structures. Recent developments in atomistic force fields have significantly improved the accuracy of all-atom computer simulations, rendering in silico prediction a promising approach to characterize the ensemble of IDP structures.26–28 The computational cost associated with atomistic simulations, however, limits their application to single and small proteins.
For more efficient simulations of IDPs, numerous groups have developed coarse-grained protein models.29–37 For example, the hydrophobicity scale model, introduced by Mittal and coworkers,29–31 treats each residue as a single particle. Non-bonded interactions between the coarse-grained particles were parameterized based on amino acid hydrophobicity to reproduce protein radius of gyrations. The Thirumalai group adopted a self-organized polymer to model IDPs with two beads per amino acid.34 Using the rescaled Betancourt-Thirumalai statistical potential38 for amino acid interactions, they showed that the model can delineate the complex interplay between protein sequence and structure. Additionally, Papoian and coworkers generalized the associative memory, water-mediated, structure and energy model (AWSEM) model developed by the Wolynes group36,39,40 for IDPs by reweighting the strength of secondary structure potentials and introducing a novel functional form to control protein size.33 Finally, in the model, ABSINTH, introduced Pappu and coworkers, interactions between amino acids are modeled with atomistic detail and solvent effect is accounted for implicitly with a novel mean-field scheme.41 The expanded range of accessible timescales in these models is valuable for an exhaustive sampling of protein conformation and computing thermodynamic quantities.
Algorithms that can systematically parameterize coarse-grained models are of great interest and can further improve the accuracy of existing IDP models. Optimization methods based on the energy landscape theory,42–46 which worked well for deriving force fields of globular proteins by maximizing the energy gap between folded and unfolded configurations,47,48 are not applicable because IDPs, by definition, lack a unique folded structure. An alternative approach to further refine the accuracy of computational models for a specific protein is to incorporate additional experimental measurements.49–51 For example, in Ref. 52, we demonstrated that a maximum entropy based algorithm can significantly improve the agreement between simulated and experimental small-angle X-ray scattering profiles by adding a linear biasing term to the model’s energy function. An obvious drawback of this algorithm is that it cannot be applied to proteins for which no experimental data is available.
In this paper, we generalize the maximum entropy algorithm introduced by us52 to parameterize a transferable force field for IDPs, which we term as maximum entropy optimized force field or MOFF. Instead of optimizing the energy gap as in folded proteins, the algorithm strives to reproduce the energy distribution for an ensemble of IDP configurations obtained from maximum entropy biasing. It involves iterations of maximum entropy optimization to derive biasing energies that reproduce experimental inputs, and least-squares fitting that converts the biasing energies into force field corrections. We demonstrate that MOFF is transferable and can be applied to predict protein structures de novo. Analysis of this force field further reveals amino acid interaction patterns not captured by statistical potentials that work well for folded proteins, explaining their lack of transferability for studying IDPs. As its quality can be continuously improved with the incorporation of more proteins and experimental measurements during parameterization, we anticipate MOFF to be useful for studying a wide range of problems related to IDPs.
Methods
Algorithm for force field parameterization
Numerous computational models and force fields have been proposed for IDPs, and they have provided molecular insight into a wide range of biological processes.29–37 Parameters in these models were often derived with phenomenological approaches based on some biophysical properties and statistics of amino acids derived from folded proteins. Systematically improving upon them to provide a better characterization of IDPs remains non-trivial. In the following, we present an algorithm to refine IDP force fields for better agreement with experimental measurements. The hydrophobic scale (HPS) model introduced by Mittal and coworkers30 was used as an example for illustration purposes, but the algorithm is general and can be directly applied to other force fields as well.
Our goal is to fine-tune force field parameters to improve the agreement between the simulated and experimental radius of gyrations (Rg) of IDPs. Here, we focus on Rg due to its availability for a broad set of proteins, but other experimental measurements can be incorporated as well. A promising approach for improving the agreement between simulation and experiment is to introduce maximum entropy biases to computational models.49–51,53 The maximum entropy approach has gained wide popularity and been applied to a variety of problems due to its simplicity and fundamental connection to the information theory.52,54–58 It suggests that for a given protein, to ensure that the Rg of simulated structures matches the experimental value, a linear bias should be added to the model Hamiltonian
| (1) |
where r represents protein configurations and UHPS(r) is the energy function defined by the hydrophobicity scale model.30
The maximum entropy approach can be applied to any protein and is guaranteed to reproduce experimental measurements. Its main drawback is that the resulting energy function is not transferable and a unique biasing energy αRg(r) needs to be determined for every protein of interest. Such repeated parameterization can be cumbersome and becomes impractical for proteins with no experimental input. We introduce the maximum entropy optimized force field (MOFF) for transferable modeling of IDPs
| (2) |
where I and J indexes over different amino acid types. counts the number of contacts between amino acid types I and J within a protein configuration r. The contact function between a pair of amino acids i and j separated at a distance rij is defined as
| (3) |
with ro = 8 Å and η = 0.7 Å−1. ϵIJ corresponds to the energetic cost for contact formation and their values will be derived from maximum entropy biases defined in Eq. 1 by solving the following set of linear equations
| (4) |
A total of M structures for a given protein can be used to provide a large set of variation of contacts for an efficient determination of the parameters.
To derive a transferable force field, however, several technical aspects need to be addressed. First, Eq. 4 needs to be generalized to include multiple proteins to ensure the transferability of ϵIJ. Second, there is an inherent conflict in Eq. 4, as the left and right side reaches 0 at different points. By definition, when there are no contacts, the left side of the equation is 0. However, in this limit, Rg is maximized, and the right side of the equation is at an extremum. We can resolve this apparent conflict by subtracting a reference point energy —the biasing energy at the experimental value—from both sides of the equation. This subtraction is justified by the realization that only energy differences are needed to reproduce the structural ensemble and the mean Rg, while the absolute energies are of no particular significance. Following Eq. 4, on the left hand side, we further converted the biasing energy into a linear combination of pair-wise contacts . can be estimated using simulated protein structures with Rg values that are within 0.5 Å of the experimental measurement. The small deviation 0.5 Å was introduced to account for experimental error and to ensure statistical convergence with enough protein structures. Third, the quality of the force field depends on the assumption used in deriving Eq. 4 that the biasing energies can be approximated with a linear combination of pairwise contacts. The validity of this assumption may be poor, especially at large biasing energies. Accuracy of the linear approximation can be systematically improved by an iterative procedure in which we find maximum entropy biasing energies to the newly derived MOFF and use them to update force field parameters further. As the difference between model predictions and experimental measurements drops, the strength of the biasing energies will decrease, and the quality of the linear approximation will improve.
With all these factors accounted for, we solve the following equations iteratively to derive parameters for MOFF
| (5) |
where n and m indexes over different proteins and the structures for a given protein. An illustration of the algorithm is provided in Figure 1.
Figure 1:
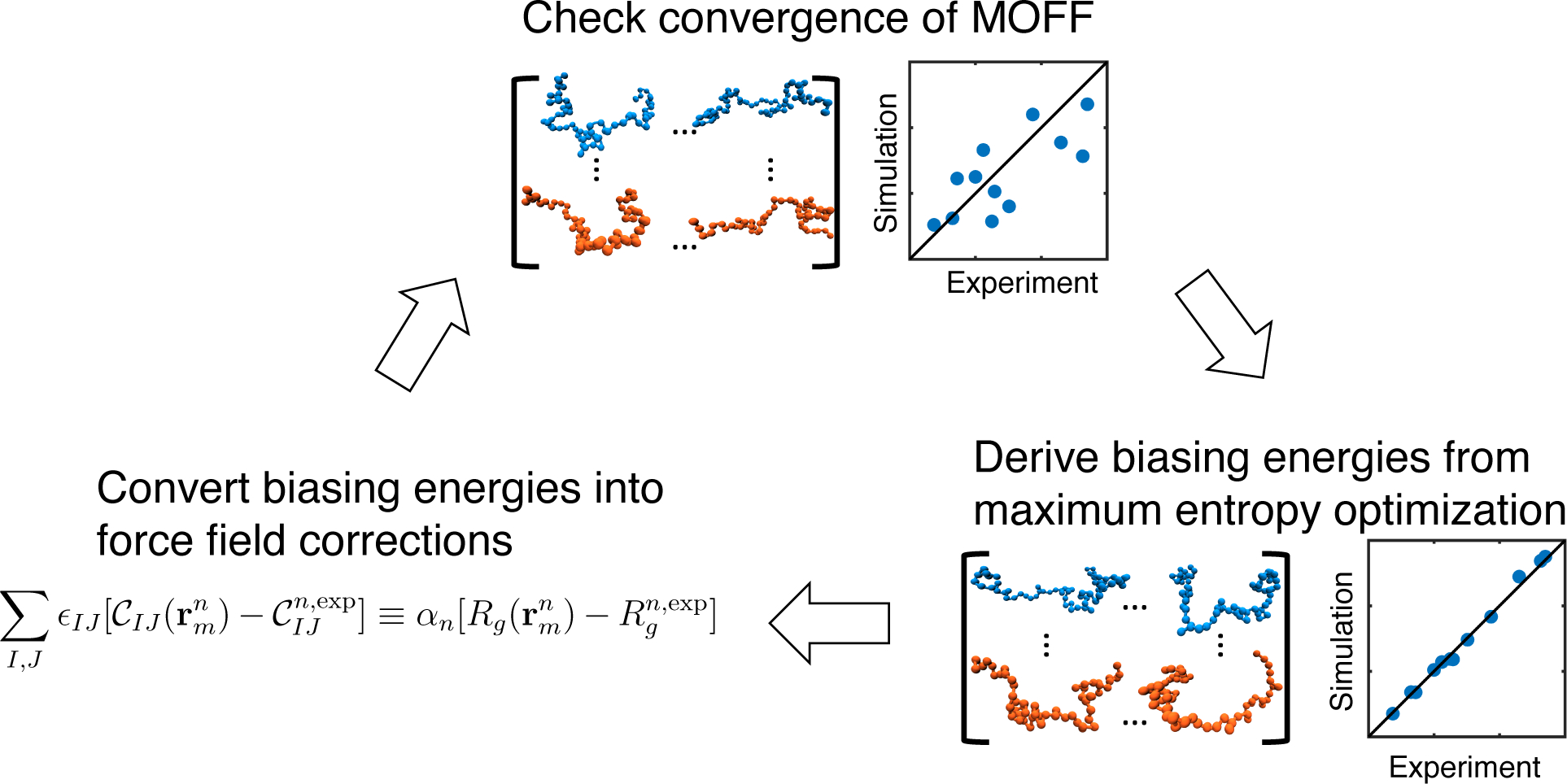
Illustration of the iterative algorithm for coarse grained force field optimization.
Solution to Eq. 5 can be found with least squares regression by recasting the equation in matrix form as
| (6) |
where ϵ, C, and Rg are matrix versions of the contact energy, contact number, and bias energy respectively. We then solve for using least squares as
| (7) |
To reduce statistical noise that originates from a finite sample of protein sequences and structures, we reconstructed CT C with the largest 60% eigenvalues before calculating its inverse.
Hydrophobic scale model of IDP
As aforementioned, we used the hydrophobicity scale model as the starting point for our force field refinement.29–31 Protein molecules in this model are described at the residue level, with one bead for each α-carbon. Interactions between amino acids were parameterized based on their hydrophobicity with a multiplicative scaling factor.
The energy function of this model consists of three terms and
| (8) |
Ub(r) is the bonding energy between adjacent amino acids and is modeled using a harmonic potential with a spring constant of 10 kJ/mol/Å2 and a bond length of 3.8 Å. Ue(r) corresponds to electrostatic interactions between charged residues modeled at the Debye-Hückle level. We used = 80 for the dielectric constant of water, and the ionic strength was set individually for each protein to match the corresponding experimental values (see Tables 1 and 2). Masses and charges of the amino acids can be found in Table S1.
Table 1:
List of proteins used for force field parameterization.
| Protein | Sequence length | Ionic Strength (mM) | a | Experimental Method |
|---|---|---|---|---|
| CspTm83 | 67 | 42 | 1.37 (0.07) | FRET |
| IN83 | 60 | 50 | 2.25 (0.11) | FRET |
| ProTα-N83 | 112 | 42 | 2.87 (0.14) | FRET |
| ProTα-C83 | 129 | 42 | 3.70 (0.19) | FRET |
| R1563 | 114 | 128 | 1.72 (0.09) | FRET |
| R1763 | 100 | 128 | 2.29 (0.11) | FRET |
| hCyp63 | 167 | 85 | 2.00 (0.10) | FRET |
| Protein-L84 | 64 | 128 | 1.65 (0.10) | FRET |
| ACTR85 | 71 | 199 | 2.51 (0.13) | SAXS |
| hNHElcdt85 | 131 | 199 | 3.63 (0.18) | SAXS |
| sNase86 | 136 | 117 | 2.12 (0.10) | SAXS |
| α-synuclein87 | 140 | 156 | 3.3 (0.3) | FRET |
Values in parentheses correspond to standard deviations.
Table 2:
List of proteins used for testing force field transferability.
Unb(r) describes nonbonded energies, and is the sum of the contact energies between all pairs of amino acids i and j separated at a distance rij, given by . Instead of the Ashbaugh-Hatch functional,30 we utilized a dampened step function with the following form
| (9) |
The first term with ϵ0 = 1.67772 × 107 kJ/mol Å12 accounts for excluded volume. measures the strength of the pairwise contact energies and I and J correspond to the amino acid types of residue i and j. Parameters in (Eq. 3) were chosen such that Vnb(rij) approximates the shape of the Lennard-Jones potential (see Figure S1). This new potential form separates out the repulsive core from specific amino acid interactions such that the model contact energies can be directly compared with the correction terms introduced in Eq. 2.
Details on computer simulations
At each iteration of the force field optimization algorithm (Figure 1), we carried out two set of replica exchange molecular dynamics simulations for each protein using UMOFF(r) and UME(r), respectively. The amino acid contact energies in UMOFF(r) were set as zero in the first iteration. We note that, except for the first iteration, UME(r) is defined differently from Eq. 1 and refers to the energy function that corrects the current version of MOFF, and
| (10) |
The biasing strength α was fine tuned manually for each protein to ensure that the average Rg for simulated protein structures is within 0.5 Å of the experimental value.
All simulations were carried out using the GROMACS software package,60 with the PLUMED plugin to incorporate biasing energies.61 Simulations were initialized from protein structures predicted by I-TASSER62 using the corresponding amino acid sequences. Each simulation trajectory consists of a total of six replicas with temperatures at 300K, 320K, 340K, 360K, 380K, and 400K, and swaps between replicas were attempted at every 100 steps. Langevin dynamics were used to control the temperature with a coupling constant of 1 ps. Proteins were placed inside cubic boxes with sides five times the maximum length of the protein. The trajectories lasted for 4 × 107 steps with a timestep of 10 fs. The first 107 steps were discarded as equilibration. We collected protein configurations at every 2000 steps for a total of 15,000 structures. Combining the two trajectories simulated with the two energy functions results in a total of 30,000 structures for each protein. To validate the convergence of our simulations, we repeated the simulations with UMOFF(r) five times in the first and final iteration and did not observe statistically significant changes in the predicted Rg.
Results and Discussion
Illustration of force field parameterization with a single protein
To illustrate key concepts in the algorithm for force field optimization, we applied it to a single protein, R15. As shown in Figure 2A (blue line), UHPS(r) overpredicts Rg with a mean of 2.23 nm compared to 1.72 nm determined by FRET.63 We then carried out the maximum entropy optimization to derive UME(r), which succeeded in reproducing Rg as shown by the orange line. Finally, we derived a force field for this individual protein, UR15(r), by solving Eq. 5 with 30,000 structures collected from the two simulations for UHPS(r) and UME(r). As shown in green, the new force field predicts a mean Rg of 1.66 nm, which is in much better agreement with the experimental value than that predicted by UHPS(r). Its variance also closely matches that from UME(r).
Figure 2:
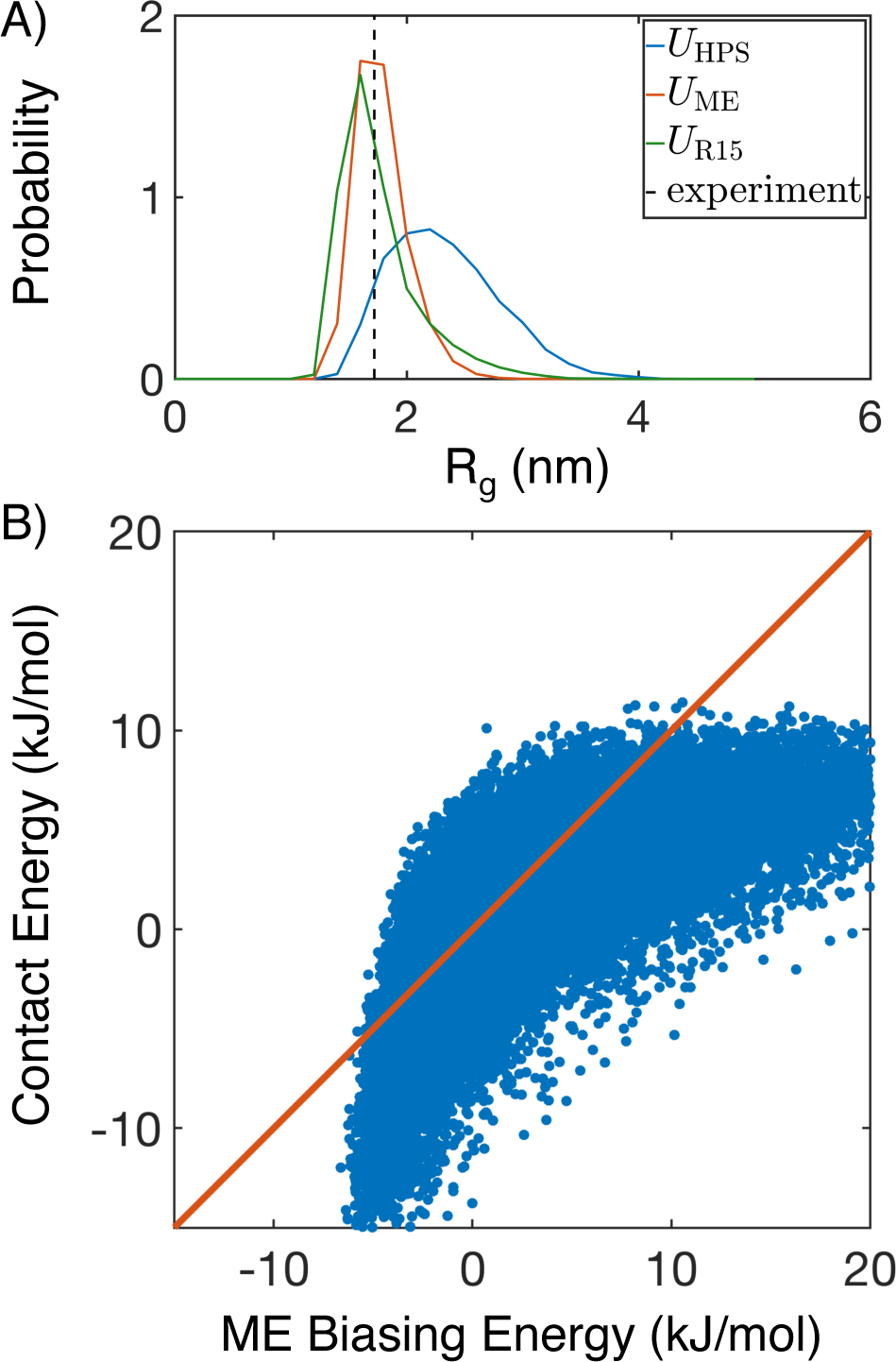
Illustration of the force field parameterization algorithm on a single protein, R15. (A) Comparison between the mean experimental value (black, dashed) for Rg and probability distributions obtained from simulating the hydrophobic scale model (UHPS(r), blue), the maximum entropy optimized model (UME(r), orange), and MOFF for this specific protein (UR15(r), green). (B) Correlation between contact energies obtained from linear fitting and the original maximum entropy biasing energy . The diagonal line (orange) is provided as a guide to the eye.
We then examined the accuracy of the linear approximation used in Eq. 5 by plotting the contact energies against the bias energies for all the 30,000 structures in Figure 2B. Though the two energies are correlated, it is evident that the linear fit is not perfect, supporting the use of an iterative algorithm for further improvement. The observed oblong shape results from different physical limits on protein size and contacts. For example, fully collapsed structures have a well defined Rg value that further gives rise to a lower bound on the biasing energy (−6 kJ/mol). On the other hand, physical contacts between residues can vary substantially without changing protein size, leading to large variations in contact energies. Similarly, for expanded configurations, the contacts will disappear first before proteins become fully stretched to reach a maximum in Rg, resulting in the heterogeneity in biasing energies at a relatively constant contact energy of 10 kJ/mol.
We further applied UR15(r) to other proteins as a test of its transferability. As shown in Figure S2, it performs worse than UHPS(r) and tends to overcollapse the proteins. This is not too surprising as the interaction energies determined for a single protein are not guaranteed to provide a good description of IDPs in general.
Parameterization and validation of MOFF for IDPs
To parameterize a transferable force field capable of modeling IDP structures, we carried out the iterative optimization algorithm over a total of twelve disordered proteins studied in Ref. 30 (Table 1). As a quantitative evaluation of the force field performance, we define
| (11) |
to measure the differences between simulated and experimental Rg. σi is the experimental standard deviation of Rg for protein i, and the sum is taken over all proteins from the training set.
A total of 15 iterations was performed to determine the final set of parameters for MOFF. As the iterations progress, χ2 declined rapidly, starting at 23.67 and reaching 3.50 by the end (Figure 3A). Rg values predicted by the converged MOFF are shown in Figure 3B, along with the results from UHPS(r). The significant improvement of MOFF over the hydrophobic scale model is encouraging and supports the usefulness of our iterative algorithm in developing more accurate protein force fields. Next, we evaluated the performance of MOFF on six additional proteins not included in our training set to test its transferability. These proteins were studied by Thirumalai and coworkers with a different model (Table 2).34
Figure 3:
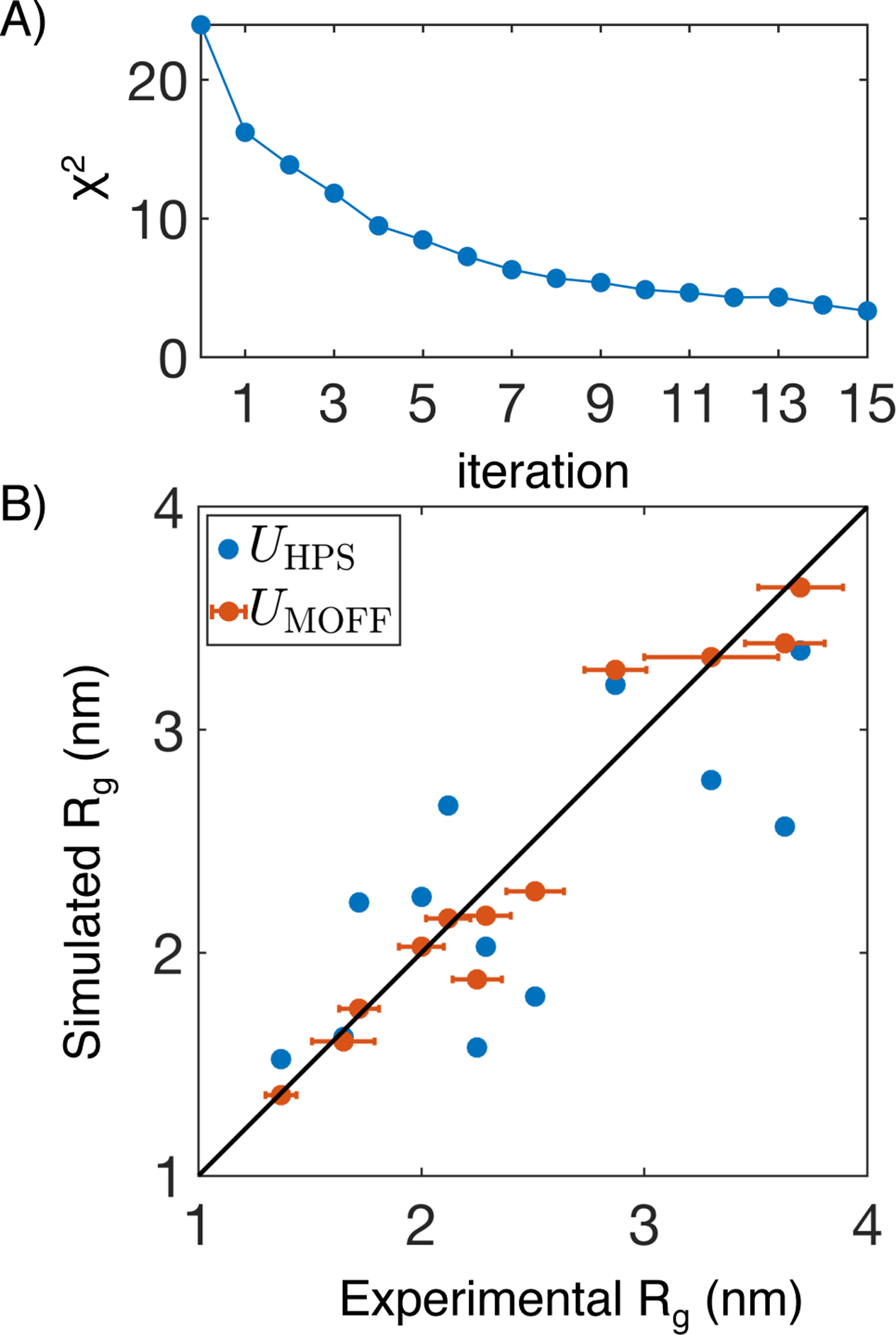
MOFF improves the prediction of Rg for proteins includes in the training set. (A) χ2 that measures the normalized difference between simulated and experimental Rg values as a function of the number of iterations for force field optimization. (B) Comparison between experimental Rg and those predicted by the hydrophobic scale model (UHPS(r), blue) and MOFF (UMOFF(r), orange). Error bars represent experimental standard deviations. Simulation errors are comparable to the symbol size.
As shown in Figure 4, MOFF significantly improves the prediction accuracy for these new proteins as well. The total sum of absolute difference drops from 5.51 to 2.30 nm when compared with predictions by the hydrophobic scale model. The monotonic decrease of this difference along MOFF optimization (see Figure S3) supports the absence of overfitting for force field parameterization. It’s worth noting that the improvement exhibits heterogeneity and the result on ERM TADn is the least satisfying, as highlighted by the arrow in Figure 4. Analysis of the underlying protein sequence shows that ERM TADn lacks polar, uncharged amino acids that are prevalent in most IDPs (Figure S4A). These amino acids constitute a much higher fraction of the training set and more successful portions of the test set. Similarly, large, apolar amino acids are less frequent in the training set than in ERM TADn. The increased levels of hydrophobic residues coupled with the decreased levels of hydrophilic ones likely explains the more collapsed configuration of ERM TADn. Therefore, MOFF is transferable, but performance can potentially be further improved with a larger training set.
Figure 4:
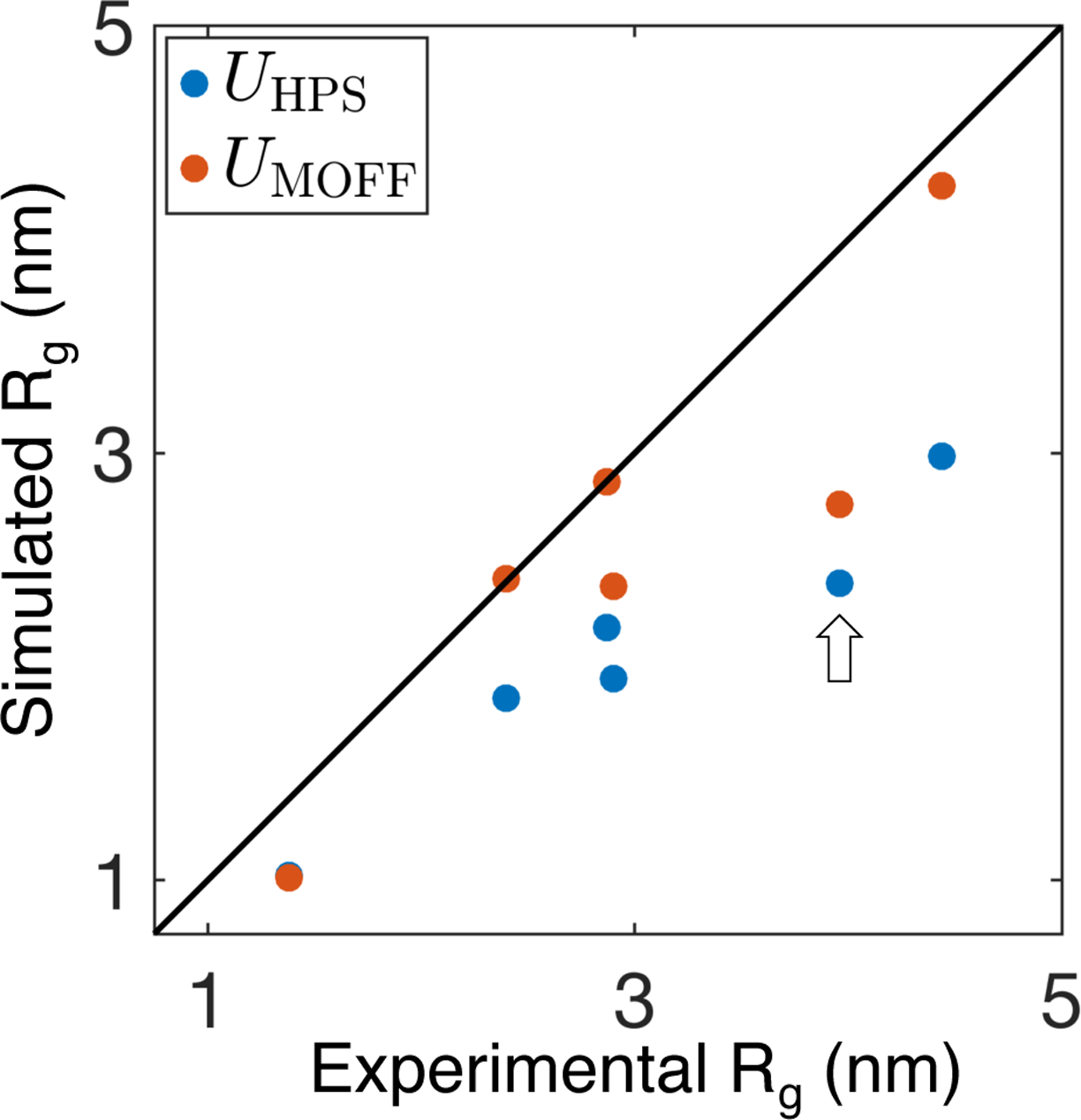
MOFF (orange) significantly improves over the hydrophobic scale model (blue) in predicting Rg for a test set of proteins not included in force field parameterization. The arrow highlights ERM TADn, which is discussed in the main text as a case where the improvement is less pronounced. Simulation errors are comparable to the symbol size.
Classification of amino acids based on MOFF interactions
Given the success of MOFF in predicting the size of IDPs, we wondered whether the derived interaction energies could provide physical insight into protein folding. When compared with the hydrophobic scale model, we found that MOFF significantly increases the contact energy between hydrophilic residues (Figure 5 and Figure S5), and even makes some of these interactions purely repulsive. A more repulsive force field is necessary to rescue the overcollapse of protein configurations observed in the hydrophobic scale model. As hydrophilic residues are over-represented in IDPs and our training set (Figure S4B),64–66 destabilizing the contacts among them is effective at expanding the configurations for most proteins.
Figure 5:
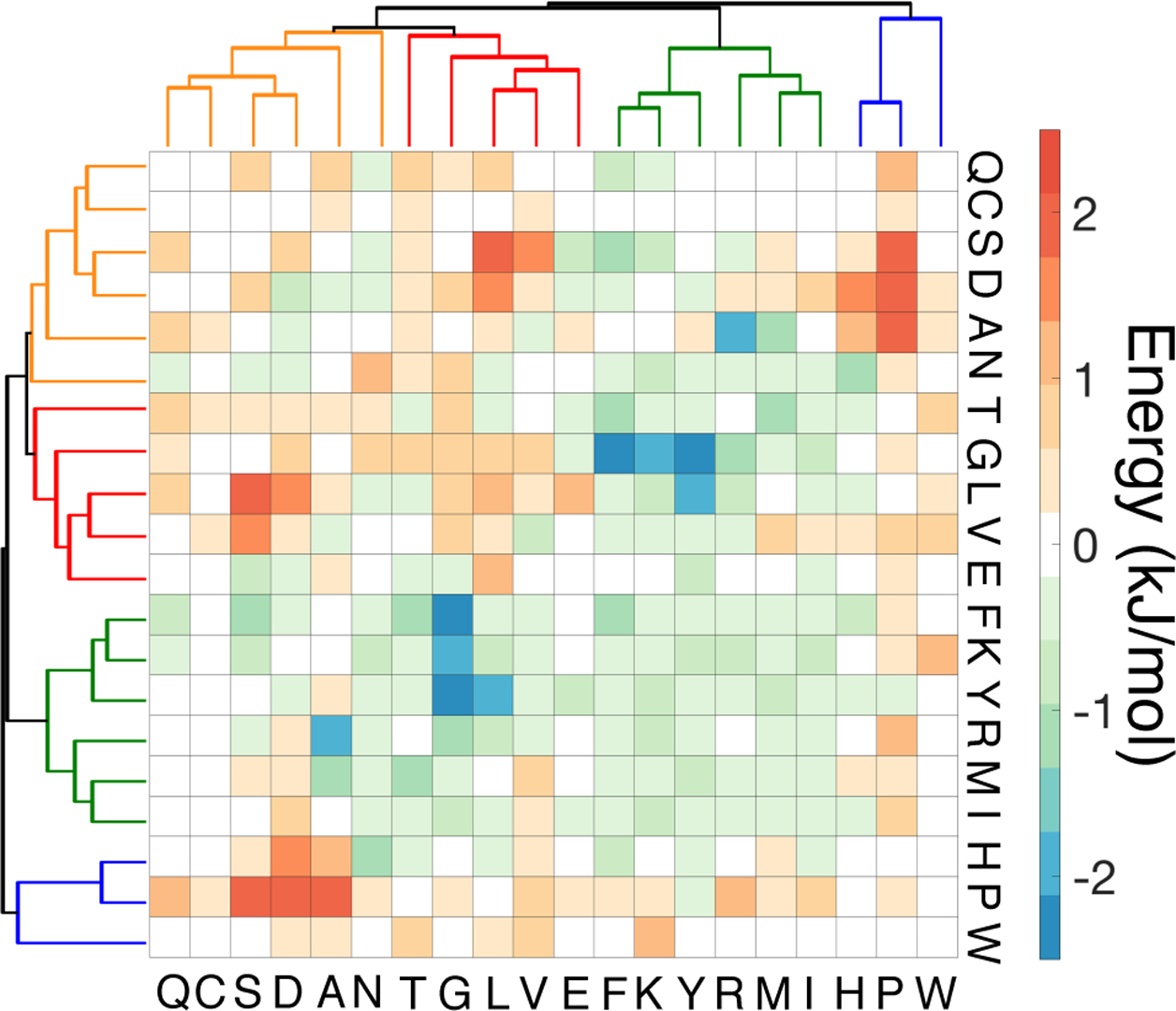
Hierarchical clustering of MOFF contact energies reveals the presence of four amino acid groups. The contact energies correspond to the sum of ϵIJ and defined in Eqs. 2 and 9 respectively. The energies between residues are shown from red (most repulsive) to blue (most attractive). The four groups are displayed as orange (GLN, CYS, SER, ASP, ALA, ASN), red (GLU, THR, GLY, LEU, VAL), green (PHE, LYS, TYR, ARG, MET, ILE), and blue (HIS, PRO, TRP), respectively.
To further untangle the complexity of the MOFF interaction matrix, we performed a hierarchical clustering to group together amino acids that share similar interaction patterns. Towards that end, we assigned each amino acid with a 20-element vector that represents its interaction energy with other residues. Distances between amino acids were then defined using the correlation coefficients of corresponding vectors. After computing the proximity between amino acids, we grouped them into four clusters using the linkage function implemented in MATLAB.67 As shown in Figure 5, hydrophilic and hydrophobic residues are generally separated from each other, partitioned into cluster 1–4 (orange and blue) and cluster 2–3 (red and green), respectively. We note that these four clusters differ from the amino acid groups derived from the analysis of the Miyazwa-Jernigan (MJ) potential that has been widely used for studying folded proteins.68–70 As shown in Table 3, MJ groups clearly separate amino acids based on hydrophobicity, but the charged residues (GLU, ARG, and LYS) are assigned into the two primarily hydrophobic clusters 2–3 by MOFF. A possible explanation for the more scattered distribution of residues in MOFF clusters is that IDPs lack a well-defined hydrophobic core and adopt more extended conformations than well-folded proteins. The hydrophobic effect “learned” from IDP sequences, therefore, may be less prominent.
Table 3:
The five groups of amino acids derived from the Miyazwa-Jernigan potential.70
| Group 1 | CYS MET PHE ILE LEU VAL TRP TYR |
| Group 2 | ALA THR HIS |
| Group 3 | GLY PRO |
| Group 4 | ASP GLU |
| Group 5 | SER ASN GLN ARG LYS |
Next, we investigated whether this difference in amino acid clustering reflects a robust feature of IDPs or a result of statistical noise in parameterization. Towards that end, we introduced two new force fields denoted as UMOFF4(r) and UMJ5(r), respectively. These two force fields share the same mathematical expression as in Eq. 2, and they only differ in the amino acid clusters used in defining the second correction term. For UMOFF4, we partitioned the 20 amino acids into the four MOFF clusters shown in Figure 5 and restricted corrections in contact energies (ϵIJ) to be identical for amino acids within the same cluster. Similarly, for UMJ5, amino acids that fall into the same MJ group share the same force field corrections. We then followed the iterative procedure as before to derive force field parameters from the twelve training proteins. If the difference between the two amino acid groups is not significant but an artifact of the current optimization algorithm, we shall anticipate comparable performance from the two simplified models in simulating IDPs.
Remarkably, we found that UMOFF4(r) essentially reproduces the success of the full model, despite its use of a much smaller number of parameters (10 compared to 210). As shown in Figures 6A and S6, UMOFF4(r) describes the size of training proteins well, and the normalized difference between simulated and experimental Rg (χ2) decreases to 7.12 after 15 iterations. Furthermore, this new force field appears to be more transferable than our original one, and the absolute Rg difference for test proteins is 2.03 nm, smaller than 2.30 from UMOFF(r) (Figure 6B). On the other hand, UMJ5(r) performs significantly worse for both training and test proteins. The χ2 plateaus around 15.64 after 15 iterations (Figures 6C and S6), and the Rg difference for test proteins is 4.03 nm (Figure 6D). Close examination of the simulated results for training proteins suggests that this force field loses the specificity required to adjust the size of individual proteins. Instead, it generally increases the size of each protein, regardless of how the initial force field performs.
Figure 6:
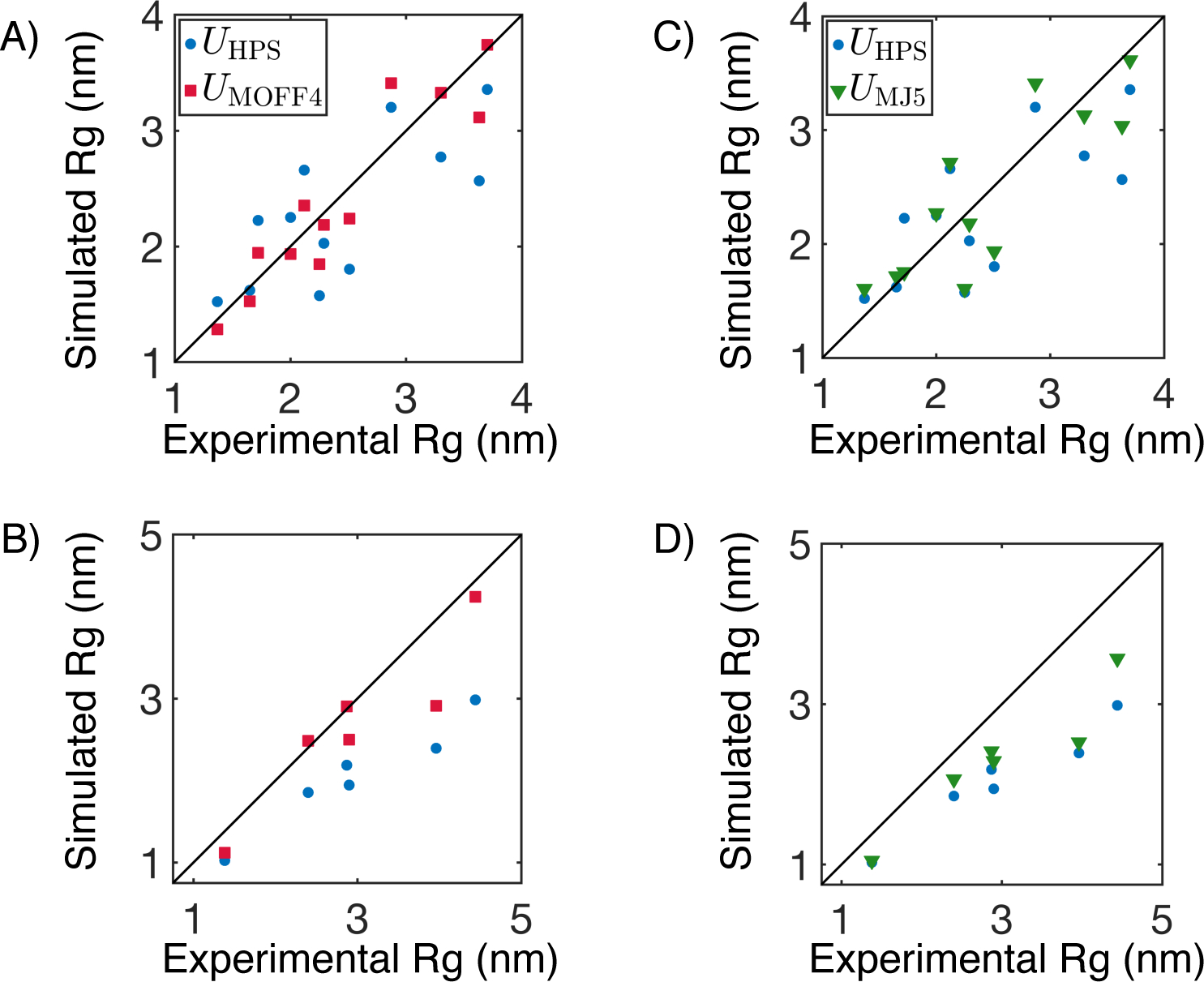
Comparison between the performance of two reduced models parameterized using amino acid clusters defined by MOFF (UMOFF4) or MJ potential (UMJ5). Simulated Rg on training and test proteins using UMOFF4 are compared with experimental values in parts A and B, respectively. The corresponding results for UMJ5 are shown in C and D. We also presented results from UHPS in blue for reference. Simulation errors are comparable to the symbol size.
The vast difference in the performance of the two simplified models strongly supports the biological importance of the four groups defined by the MOFF energies. It also highlights the challenge for transferring models that work well with folded proteins to IDPs. We note that the presence of two different amino acid clustering schemes does not necessarily suggest the existence of two separate folding codes for disordered and globular proteins. Instead, it may indicate that the protein sequences used in parameterization are not broad enough to cover the whole spectrum of biological diversity. As the size of a protein is dictated by the subtle balance of several factors, including hydrophobic effect, water-mediated interactions, etc., a limited coverage of the sequence space may result in over- or under-emphasis of one mechanism versus the other. Optimizing parameters using both disordered and ordered sequences will be crucial to ensure the consistency and transferability of computational models across protein families.
Conclusions
We presented an algorithm for parameterizing the force field of coarse-grained models. It is most effective at refining existing force fields while other top-down71,72 and bottom-up73,74 approaches are potentially more appropriate for deriving new force fields from the scratch.
Our algorithm improves force field quality by introducing additional correction terms to reproduce experimental data. It, therefore, differs from many approaches that directly adjust force field parameters.75,76 In particular, existing approaches often involve iterations of molecular dynamics simulations to compute ensemble averages and parameter fine tuning to improve the agreement between model and experiment. Modern force fields for protein molecules, especially those with atomistic details,26–28 often involve more than hundreds of parameters. Fine tuning them is, therefore, a challenging numerical problem in a high-dimensional space, and many iterations will be needed to reach convergence. Our algorithm breaks this difficult parameter search problem into two steps consisting of maximum entropy optimization and least squares regression. Instead of directly adjusting parameters of the original force field, we introduce correction terms that can be defined independently from these parameters, effectively reducing the complexity of the optimization. For example, parameters in the maximum entropy optimization step share the same number as experimental constraints, which is often much smaller than those involved in defining the force field, and can be determined very efficiently. The second step does not involve molecular dynamics simulations and can be solved with negligible computational cost. In its current version, we used linear regression to fit maximum entropy biases for its simplicity of implementation. Non-linear fitting, however, can be adopted straightforwardly to improve the accuracy of the second step. A more accurate regression can potentially abolish the need of iteration used in the current algorithm, further reducing its computational cost.
We applied the algorithm to parameterize an IDP model using the radius of gyration for a set of proteins. The resulting force field, which we term as MOFF, succeeded in de novo structural prediction for disordered proteins. Further analysis of interaction energies from MOFF suggested a classification of amino acids that differs significantly from the five types defined by the MJ potential, a model that has found great success for studying folded proteins. We demonstrated that this difference is not a result of statistical noise in parameters and is critical for the success of MOFF in modeling IDPs. Given its accuracy and efficiency, we anticipate MOFF to be useful for elucidating sequence-specific features of IDPs that drive their phase separation and improving our understanding of the emerging disorder-function paradigm.
We note that the quality of the force field introduced here can be continuously improved. For example, due to the limited availability of experimental data, the number of proteins included in the training and test set is small. Increasing the size of the training set will, in general, lead to better force field accuracy and transferability. Furthermore, though we mainly used the radius of gyration derived from SAXS and FRET as constraints, the general framework for maximum entropy biasing makes the incorporation of data from other types of experiments such as nuclear magnetic resonance straightforward. Finally, experimental data are noisy and their interpretation is not always straightforward. Using more rigorous protocols to process experimental measurements77–79 and accounting for errors in these data with a Bayesian optimization approach80–82 are important future directions to further improve force field quality.
It is worth pointing out that while we restricted the discussion to disordered proteins, one can incorporate well-folded proteins into the training set for parameter optimization as well. Deriving a consistent force field for both disordered and globular proteins has been challenging. A simultaneous optimization for both folded and disordered proteins using the algorithm outlined here would be an exciting future direction. It may shed light on general principles of protein folding by resolving the difference in amino acid clustering derived from MOFF and MJ potential.
Supplementary Material
Acknowledgement
This work was supported by the National Institutes of Health (Grant 1R35GM133580-01). A.L. was supported by the National Science Foundation Graduate Research Fellowship Program.
Footnotes
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website.
• Illustration of new energy function; Test of UR15 to our test set; Amino acid frequency data; Illustration of UHPS contact energies; χ2 from optimization of UMOFF4 and UMJ5; Sequences and experimental details of test and training proteins; Amino acid dependent parameters in UMOFF; Amino acid groups from the Miyazwa-Jernigan potential.
References
- (1).Lodish H; Berk A; Kaiser CA; Matsudaira P; Krieger M; Scott MP; Darnell J; Others, Molecular Cell Biology; Freeman WH, 2004. [Google Scholar]
- (2).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The protein data bank. Nucleic Acids Res 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Rodrigues RC; Ortiz C; Berenguer-Murcia A; Torres R; Fernández-Lafuente, R.´ Modifying enzyme activity and selectivity by immobilization. Chem. Soc. Rev 2013, 42, 6290–6307. [DOI] [PubMed] [Google Scholar]
- (4).DePristo MA; Weinreich DM; Hartl DL Missense meanderings in sequence space: A biophysical view of protein evolution. Nat. Rev. Genet 2005, 6, 678–687. [DOI] [PubMed] [Google Scholar]
- (5).Ward JJ; Sodhi JS; McGuffin LJ; Buxton BF; Jones DT Prediction and Functional Analysis of Native Disorder in Proteins from the Three Kingdoms of Life. J. Mol. Biol 2004, 337, 635–645. [DOI] [PubMed] [Google Scholar]
- (6).Kroschwald S; Munder MC; Maharana S; Franzmann TM; Richter D; Ruer M; Hyman AA; Alberti S Different Material States of Pub1 Condensates Define Distinct Modes of Stress Adaptation and Recovery. Cell Rep 2018, 23, 3327–3339. [DOI] [PubMed] [Google Scholar]
- (7).Brangwynne CP; Eckmann CR; Courson DS; Rybarska A; Hoege C; Gharakhani J; Julicher F; Hyman AA Germline P Granules Are Liquid Droplets That Localize by Controlled Dissolution/Condensation. Science 2009, 324, 1729–1732. [DOI] [PubMed] [Google Scholar]
- (8).Sabari BR et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018, 361, eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Larson AG; Narlikar GJ The Role of Phase Separation in Heterochromatin Formation, Function, and Regulation. Biochemistry 2018, 57, 2540–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Strom AR; Emelyanov AV; Mir M; Fyodorov DV; Darzacq X; Karpen GH Phase separation drives heterochromatin domain formation. Nature 2017, 547, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Gao A; Shrinivas K; Lepeudry P; Suzuki HI; Sharp PA; Chakraborty AK Evolution of weak cooperative interactions for biological specificity. Proc. Natl. Acad. Sci. U.S.A 2018, 115, 201815912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Van Der Lee R et al. Classification of intrinsically disordered regions and proteins. Chem. Rev 2014, 114, 6589–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Jacobs WM; Frenkel D Phase Transitions in Biological Systems with Many Components. Biophys. J 2017, 112, 683–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Boeynaems S; Alberti S; Fawzi NL; Mittag T; Polymenidou M; Rousseau F; Schymkowitz J; Shorter J; Wolozin B; Van Den Bosch L; Tompa P; Fuxreiter M Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol 2018, 28, 420–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Conicella AE; Zerze GH; Mittal J; Fawzi NL ALS Mutations Disrupt Phase Separation Mediated by α-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 2016, 24, 1537–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Burke KA; Janke AM; Rhine CL; Fawzi NL Residue-by-Residue View of In Vitro FUS Granules that Bind the C-Terminal Domain of RNA Polymerase II. Mol. Cell 2015, 60, 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Shakya A; King JT DNA Local-Flexibility-Dependent Assembly of Phase-Separated Liquid Droplets. Biophys. J 2018, 115, 1840–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Elbaum-Garfinkle S; Kim Y; Szczepaniak K; Chen CC-H; Eckmann CR; Myong S; Brangwynne CP The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc. Natl. Acad. Sci. U.S.A 2015, 112, 7189–7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wright PE; Dyson HJ Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol 2015, 16, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Habchi J; Tompa P; Longhi S; Uversky VN Introducing Protein Intrinsic Disorder. Chem. Rev 2014, 114, 6561–6588. [DOI] [PubMed] [Google Scholar]
- (21).Oldfield CJ; Dunker AK Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions. Annu. Rev. Biochem 2014, 83, 553–584. [DOI] [PubMed] [Google Scholar]
- (22).Bernadó P; Svergun DI Analysis of intrinsically disordered proteins by small-angle X-ray scattering. Methods Mol. Biol 2012, 896, 107–122. [DOI] [PubMed] [Google Scholar]
- (23).Putnam CD; Hammel M; Hura GL; Tainer JA X-ray solution scattering (SAXS) combined with crystallography and computation: Defining accurate macro-molecular structures, conformations and assemblies in solution. Q. Rev. Biophys 2007, 40, 191–285. [DOI] [PubMed] [Google Scholar]
- (24).Schuler B; Sonja M; Soranno A; Nettels D Intrinsically Disordered Protein Analysis; 2012; Vol. 895; pp 21–45. [Google Scholar]
- (25).Leblanc SJ; Kulkarni P; Weninger KR Single molecule FRET: A powerful tool to study intrinsically disordered proteins. Biomolecules 2018, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Robustelli P; Piana S; Shaw DE Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. U.S.A 2018, 115, E4758–E4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Best RB; Zheng W; Mittal J Balanced Protein-Water Interactions Improve Properties of Disorderd Proteins and Non-Specific Protein Association. J. Chem. Theory Comput 2014, 10, 5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmüller H; MacKerell AD CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Dignon GL; Zheng W; Best RB; Kim YC; Mittal J Relation between single-molecule properties and phase behavior of intrinsically disordered proteins. Proc. Natl. Acad. Sci. U.S.A 2018, 201804177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Dignon GL; Zheng W; Kim YC; Best RB; Mittal J Sequence determinants of protein phase behavior from a coarse-grained model. PLOS Comput. Biol 2018, 14, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Dignon GL; Zheng W; Kim YC; Mittal J Temperature-Controlled Liquid-Liquid Phase Separation of Disordered Proteins. ACS Cent. Sci 2019, 5, 821–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Das S; Amin AN; Lin YH; Chan HS Coarse-grained residue-based models of disordered protein condensates: Utility and limitations of simple charge pattern parameters. Phys. Chem. Chem. Phys 2018, 20, 28558–28574. [DOI] [PubMed] [Google Scholar]
- (33).Wu H; Zhao H; Wolynes PG; Papoian GA AWSEM-IDP: A Coarse-Grained Force Field for Intrinsically Disordered Proteins. J. Phys. Chem. B 2018, 122, 11115–11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Baul U; Chakraborty D; Mugnai ML; Straub JE; Thirumalai D Sequence Effects on Size, Shape, and Structural Heterogeneity in Intrinsically Disordered Proteins. J. Phys. Chem. B 2019, 123, 3462–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Cragnell C; Rieloff E; Skepö M Utilizing Coarse-Grained Modeling and Monte Carlo Simulations to Evaluate the Conformational Ensemble of Intrinsically Disordered Proteins and Regions. J. Mol. Biol 2018, 430, 2478–2492. [DOI] [PubMed] [Google Scholar]
- (36).Lin X; Roy S; Jolly MK; Bocci F; Schafer NP; Tsai MY; Chen Y; He Y; Grishaev A; Weninger K; Orban J; Kulkarni P; Rangarajan G; Levine H; Onuchic JN PAGE4 and Conformational Switching: Insights from Molecular Dynamics Simulations and Implications for Prostate Cancer. J. Mol. Biol 2018, 430, 2422–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ramis R; Ortega-Castro J; Casasnovas R; Marino L; Vilanova B; Adrover M; Frau J A Coarse-Grained Molecular Dynamics Approach to the Study of the Intrinsically Disordered Protein α-Synuclein. J. Chem. Inf. Model 2019, 59, 1458–1471. [DOI] [PubMed] [Google Scholar]
- (38).Betancourt MR; Thirumalai D Pair potentials for protein folding: Choice of reference states and sensitivity of predicted native states to variations in the interaction schemes. Protein Sci 1999, 8, 361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Davtyan A; Schafer NP; Zheng W; Clementi C; Wolynes PG; Papoian GA AWSEM-MD: Protein structure prediction using coarse-grained physical potentials and bioinformatically based local structure biasing. J. Phys. Chem. B 2012, 116, 8494–8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Schafer NP; Kim BL; Zheng W; Wolynes PG Learning to fold proteins using energy landscape theory. Isr. J. Chem 2014, 54, 1311–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Vitalis A; Pappu RV ABSINTH: A new continuum solvation model for simulations of polypeptides in aqueous solutions. J. Comput. Chem 2009, 30, 673–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Bryngelson JDD; Wolynes PGG Spin glasses and the statistical mechanics of protein folding. Proc. Natl. Acad. Sci. U.S.A 1987, 84, 7524–7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Onuchic JN; Wolynes PG Theory of protein folding. Curr. Opin. Struct. Biol 2004, 14, 70–75. [DOI] [PubMed] [Google Scholar]
- (44).Bryngelson JD; Onuchic JN; Socci ND; Wolynes PG Funnels, pathways, and the energy landscape of protein folding: A synthesis. Proteins 1995, 21, 167–195. [DOI] [PubMed] [Google Scholar]
- (45).Onuchic JN; Luthey-Schulten Z; Wolynes PG THEORY OF PROTEIN FOLDING: The Energy Landscape Perspective. Annu. Rev. Phys. Chem 1997, 48, 545–600. [DOI] [PubMed] [Google Scholar]
- (46).Dill KA; Chan HS From Levinthal to pathways to funnels. Nat. Struct. Mol. Biol 1997, 4, 10–19. [DOI] [PubMed] [Google Scholar]
- (47).Liwo A; Pincus MR; Wawak RJ; Rackovsky S; O ldziej S; Scheraga HA A united-residue force field for off-lattice protein-structure simulations. II. Parameterization of short-range interactions and determination of weights of energy terms by Z-score optimization. J. Comput. Chem 1997, [Google Scholar]
- (48).Eastwood MP; Hardin C; Luthey-Schulten Z; Wolynes PG Statistical mechanical refinement of protein structure prediction schemes: Cumulant expansion approach. J. Chem. Phys 2002, 117, 4602–4615. [Google Scholar]
- (49).Pitera JW; Chodera JD On the use of experimental observations to bias simulated ensembles. J. Chem. Theo. Comput 2012, 8, 3445–3451. [DOI] [PubMed] [Google Scholar]
- (50).Roux B; Weare J On the statistical equivalence of restrained-ensemble simulations with the maximum entropy method. J. Chem. Phys 2013, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Cesari A; Reißer S; Bussi G Using the Maximum Entropy Principle to Combine Simulations and Solution Experiments. Computation 2018, 1–26. [Google Scholar]
- (52).Latham AP; Zhang B Improving Coarse-Grained Protein Force Fields with Small-Angle X-Ray Scattering Data. J. Phys. Chem. B 2019, 1026–1034. [DOI] [PubMed] [Google Scholar]
- (53).Dannenhoffer-Lafage T; White AD; Voth GA A Direct Method for Incorporating Experimental Data into Multiscale Coarse-Grained Models. J. Chem. Theo. Comput 2016, 12, 2144–2153. [DOI] [PubMed] [Google Scholar]
- (54).Zhang B; Wolynes PG Topology, structures, and energy landscapes of human chromosomes. Proc. Natl. Acad. Sci. U.S.A 2015, 112, 6062–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Zhang B; Wolynes PG Shape Transitions and Chiral Symmetry Breaking in the Energy Landscape of the Mitotic Chromosome. Phys. Rev. Lett 2016, 116, 1–6. [DOI] [PubMed] [Google Scholar]
- (56).Zhang B; Wolynes PG Genomic Energy Landscapes. Biophys. J 2017, 112, 427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Qi Y; Zhang B Predicting three-dimensional genome organization with chromatin states. PLOS Comput. Biol 2019, 15, e1007024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Xie WJ; Zhang B Learning the Formation Mechanism of Domain-Level Chromatin States with Epigenomics Data. Biophys. J 2019, 116, 2047–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Clementi C; Nymeyer H; Onuchic JN Topological and energetic factors: What determines the structural details of the transition state ensemble and ‘en-route’ intermediates for protein folding? An investigation for small globular proteins. J. Mol. Biol 2000, 298, 937–953. [DOI] [PubMed] [Google Scholar]
- (60).Berendsen HJ; van der Spoel D; van Drunen R GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun 1995, 91, 43–56. [Google Scholar]
- (61).Bonomi M; Branduardi D; Bussi G; Camilloni C; Provasi D; Raiteri P; Donadio D; Marinelli F; Pietrucci F; Broglia RA; Parrinello M PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun 2009, 180, 1961–1972. [Google Scholar]
- (62).Roy A; Kucukural A; Zhang Y I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc 2010, 5, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Hofmann H; Soranno A; Borgia A; Gast K; Nettels D; Schuler B Polymer scaling laws of unfolded and intrinsically disordered proteins quantified with single-molecule spectroscopy. Proc. Natl. Acad. Sci. U.S.A 2012, 109, 16155–16160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Athey J; Alexaki A; Osipova E; Rostovtsev A; Santana-Quintero LV; Katneni U; Simonyan V; Kimchi-Sarfaty C A new and updated resource for codon usage tables. BMC Bioinform 2017, 18, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Piovesan D et al. DisProt 7.0: A major update of the database of disordered proteins. Nucleic Acids Res 2017, 45, D219–D227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Uversky VN The alphabet of intrinsic disorder: II. Various roles of glutamic acid in ordered and intrinsically disordered proteins. Intrinsically Disord. Proteins 2013, 1, e24684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Bioinformatics Toolbox User’s Guide. MATLAB: 1 Apple Hill Dr, Natick, MA 01760–2098, 2019.
- (68).Miyazawa S; Jernigan RL Residue-Residue Potentials with a Favorable Contact Pair Term and an Unfavorable High Packing Density Term, for Simulation and Threading. J. Mol. Biol 1996, 623–644. [DOI] [PubMed] [Google Scholar]
- (69).Truong HH; Kim BL; Schafer NP; Wolynes PG Funneling and frustration in the energy landscapes of some designed and simplified proteins. J. Chem. Phys 2013, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Wang J; Wang W A computational approach to simplifying the protein folding alphabet. Nat. Struct. Mol. Biol 1999, 6, 1033–1038. [DOI] [PubMed] [Google Scholar]
- (71).Marrink SJ; Risselada HJ; Yefimov S; Tieleman DP; De Vries AH The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [DOI] [PubMed] [Google Scholar]
- (72).Shinoda W; Devane R; Klein ML Coarse-grained molecular modeling of non-ionic surfactant self-assembly. Soft Matter 2008, 4, 2454–2462. [Google Scholar]
- (73).Izvekov S; Voth GA A multiscale coarse-graining method for biomolecular systems. J. Phys. Chem. B 2005, 109, 2469–2473. [DOI] [PubMed] [Google Scholar]
- (74).Dunn NJ; Lebold KM; Delyser MR; Rudzinski JF; Noid WG BOCS: Bottom-up Open-source Coarse-graining Software. J. Phys. Chem. B 2018, 122, 3363–3377. [DOI] [PubMed] [Google Scholar]
- (75).Faller R; Schmitz H; Biermann O; Müller-Plathe, F. Automatic parameterization of force fields for liquids by simplex optimization. J. Comput. Chem 1999, 20, 1009–1017. [Google Scholar]
- (76).Wang LP; Head-Gordon T; Ponder JW; Ren P; Chodera JD; Eastman PK; Martinez TJ; Pande VS Systematic improvement of a classical molecular model of water. J. Phys. Chem. B 2013, 117, 9956–9972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Zheng W; Best RB An Extended Guinier Analysis for Intrinsically Disordered Proteins. J. Mol. Biol 2018, 430, 2540–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Zheng W; Zerze GH; Borgia A; Mittal J; Schuler B; Best RB Inferring properties of disordered chains from FRET transfer efficiencies. J. Chem. Phys 2018, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Riback JA; Bowman MA; Zmyslowski AM; Knoverek CR; Jumper JM; Hinshaw JR; Kaye EB; Freed KF; Clark PL; Sosnick TR Innovative scattering analysis shows that hydrophobic disordered proteins are Expanded in water. Science 2017, 358, 238–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Cesari A; Gil-Ley A; Bussi G Combining Simulations and Solution Experiments as a Paradigm for RNA Force Field Refinement. J. Chem. Theory Comput 2016, 12, 6192–6200. [DOI] [PubMed] [Google Scholar]
- (81).Beauchamp KA; Pande VS; Das R Bayesian energy landscape tilting: Towards concordant models of molecular ensembles. Biophys. J 2014, 106, 1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Bottaro S; Bengtsen T; Lindorff-Larsen K Integrating Molecular Simulation and Experimental Data: A Bayesian/Maximum Entropy Reweighting Approach. bioRxiv 2018, 457952. [DOI] [PubMed] [Google Scholar]
- (83).Müller-Späth S; Soranno A; Hirschfeld V; Hofmann H; Rüegger S; Reymond L; Nettels D; Schuler B Charge interactions can dominate the dimensions of intrinsically disordered proteins. Proc. Natl. Acad. Sci. U.S.A 2013, 110, 16693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Sherman E; Haran G Coil-globule transition in the denatured state of a small protein. Proc. Natl. Acad. Sci. U.S.A 2006, 103, 11539–11543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Kjaergaard M; Nørholm AB; Hendus-Altenburger R; Pedersen SF; Poulsen FM; Kragelund BB Temperature-dependent structural changes in intrinsically disordered proteins: Formation of α-helices or loss of polyproline II? Protein Sci 2010, 19, 1555–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Flanagan JM; Kataoka M; Shortle D; Engelman DM Truncated staphylococcal nuclease is compact but disordered. Proc. Natl. Acad. Sci. U.S.A 2006, 89, 748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Nath A; Sammalkorpi M; Dewitt DC; Trexler AJ; Elbaum-Garfinkle S; O’Hern CS; Rhoades E The conformational ensembles of α-synuclein and tau: Combining single-molecule FRET and simulations. Biophys. J 2012, 103, 1940–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Balu R; Dutta NK; Choudhury NR; Elvin CM; Lyons RE; Knott R; Hill AJ An16-resilin: An advanced multi-stimuli-responsive resilin-mimetic protein polymer. Acta Biomater 2014, 10, 4768–4777. [DOI] [PubMed] [Google Scholar]
- (89).Lens Z; Dewitte F; Monté D; Baert JL; Bompard C; Sénéchal M; Van Lint C; de Launoit Y; Villeret V; Verger A Solution structure of the N-terminal transactivation domain of ERM modified by SUMO-1. Biochem. Biophys. Res. Commun 2010, 399, 104–110. [DOI] [PubMed] [Google Scholar]
- (90).Cragnell C; Durand D; Cabane B; Skepö M Coarse-grained modeling of the intrinsically disordered protein Histatin 5 in solution: Monte Carlo simulations in combination with SAXS. Proteins 2016, 84, 777–791. [DOI] [PubMed] [Google Scholar]
- (91).Mercadante D; Milles S; Fuertes G; Svergun DI; Lemke EA; Gräter F Kirkwood-Buff Approach Rescues Overcollapse of a Disordered Protein in Canonical Protein Force Fields. J. Phys. Chem. B 2015, 119, 7975–7984. [DOI] [PubMed] [Google Scholar]
- (92).Wells M; Tidow H; Rutherford TJ; Markwick P; Jensen MR; Mylonas E; Svergun DI; Blackledge M; Fersht AR Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. U.S.A 2008, 105, 5762–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Arbesú M; Maffei M; Cordeiro TN; Teixeira JM; Pèrez Y; Bernadó P; Roche S; Pons M The Unique Domain Forms a Fuzzy Intramolecular Complex in Src Family Kinases. Structure 2017, 25, 630–640.e4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.