

Abstract
Disrupted clearance of all-trans-retinal (atRAL), a component of the visual (retinoid) cycle in the retina, may cause photoreceptor atrophy in autosomal recessive Stargardt disease (STGD1) and dry age-related macular degeneration (AMD). However, the mechanisms underlying atRAL-induced photoreceptor loss remain elusive. Here, we report that atRAL activates c-Jun N-terminal kinase (JNK) signaling at least partially through reactive oxygen species production, which promoted mitochondria-mediated caspase- and DNA damage-dependent apoptosis in photoreceptor cells. Damage to mitochondria in atRAL-exposed photoreceptor cells resulted from JNK activation, leading to decreased expression of Bcl2 apoptosis regulator (Bcl2), increased Bcl2 antagonist/killer (Bak) levels, and cytochrome c (Cyt c) release into the cytosol. Cytosolic Cyt c specifically provoked caspase-9 and caspase-3 activation and thereby initiated apoptosis. Phosphorylation of JNK in atRAL-loaded photoreceptor cells induced the appearance of γH2AX, a sensitive marker for DNA damage, and was also associated with apoptosis onset. Suppression of JNK signaling protected photoreceptor cells against atRAL-induced apoptosis. Moreover, photoreceptor cells lacking Jnk1 and Jnk2 genes were more resistant to atRAL-associated cytotoxicity. The Abca4−/− Rdh8−/− mouse model displays defects in atRAL clearance that are characteristic of STGD1 and dry AMD. We found that JNK signaling was activated in the neural retina of light-exposed Abca4−/−Rdh8−/− mice. Of note, intraperitoneal administration of JNK–IN-8, which inhibits JNK signaling, effectively ameliorated photoreceptor degeneration and apoptosis in light-exposed Abca4−/−Rdh8−/− mice. We propose that pharmacological inhibition of JNK signaling may represent a therapeutic strategy for preventing photoreceptor loss in retinopathies arising from atRAL overload.
Keywords: retina, retinal metabolism, photoreceptor, apoptosis, DNA damage
Introduction
Timely clearance of all-trans-retinal (atRAL)2 released from photoactivated rhodopsin is very important for sustaining vision (1, 2). Interruptions in the clearing of atRAL in outer segments of photoreceptors result in an accumulation of atRAL in both photoreceptors and the retinal pigment epithelium (RPE), which may cause typical manifestations of retinal dystrophy in patients with autosomal recessive Stargardt disease (STGD1) and dry age-related macular degeneration (AMD) (3, 4). Moreover, several lines of evidence demonstrate that transient amassing of atRAL by delayed clearance from the retina is one of the key mechanisms in light-induced retinal degeneration (2, 5, 6). During the visual (retinoid) cycle, light-activated rhodopsin releases atRAL (1, 7). Subsequently, atRAL is transported by ABC transporter 4 (ABCA4) or diffuses from the disk lumen to the cytoplasm of photoreceptor outer segments, followed by reduction of atRAL to all-trans-retinol, also known as vitamin A, by all-trans-retinol dehydrogenase 8 (RDH8) (8–14). Therefore, the responsibility of ABCA4 and RDH8 in the visual (retinoid) cycle is to clear atRAL in the retina. Mice lacking Abca4 and Rdh8 genes (Abca4−/−Rdh8−/− mice) exhibit the primary features of STGD1 and dry AMD, such as photoreceptor loss and RPE dystrophy (2, 5, 6). These findings suggest a link between atRAL toxicity and photoreceptor/RPE degeneration. Previous studies have indicated that atRAL provokes apoptosis in the human RPE cell line ARPE-19 via mitochondrial injury (15, 16), Bax activation by phosphorylated p53 (17), DNA damage (17), and endoplasmic reticulum stress (15). Most recently, we also show that atRAL activates the NLRP3 inflammasome to induce cell death and stimulates caspase-3/GSDME-mediated pyroptosis in ARPE-19 cells (4). In addition, Maeda and co-workers (17) report that atRAL activates Bax and elicits cell death in a murine 661W photoreceptor cell line.
The c-Jun N-terminal kinases (JNKs), which are members of the mitogen-activated protein kinase superfamily, are activated by stress stimuli, inflammatory cytokines, and growth factors (18–24). Activation of JNK signaling is linked to apoptosis (23), and it involves the development of several kinds of degenerative diseases, including Alzheimer's disease and Parkinson's disease (25, 26). However, whether aberrant aggregation of atRAL activates JNK signaling in photoreceptor cells and its potential relationship to photoreceptor loss remain unclarified. In this study, we further elucidated the mechanisms underlying photoreceptor degeneration in retinopathies correlated with disrupted atRAL clearance.
Results
atRAL induces apoptosis in 661W photoreceptor cells via activating the mitochondria-dependent pathway
The health of 661W photoreceptor cells exposed to atRAL for 6 h was examined. atRAL decreased viability of 661W photoreceptor cells in a concentration-dependent manner (Fig. 1A). The IC50 value for atRAL incubated with 661W photoreceptor cells for 6 h was 5.68 μm. Additionally, treatment of 661W photoreceptor cells with atRAL for 6 h at concentrations of 5, 10, and 20 μm caused significant decreases in cell viability of ∼40.5, 80.5, and 92.4%, respectively. Based on the results of these cytotoxicity tests, 661W photoreceptor cells were treated for 6 h with 5 μm atRAL in subsequent experiments. TUNEL staining, 6 h after incubating 661W photoreceptor cells with 5 μm atRAL, revealed a significant increase in the number of apoptotic cells (Fig. 1, B and C). Imaging of mitochondrial membrane potential (ΔΨm) using rhodamine-123 showed that the treatment of 661W photoreceptor cells with 5 μm atRAL for 6 h significantly reduced the levels of ΔΨm (Fig. 1D). Immunoblot analysis indicated that atRAL stimulated a significant decline in the expression of antiapoptotic protein Bcl2 in lysates of 661W photoreceptor cells (Fig. 1, E and F). 661W photoreceptor cells treated for 6 h with 5 μm atRAL or an equal volume of dimethyl sulfoxide (DMSO) serving as a vehicle control were fractionated to collect mitochondrial and cytosolic fractions for Western blot analysis of Bak and cytochrome c (Cyt c), respectively. The data manifested significant increases in protein levels of mitochondrial Bak (Fig. 1G) and cytosolic Cyt c (Fig. 1H) in atRAL-loaded 661W photoreceptor cells, suggesting that increased Bak protein may escape suppression by Bcl2 protein (27), leading to Bak activation and Cyt c release from mitochondria to the cytosol. Protein levels of cleaved caspase-9 and cleaved caspase-3 were found be clearly elevated in lysates of atRAL-loaded 661W photoreceptor cells (Fig. 1I). Immunofluorescence staining also showed that 5 μm atRAL significantly increased protein levels of cleaved caspase-3 in 661W photoreceptor cells after 6 h of exposure (Fig. 1J). Overexpression of Bcl2 protein in 661W photoreceptor cells was done at a level that was very far above the level in control cells (Fig. 1K), and attenuated protein levels of cleaved caspase-3 in lysates of atRAL-loaded 661W photoreceptor cells (Fig. 1K). However, with such a huge overexpression of Bcl2 protein, we still observed a certain level of cleaved caspase-3 (Fig. 1K) in atRAL-loaded 661W photoreceptor cells and only a marginal effect on restoration of cell viability (Fig. 1L), thus suggesting that the role of Bcl-2 decrease may not be crucial in this particular mitochondrial apoptotic process. Moreover, treatment with 20 μm caspase inhibitor Z-VAD-FMK decreased protein levels of cleaved caspase-3 by ∼28% in lysates of atRAL-loaded 661W photoreceptor cells (Fig. 1M) and increased cell viability by ∼29% (Fig. 1N). Taken together, these findings indicate that atRAL stimulates an apoptotic cell death in photoreceptor cells through the mitochondria-dependent pathway.
Figure 1.

Evidence that atRAL induces apoptosis of 661W photoreceptor cells. A, cell viability, 6 h after exposure of 661W photoreceptor cells to serial concentrations of atRAL (2.5, 5, 10 and 20 μm), was measured using MTS assay. Control cells were treated with vehicle (DMSO) alone. Bar heights indicate MTS absorbance at 490 nm and reflect cell viability. Data are presented as mean ± S.D. (error bars) of three independent experiments, and statistical analyses were performed by using one-way ANOVA with Tukey's post-test. ***, p < 0.001. B, TUNEL staining of 661W photoreceptor cells treated with 5 μm atRAL or vehicle (DMSO) alone for 6 h. Stained cells were visualized by using an Olympus FV1000 confocal fluorescence microscope. Red arrows indicate TUNEL-positive cells. Scale bars, 20 μm. Ctrl, control. C, percentage of TUNEL-positive cells relative to the total number of cells. 661W photoreceptor cells were incubated with 5 μm atRAL for 6 h. Cells treated for 6 h with an equal volume of DMSO served as a vehicle control. Data are presented as mean ± S.D. (error bars) of three independent experiments, and statistical analyses were performed by using Student's t test. ***, p < 0.001. D, fluorescence measurement of ΔΨm in 661W photoreceptor cells treated with 5 μm atRAL or vehicle (DMSO) alone for 6 h. The cells were stained by rhodamine-123 for 20 min and observed by using the confocal fluorescence microscope. Scale bars, 10 μm. E, Western blot analysis of Bcl2 in lysates of 661W photoreceptor cells exposed to 5 μm atRAL or vehicle (DMSO) alone for 6 h. GAPDH was used as an internal control. Molecular mass markers (kDa) are indicated to the left of the blots. F, expression levels of Bcl2 protein were normalized to those of GAPDH and presented as fold changes relative to vehicle (DMSO) controls. Data are presented as mean ± S.D. (error bars) of three independent experiments, and statistical analyses were performed by using Student's t test. **, p < 0.01. G, Western blotting was used to analyze protein levels of mitochondrial Bak in 661W photoreceptor cells incubated with 5 μm atRAL for 6 h. Control cells were treated with vehicle (DMSO) alone. COX IV was used as a loading control. H, protein levels of Cyt c in the cytosol of 661W photoreceptor cells exposed to 5 μm atRAL for 6 h were detected by Western blotting. Control cells were incubated with vehicle (DMSO) alone. GAPDH was used as a loading control. I, Western blot analysis of pro-caspase-9, cleaved caspase-9, and cleaved caspase-3 in lysates of 661W photoreceptor cells exposed to 5 μm atRAL or vehicle (DMSO) alone for 6 h. GAPDH was used as an internal control. J, immunofluorescence staining of cleaved caspase-3 in 661W photoreceptor cells incubated for 6 h with 5 μm atRAL or vehicle (DMSO) alone. Nuclei were stained blue with DAPI. Scale bars, 20 μm. K, immunoblot analysis of Bcl2, Flag (Bcl2), and cleaved caspase-3 in lysates of Bcl2 overexpressing 661W photoreceptor cells incubated with 5 μm atRAL or vehicle (DMSO) alone for 6 h, respectively. Cells transfected with vector pcDNA3.1 served as a control. GAPDH was used as a loading control. L, cell viability, 6 h after exposure of Bcl2 overexpressing 661W photoreceptor cells to 5 μm atRAL or vehicle (DMSO) alone, was determined by MTS assay. Cells transfected with vector pcDNA3.1 were used as a control. M, immunoblot analysis of cleaved caspase-3 in 661W photoreceptor cells exposed to 5 μm atRAL for 6 h in the absence or presence of 20 μm caspase inhibitor Z-VAD-FMK. Note that cells were pretreated with 20 μm Z-VAD-FMK for 1 h. Cells treated with Z-VAD-FMK or vehicle (DMSO) served as controls. GAPDH was utilized as an internal control. N, cell viability was probed by MTS assay. 661W photoreceptor cells were pretreated with caspase-3 inhibitor Z-VAD-FMK (20 μm) for 1 h, followed by incubation with or without 5 μm atRAL for 6 h. Control cells were cultured with atRAL or vehicle (DMSO) alone. Each value represents mean ± S.D. (error bars) of three independent experiments, and statistical analyses were performed by using one-way ANOVA with Tukey's post-test.
atRAL activates JNK signaling in 661W photoreceptor cells
Analyses using Western blotting and confocal microscopy with immunofluorescence staining demonstrated that protein levels of phosphorylated JNK (p-JNK), an active form of JNK, were significantly up-regulated in lysates of 661W photoreceptor cells treated for 6 h with 5 μm atRAL (Fig. 2, A and B), indicative of the ability of atRAL to activate JNK signaling in 661W photoreceptor cells. Given that the phosphorylation of c-Jun, a direct substrate of JNK, is an indicator of JNK activation, we also examined c-Jun activation status in 661W photoreceptor cells in response to atRAL by immunoblotting and immunofluorescence staining. As expected, protein levels of phosphorylated c-Jun (p-c-Jun), a downstream transcription factor directly activated by JNK, were clearly elevated in atRAL-treated 661W photoreceptor cells (Fig. 2, C and D). Because JNK–IN-8 is considered as an efficient, specific, and irreversible JNK inhibitor that inhibits c-Jun phosphorylation (28), we used JNK–IN-8 to suppress JNK signaling for determining whether JNK activation by atRAL contributed to apoptosis of 661W photoreceptor cells. Immunoblot analysis demonstrated that JNK–IN-8 at a concentration of 1 μm clearly decreased protein levels of p-c-Jun and cleaved caspase-3 in lysates of atRAL-loaded 661W photoreceptor cells (Fig. 2E). Notably, the results of the CellTiter 96 AQueous One-Solution Cell Proliferation assay (MTS) showed that treatment with 1 μm JNK–IN-8 effectively rescued 661W photoreceptor cells from apoptosis caused by atRAL (Fig. 2F). Collectively, these findings disclose that atRAL evokes photoreceptor apoptosis at least in part via activating JNK signaling.
Figure 2.
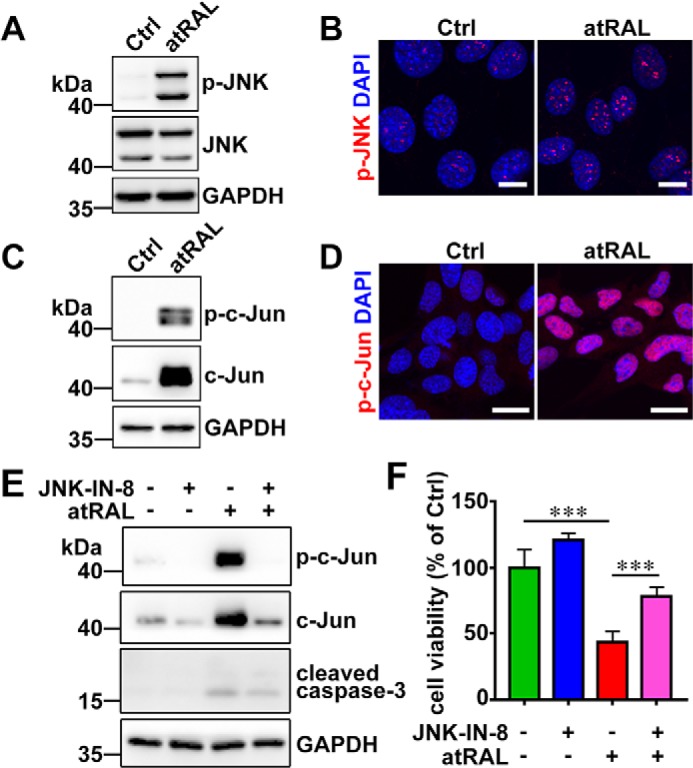
atRAL activates JNK signaling in 661W photoreceptor cells. A, immunoblot analysis of p-JNK and JNK in lysates of 661W photoreceptor cells treated with 5 μm atRAL or vehicle (DMSO) alone for 6 h. GAPDH was used as an internal control. Molecular mass markers (kDa) are indicated to the left of the blots. Ctrl, control. B, immunofluorescence staining for p-JNK in 661W photoreceptor cells incubated with 5 μm atRAL or vehicle (DMSO) alone for 6 h. Nuclei were stained blue with DAPI. Scale bars, 10 μm. C, Western blot analysis of p-c-Jun and c-Jun in lysates of 661W photoreceptor cells exposed to 5 μm atRAL or vehicle (DMSO) alone for 6 h. GAPDH was used as an internal control. D, immunofluorescence staining for p-c-Jun in 661W photoreceptor cells exposed to 5 μm atRAL or vehicle (DMSO) alone for 6 h. Nuclei were stained blue with DAPI. Scale bars, 10 μm. E, immunoblot analysis of p-c-Jun, c-Jun, and cleaved caspase-3 in 661W photoreceptor cells treated with 5 μm atRAL for 6 h in the absence or presence of 1 μm JNK-specific inhibitor JNK–IN-8. Note that cells were pretreated with 1 μm JNK–IN-8 for 1 h. Cells treated with vehicle (DMSO) or JNK–IN-8 served as controls. GAPDH was utilized as an internal control. F, cell viability was detected by MTS assay. 661W photoreceptor cells were pretreated with JNK-specific inhibitor JNK–IN-8 (1 μm) for 1 h, followed by incubation with and without 5 μm atRAL for 6 h. Control cells were treated with atRAL or vehicle (DMSO) alone. Each value represents mean ± S.D. (error bars) of three independent experiments, and statistical analyses were performed by using one-way ANOVA with Tukey's post-test.
atRAL causes DNA damage response in 661W photoreceptor cells
Protein levels of γH2AX, a marker of DNA double-strand breaks, were significantly increased in 661W photoreceptor cells treated with 5 μm atRAL for 6 h, as determined by immunoblotting and immunofluorescence staining (Fig. 3, A and B). Given that ataxia telangiectasia–mutated (ATM), a serine/threonine protein kinase, is recruited, autophosphorylated, and activated at sites of DNA double-strand breaks (29, 30), protein levels of phosphorylated ATM (p-ATM) in atRAL-loaded 661W photoreceptor cells were assayed using immunofluorescence staining. As expected, the p-ATM fluorescence intensity was markedly elevated in 661W photoreceptor cells exposed to 5 μm atRAL for 6 h (Fig. 3C). These results suggest that atRAL induces DNA damage response in photoreceptor cells. Furthermore, immunoblot analysis demonstrated that treatment with 1 μm JNK–IN-8 significantly reduced protein levels of γH2AX in atRAL-loaded 661W photoreceptor cells (Fig. 3D).
Figure 3.
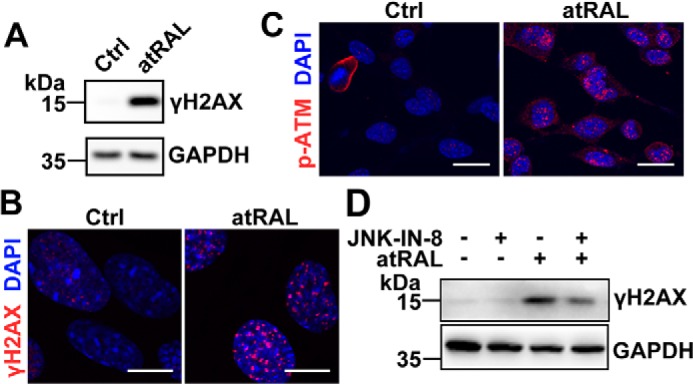
atRAL induces DNA damage response in 661W photoreceptor cells. A, immunoblot analysis of γH2AX in 661W photoreceptor cells treated with 5 μm atRAL or vehicle (DMSO) alone for 6 h. GAPDH was used as an internal control. Molecular mass markers (kDa) are indicated to the left of the blots. Ctrl, control. B and C, immunofluorescence staining for γH2AX (B) and p-ATM (C) in 661W photoreceptor cells incubated for 6 h with 5 μm atRAL or vehicle (DMSO) alone. Nuclei were stained blue with DAPI. Scale bars, 10 μm. D, Western blot analysis of γH2AX in 661W photoreceptor cells treated with 5 μm atRAL for 6 h in the absence or presence of 1 μm JNK-specific inhibitor JNK–IN-8. Note that cells were pretreated with 1 μm JNK–IN-8 for 1 h. Cells treated with vehicle (DMSO) or JNK–IN-8 served as controls. GAPDH was utilized as an internal control.
Knockout of Jnk1 and Jnk2 genes rescues 661W photoreceptor cells from atRAL-induced apoptosis via blocking JNK signaling
To further ascertain the role of JNK activation in apoptosis caused by atRAL, Jnk1 and Jnk2 genes were knocked out in 661W photoreceptor cells using the CRISPR–CAS9 system. As expected, immunoblot analysis revealed a deficient expression of JNK protein in lysates of 661W photoreceptor cells lacking Jnk1 and Jnk2 genes (Jnk1−/−Jnk2−/− 661W photoreceptor cells) (Fig. 4A). Also by Western blotting, a significant decrease in protein levels of cleaved caspase-3, cleaved poly-ADP-ribose polymerase (PARP), and γH2AX (Fig. 4, B and C), and a deficiency of p-JNK, JNK, and p-c-Jun (Fig. 4, B and D) were observed in lysates of atRAL-loaded Jnk1−/−Jnk2−/− versus wildtype (WT) 661W photoreceptor cells. Moreover, in contrast to the finding that atRAL stimulated a significant decline in protein levels of Bcl2 in atRAL-loaded 661W photoreceptor cells (Fig. 1, E and F), protein expression of Bcl2 was remarkably elevated in Jnk1−/−Jnk2−/− over WT 661W photoreceptor cells regardless of atRAL exposure (Fig. 4E). Conversely, exposure of Jnk1−/−Jnk2−/− 661W photoreceptor cells to 5 μm atRAL for 6 h resulted in significant decreases in protein levels of mitochondrial Bak and cytosolic Cyt c (Fig. 4, F and G), revealing that Bak is the downstream factor that damages mitochondria after JNK activation. More importantly, the results of MTS assay showed that the viability of Jnk1−/−Jnk2−/− 661W photoreceptor cells, 6 h after exposure to 5 μm atRAL, was clearly increased compared with that of WT 661W photoreceptor cells (Fig. 4H). These findings indicate that, after atRAL loading, inactivation of JNK signaling in 661W photoreceptor cells attenuates mitochondria-mediated caspase-/DNA damage-dependent apoptosis.
Figure 4.

Genetic deletion of Jnk1 and Jnk2 genes rescues 661W photoreceptor cells from apoptosis caused by atRAL via blocking JNK signaling. A, Western blot analysis of JNK in lysates of WT and Jnk1−/−Jnk2−/− 661W photoreceptor cells. GAPDH was utilized as an internal control. Molecular mass markers (kDa) are indicated to the left of the blots. B, immunoblot analysis of p-JNK, JNK, cleaved caspase-3, PARP, cleaved PARP and γH2AX in lysates of WT and Jnk1−/−Jnk2−/− 661W photoreceptor cells cultured with 5 μm atRAL or vehicle (DMSO) for 6 h, respectively. GAPDH was used as an internal control. C, protein levels of cleaved caspase-3, cleaved PARP, and γH2AX were normalized to those of GAPDH and presented as fold changes relative to vehicle (DMSO) controls. Ctrl, control. Data are presented as mean ± S.D. (error bars) of three independent experiments, and statistical analyses were performed by using two-way ANOVA with Sidak's post-test. ***, p < 0.001. D, immunoblot analysis of p-c-Jun and c-Jun in lysates of WT and Jnk1−/−Jnk2−/− 661W photoreceptor cells exposed to 5 μm atRAL or vehicle (DMSO) for 6 h, respectively. GAPDH was used as an internal control. E, Western blot analysis of Bcl2 in lysates of WT and Jnk1−/−Jnk2−/− 661W photoreceptor cells exposed to 5 μm atRAL or vehicle (DMSO) for 6 h, respectively. GAPDH was used as an internal control. F, Western blotting was used to analyze protein expression of mitochondrial Bak in WT and Jnk1−/−Jnk2−/− 661W photoreceptor cells treated with 5 μm atRAL or vehicle (DMSO) for 6 h, respectively. GAPDH was used as an internal control. G, immunoblot analysis of Cyt c in the cytosol of WT and Jnk1−/−Jnk2−/− 661W photoreceptor cells exposed to 5 μm atRAL or vehicle (DMSO) for 6 h, respectively. GAPDH was used as an internal control. H, cell viability, 6 h after incubating WT and Jnk1−/−Jnk2−/− 661W photoreceptor cells with 5 μm atRAL or vehicle (DMSO) alone, respectively, was measured by MTS assay. Each value represents mean ± S.D. (error bars) of three independent experiments, and statistical analyses were performed by using two-way ANOVA with Sidak's post-test. **, p < 0.01. I, Western blot analysis of p-JNK, JNK, Flag (JNK), cleaved caspase-3, and γH2AX in atRAL-loaded Jnk1−/−Jnk2−/− 661W photoreceptor cells re-expressing JNK1, JNK2, or both of them. Jnk1−/−Jnk2−/− 661W photoreceptor cells transfected with vector pcDNA3.1 were used as a control. Cells were treated with 5 μm atRAL or vehicle (DMSO) alone for 6 h. GAPDH served as a loading control. J, immunoblot analysis of p-c-Jun and c-Jun in atRAL-loaded Jnk1−/−Jnk2−/− 661W photoreceptor cells re-expressing JNK1, JNK2, or both of them. Jnk1−/−Jnk2−/− 661W photoreceptor cells transfected with vector pcDNA3.1 were used as a control. Cells were incubated with 5 μm atRAL or vehicle (DMSO) alone for 6 h. GAPDH was utilized as a loading control. K, cell viability was examined by MTS assay. Jnk1−/−Jnk2−/− 661W photoreceptor cells re-expressing JNK1, JNK2, or both of them were treated with 5 μm atRAL or vehicle (DMSO) alone for 6 h. Jnk1−/−Jnk2−/− 661W photoreceptor cells transfected with vector pcDNA3.1 were used as a control. Each value represents mean ± S.D. (error bars) of three independent experiments, and statistical analyses were performed by using two-way ANOVA with Sidak's post-test. ***, p < 0.001.
To provide further evidence for the involvement of JNK activation in atRAL-induced apoptosis of 661W photoreceptor cells, we re-expressed JNK1, JNK2, or both in Jnk1−/−Jnk2−/− 661W photoreceptor cells. Immunoblot analysis revealed that protein levels of p-JNK, cleaved caspase-3, γH2AX, and p-c-Jun were significantly increased in atRAL-loaded Jnk1−/−Jnk2−/− 661W photoreceptor cells re-expressing JNK1, JNK2, or both (Fig. 4, I and J). Re-expression of JNK1, JNK2, or both caused a decreased viability in atRAL-loaded Jnk1−/−Jnk2−/− 661W photoreceptor cells (Fig. 4K). Interestingly, re-expression of JNK1 and JNK2 significantly aggravated apoptotic cell death caused by atRAL in Jnk1−/−Jnk2−/− 661W photoreceptor cells than that of either JNK1 or JNK2 (Fig. 4K).
atRAL provokes reactive oxygen species (ROS) generation in 661W photoreceptor cells
Considering that ROS can induce apoptosis by activating JNK signaling (31), intracellular ROS generation was measured by flow cytometry and confocal laser-scanning microscopy after atRAL-loaded 661W photoreceptor cells were incubated with the fluorescent probe H2DCFDA. Exposure of 5 μm atRAL to 661W photoreceptor cells for 6 h dramatically stimulated the production of intracellular ROS (Fig. 5, A–C), with an ∼7.8-fold increase over control cells (Fig. 5B). MitoSOX Red is a specific dye for mitochondrial ROS (32). After incubating atRAL-loaded 661W photoreceptor cells with MitoSOX Red, confocal microscopy was used to examine fluorescently-stained cells and revealed that ROS induced by atRAL were at least in part localized to mitochondria (Fig. 5D).
Figure 5.

atRAL provokes ROS (including mitochondrial ROS) generation in 661W photoreceptor cells. A, intracellular ROS production, 6 h after incubating 661W photoreceptor cells with 5 μm atRAL or vehicle (DMSO) alone, was measured by H2DCFDA staining coupled with flow cytometry. B, quantification of H2DCFDA-positive cells by flow cytometry. Each value is shown as a percentage of the total cell population and presented as mean ± S.D. (error bars) of three independent experiments. Statistical analyses were performed by using Student's t test. **, p < 0.01. C, intracellular ROS generation, 6 h after exposure of 661W photoreceptor cells to 5 μm atRAL, was visualized using the fluorescent probe H2DCFDA by confocal laser-scanning fluorescence microscopy. H2DCFDA emits green fluorescence upon reaction with ROS. Cells treated with DMSO alone served as a vehicle control (Ctrl). Nuclei were stained with Hoechst 33342 (blue). Scale bars, 10 μm. D, mitochondrial ROS production in 661W photoreceptor cells exposed to 5 μm atRAL for 6 h was determined by MitoSOX Red staining. Control cells were incubated with DMSO alone. Nuclei were labeled by Hoechst 33342 (blue). Scale bars, 10 μm.
Inhibiting ROS generation protects 661W photoreceptor cells from apoptosis induced by atRAL via attenuating activation of JNK signaling
Treatment with antioxidant N-acetyl-l-cysteine (NAC) at a concentration of 2 mm effectively reduced ROS production in 661W photoreceptor cells after 6 h of exposure to 5 μm atRAL (Fig. 6, A and B), and it promoted cellular survival (Fig. 6C) against apoptosis caused by atRAL. Western blot analysis indicated that 2 mm NAC significantly decreased the ratio of the p-JNK/JNK protein level and protein levels of cleaved caspase-9, cleaved caspase-3, cleaved PARP, γH2AX, and p-c-Jun in lysates of atRAL-loaded 661W photoreceptor cells (Fig. 6, D–F). Moreover, treatment with 2 mm NAC resulted in a significant decrease in protein levels of Cyt c in the cytosol of atRAL-exposed 661W photoreceptor cells (Fig. 6G). These findings suggest that attenuation of JNK signaling in atRAL-exposed 661W photoreceptor cells by suppressing the ROS generation alleviates mitochondria-mediated caspase-/DNA damage-dependent apoptosis and thereby increases cell viability.
Figure 6.

Antioxidant NAC ameliorates atRAL-induced apoptosis of 661W photoreceptor cells by suppressing JNK signaling. A, intracellular ROS levels were measured by flow cytometry using the fluorescence probe H2DCFDA. 661W photoreceptor cells were cultured for 6 h with 5 μm atRAL or vehicle (DMSO) alone. Alternatively, 661W photoreceptor cells were pretreated with 2 mm NAC for 2 h and incubated with 5 μm atRAL for 6 h. Subsequently, the cells were stained by 10 μm H2DCFDA for 10 min at 37 °C, and the levels of intracellular ROS were determined by flow cytometry. B, quantity of H2DCFDA-positive cells was assayed by flow cytometry and expressed as a percentage of the total cell population. Each value represents mean ± S.D. (error bars) of three independent experiments, and statistical analyses were performed by using one-way ANOVA with Tukey's post-test. **, p < 0.01; ***, p < 0.001. C, cell viability was examined by MTS assay. 661W photoreceptor cells were pretreated with antioxidant NAC (2 mm) for 2 h, followed by incubation with or without 5 μm atRAL for 6 h. Control cells were treated with atRAL or vehicle (DMSO) alone. Each value represents mean ± S.D. (error bars) of three independent experiments, and statistical analyses were performed by using one-way ANOVA with Tukey's post-test. D, immunoblot analysis of p-JNK, JNK, pro-caspase-9, cleaved caspase-9, cleaved caspase-3, PARP, cleaved PARP, and γH2AX in lysates of 661W photoreceptor cells treated for 6 h with 5 μm atRAL in the absence or presence of 2 mm NAC. Note that cells were pretreated with 2 mm NAC for 2 h. Cells treated with NAC or vehicle (DMSO) alone served as controls. GAPDH was utilized as an internal control. Molecular mass markers (kDa) are indicated to the left of the blots. E, ratios of p-JNK/JNK protein level and protein levels of cleaved caspase-9, cleaved caspase-3, cleaved PARP, and γH2AX were shown as fold changes relative to vehicle (DMSO) controls. Levels of each protein were normalized to those of GAPDH. Note that the normalization for band intensity of p-JNK or JNK against that of GAPDH was individually made before calculating the p-JNK/JNK ratio. Each value represents mean ± S.D. (error bars) of three independent experiments, and statistical analyses were performed by using one-way ANOVA with Tukey's post-test. **, p < 0.01; ***, p < 0.001. F, immunoblot analysis of p-c-Jun and c-Jun in lysates of 661W photoreceptor cells incubated with 5 μm atRAL for 6 h in the presence or absence of 2 mm NAC. Note that cells were pretreated with 2 mm NAC for 2 h. Control cells were treated with NAC or vehicle (DMSO) alone. GAPDH served as an internal control. G, Western blot analysis of cytosolic Cyt c in 661W photoreceptor cells exposed to 5 μm atRAL for 6 h with or without 2 mm NAC. Note that cells were pretreated with 2 mm NAC for 2 h. Cells treated with NAC or vehicle (DMSO) alone served as controls. GAPDH was used as an internal control.
Light-induced photoreceptor degeneration and apoptosis in Abca4−/−Rdh8−/− mice involves activation of JNK signaling
Previous studies have indicated that exposure of Abca4−/− Rdh8−/− mice featuring defective atRAL clearance to intense light increases atRAL levels in the retina and leads to severe photoreceptor degeneration (5, 6). In this investigation, 2-day dark-adapted 4-week-old C57BL/6J WT and Abca4−/− Rdh8−/− mice were exposed to light-emitting diode (LED) light with an intensity of 10,000 lx for 2 h and then were raised in the dark for 1, 3, and 5 days. Control C57BL/6J WT and Abca4−/− Rdh8−/− mice were normally raised in the dark for 7 days in the absence of light exposure. As expected, hematoxylin and eosin (H&E) staining showed that the morphology of the neural retina of light-exposed Abca4−/−Rdh8−/− mice was altered obviously and damaged severely, and manifested marked thinning of the photoreceptor layer, particularly the outer segment (OS) layer and outer nuclear layer (ONL) (Fig. 7A). Interestingly, TUNEL staining of the neural retina of Abca4−/−Rdh8−/− mice exposed to light displayed a significant increase in the number of apoptotic photoreceptor cells (Fig. 7B). Immunoblot analysis revealed a clear elevation of p-JNK protein levels in extracts from the neural retina of light-exposed Abca4−/− Rdh8−/− mice (Fig. 7C). In addition, protein levels of γH2AX and cleaved caspase-3, which function downstream of JNK signaling (Fig. 4B), were notably up-regulated in extracts from the neural retina of light-exposed Abca4−/−Rdh8−/− mice (Fig. 7C). On the contrary, C57BL/6J WT mice illuminated at 10,000 lx for 2 h exhibited no obvious photoreceptor degeneration and apoptosis (Fig. 7, A and B). Likewise, protein levels of p-JNK, γH2AX, and cleaved caspase-3 remain almost intact in extracts from the neural retina of light-exposed C57BL/6J WT mice (Fig. 7C). The results imply that photoinduced photoreceptor injury in Abca4−/−Rdh8−/− mice is at least in part associated with atRAL overload and JNK activation.
Figure 7.

Light-induced photoreceptor degeneration in Abca4−/−Rdh8−/− mice involves activation of JNK signaling. Four-week-old C57BL/6J WT and Abca4−/−Rdh8−/− mice were exposed to LED light with an intensity of 10,000 lx for 2 h after their pupils were dilated with 1% tropicamide. Eyeballs, 1, 3, and 5 days after light exposure, were harvested for subsequent studies. C57BL/6J WT and Abca4−/−Rdh8−/− mice raised normally in the dark for 7 days in the absence of 10,000 lx LED light served as controls. A, histology of mouse retina was examined by H&E staining. Scale bars, 20 μm. B, apoptotic cells in mouse retina were detected by TUNEL staining. Scale bars, 20 μm. C, immunoblot analysis of p-JNK, JNK, γH2AX and cleaved caspase-3 in extracts from mouse neural retina. GAPDH was used as an internal control. Molecular mass markers (kDa) are indicated to the left of the blots. D, genotyping of Abca4−/−Rdh8−/− mice on a C57BL/6J genetic background was carried out by PCR amplification of tail DNAs with specific primers. Abca4 deletion in Abca4−/−Rdh8−/− mice was identified by producing a 455-bp amplicon with the A0 and N1 primers, yet C57BL/6J WT mice generated a 619-bp amplicon with the ABCR1 and ABCR2 primers. Rdh8 deletion in Abca4−/−Rdh8−/− mice was corroborated by giving rise to a 450-bp amplicon with the Neo1 and DMR11 primers, whereas C57BL/6J WT mice produced an 800-bp amplicon with the DMR5 and DMR6 primers. To further confirm the absence of Rd8 mutation of the Crb1 gene in C57BL/6J WT and Abca4−/−Rdh8−/− mice used in the study, DNA samples extracted from mouse tail biopsies were amplified for Wt allele and mutant Rd8 allele using primers mCrb1 mF1, mCrb1 mF2, and mCrb1 mR. C57BL/6N mice carrying Rd8 mutation in Crb1 gene served as a control. Amplicon sizes were 220 bp for Wt allele and 244 bp for Rd8 allele. A 100-bp DNA ladder was used as a size standard. OS, outer segment; IS, inner segment; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer.
Suppression of JNK signaling by JNK–IN-8 ameliorates photoreceptor degeneration and apoptosis in light-exposed Abca4−/−Rdh8−/− mice
Histological assessment demonstrated that intraperitoneal treatment with JNK-specific inhibitor JNK–IN-8 (4 mg/kg body weight) relieved photoreceptor degeneration in light-exposed Abca4−/−Rdh8−/− mice (Fig. 8A). TUNEL staining also indicated that in vivo administration of JNK–IN-8 alleviated photoreceptor apoptosis of light-exposed Abca4−/−Rdh8−/− mice (Fig. 8B). To validate that JNK signaling and its downstream events (caspase-3 activation and DNA damage response) were indeed inhibited by JNK–IN-8 in neural retina of light-exposed Abca4−/−Rdh8−/− mice, Western blotting was adopted to examine protein levels of p-c-Jun, cleaved caspase-3, and γH2AX. The results revealed that JNK–IN-8 administration significantly decreased protein levels of p-c-Jun, cleaved caspase-3, and γH2AX in extracts from neural retina of light-exposed Abca4−/−Rdh8−/− mice (Fig. 8, C and D).
Figure 8.

JNK-specific inhibitor JNK–IN-8 alleviates light-induced photoreceptor degeneration and apoptosis in Abca4−/−Rdh8−/− mice. 48-h dark-adapted Abca4−/−Rdh8−/− mice at 4 weeks of age were intraperitoneally injected with JNK–IN-8 or DMSO (vehicle) (4 mg/kg body weight). One hour later, pupils of mice were dilated with 1% tropicamide, and the mice were exposed to LED light with the intensity of 10,000 lx for 2 h, followed by once-daily treatment with JNK–IN-8 or DMSO (vehicle) (4 mg/kg body weight) for 4 days. Eyeballs, 5 days after light illumination, were collected for subsequent studies. Control Abca4−/−Rdh8−/− mice were administered intraperitoneally with JNK–IN-8 or DMSO (vehicle) (4 mg/kg body weight) in the absence of light exposure. A, morphology of mouse retina was analyzed by H&E staining. Scale bars, 20 μm. B, apoptotic cells in mouse retina were examined by TUNEL staining. Scale bars, 20 μm. C and D, immunoblot analysis of p-c-Jun, c-Jun, cleaved caspase-3, and γH2AX in extracts from mouse neural retina. GAPDH served as a loading control. Molecular mass markers (kDa) are indicated to the left of the blots.
Discussion
AMD is broadly divided into two types: dry AMD and wet AMD (33, 34). Wet AMD is characterized by the formation of choroidal neovascularization mediated by vascular endothelial growth factor (VEGF) (35, 36). Previous studies indicate that suppressing JNK signaling reduces choroidal VEGF expression and neovascularization in a murine model of wet AMD (37), and hypoxia-induced JNK activation promotes retinal VEGF production and pathological angiogenesis in a murine model of retinopathy of prematurity (38). Dry AMD affects up to 90% of AMD cases, for which there is no effective cure. Photoreceptor degeneration is a common feature of STGD1 and dry AMD (39, 40). However, the correlation of JNK activation with photoreceptor loss in STGD1 and dry AMD is still unclear. Abca4−/−Rdh8−/− mice, a model of STGD1 and dry AMD characterized by disrupted clearance of atRAL, develop severe photoreceptor dystrophy (2). Exposure of Abca4−/−Rdh8−/− mice to 10,000 lx of fluorescent light leads to rapid accumulation of atRAL in the retina (6), and it provokes retinal damage and photoreceptor cell death (5). In this study, we showed that JNK signaling was activated in neural retina of Abca4−/−Rdh8−/− mice versus age-matched C57BL/6J WT mice after 10,000-lx LED light exposure for 2 h (Fig. 7C). Nevertheless, it is reported that JNK activation is associated with apoptosis (31). Evidently, these lines of investigation disclose that atRAL accumulating in photoreceptor cells may activate JNK signaling to cause apoptosis. With the use of in vitro cell-based assays, we confirmed that atRAL at the concentration of 5 μm significantly induced apoptosis of 661W photoreceptor cells via activating JNK signaling (Figs. 1, 2, and 4). Moreover, JNK activation by atRAL stimulated mitochondria-mediated caspase-/DNA damage-dependent apoptotic cell death in 661W photoreceptor cells (Figs. 1–4).
We found that protein levels of p-JNK were almost intact in Bcl2-overexpressing 661W photoreceptor cells in response to atRAL versus atRAL-loaded vector-control cells (Fig. S1), suggesting that expression changes of Bcl2 protein do not affect atRAL-mediated JNK activation. Moreover, treatment with JNK–IN-8 increased protein expression of Bcl2 by ∼24% in atRAL-loaded 661W photoreceptor cells (Fig. S2), implying that suppressing JNK signaling attenuates Bcl2 decrease. In Fig. 4E, we provided evidence that protein expression of Bcl2 was significantly elevated in atRAL-loaded Jnk1−/−Jnk2−/− over WT 661W photoreceptor cells. Taken together, these findings reveal that Bcl2 functions downstream of JNK. In atRAL-loaded 661W photoreceptor cells, increased Bak protein (Fig. 1G) likely caused Bak activation and damage to mitochondria, thereby eliciting the release of Cyt c into the cytosol (Fig. 1H). Previous evidence has revealed that cytosolic Cyt c functions as a trigger for caspase-9 activation, and the active form of caspase-9 (cleaved caspase-9) cleaves/activates caspase-3, followed by activated caspase-3–mediated cleavage of PARP (41–43). As expected, we observed that protein levels of cleaved caspase-9, cleaved caspase-3, and cleaved PARP were significantly increased in lysates of atRAL-loaded 661W photoreceptor cells (Figs. 1, I and J, 4B, and 6D).
Phosphorylation of H2AX at the sites of DNA damage is a very early step in the cellular response to DNA double-strand breaks (44, 45). Either JNK or ATM involves the generation of phosphorylated H2AX (γH2AX) (46, 47). From the results shown in Figs. 2 and 3, increased protein levels of p-JNK, γH2AX, and p-ATM were clearly detected in atRAL-loaded 661W photoreceptor cells, implying that activation of JNK and ATM contributes to DNA damage. Moreover, protein levels of γH2AX were significantly decreased in atRAL-loaded Jnk1−/−Jnk2−/− over WT 661W photoreceptor cells (Fig. 4, B and C), further corroborating that JNK activation by atRAL evokes DNA damage response.
After incubating with 661W photoreceptor cells, atRAL induced a marked increase in levels of ROS (Fig. 5, A–C), partially generated inside mitochondria (Fig. 5D). As reported previously, ROS are involved in activation of JNK signaling (48). Indeed, we showed that treatment with NAC, a general ROS inhibitor, attenuated JNK activation in atRAL-loaded 661W photoreceptor cells (Fig. 6, D and E). It was worth noting that NAC only partially rescued 661W photoreceptor cells from apoptosis caused by atRAL (Fig. 6C), probably because NAC limitedly reduces the ROS production (Fig. 6, A and B), and intracellular generation of ROS by atRAL may not be the only way for JNK activation, DNA damage, and apoptosis. Moreover, nonapoptotic processes may also involve the death of 661W photoreceptor cells induced by atRAL. Treatment of atRAL-loaded 661W photoreceptor cells with NAC prevented the release of Cyt c into the cytosol (Fig. 6G), indicating that the ROS production is an upstream factor of the mitochondria-dependent apoptotic pathway.
Double-knockout of Jnk1 and Jnk2 genes in 661W photoreceptor cells also partially alleviated apoptosis caused by atRAL through reduction in protein levels of cleaved caspase-3, cleaved PARP, and γH2AX (Fig. 4, B, C and H), reflecting that JNK activation functions upstream of caspase-3 activation and PARP cleavage and serves as an initiator for DNA damage. In addition, increased expression of Bcl2 protein and reduced protein levels of mitochondrial Bak and cytosolic Cyt c were observed in atRAL-loaded Jnk1−/−Jnk2−/− 661W photoreceptor cells (Fig. 4, E–G), which reveal that the deletion of JNK protein inhibits the mitochondrial events linked to apoptotic cell death.
Each murine retina is estimated to contain 4.62 mm rhodopsin in disk membrane and 8.23 mm with respect to the rod outer segment cytoplasm (49). Because bleaching of rhodopsin by light generates an equivalent amount of atRAL (50), the concentration of atRAL used in our experiments could be physiologically significant.
Treating atRAL-loaded 661W photoreceptor cells with JNK-specific inhibitor JNK–IN-8 significantly enhanced cell survival via inhibiting JNK activation and its downstream effectors caspase-3 (Fig. 2, E and F) and γH2AX (Fig. 3D). More importantly, the results of animal experiments showed that intraperitoneal injection of JNK–IN-8 effectively alleviated photoreceptor degeneration and apoptosis by suppressing JNK signaling in Abca4−/−Rdh8−/− mice after light exposure (Fig. 8). Therefore, pharmacological inhibition of JNK signaling may ameliorate atRAL-related retinopathies, such as STGD1 and dry AMD, through interference with apoptotic photoreceptor cell death.
Considered collectively, our data suggest that atRAL promotes mitochondria-mediated caspase-/DNA damage-dependent apoptosis by triggering activation of JNK signaling in photoreceptor cells. A plausible mechanism for these findings is depicted in Fig. 9. atRAL accumulating in photoreceptor cells activates JNK signaling at least partially by ROS (including mitochondrial ROS) and reduces ΔΨm. JNK activation by atRAL decreases Bcl2 protein expression and produces increased Bak protein that may cause Bak activation, thereby adversely affecting mitochondrial integrity. Cyt c is liberated from ruptured mitochondria into the cytoplasm where it stimulates the formation of apoptosomes consisting of Cyt c, caspase-9, and Apaf-1 (51, 52). This complex initiates caspase-9 and caspase-3 cascade activation to provoke apoptotic cell death. Alternatively, the active form of JNK (p-JNK) in the cytosol partially translocates into the nucleus where it phosphorylates H2AX to produce γH2AX, a marker of DNA double-strand breaks, thus leading to apoptosis induced by DNA damage (53). This is the first report describing a relationship between atRAL-mediated apoptosis of photoreceptor cells and activation of JNK signaling. The results of this study suggest a key role of JNK signaling in photoreceptor dystrophy arising from atRAL accumulation beyond a threshold level and expand the understanding of atRAL-associated toxicity in photoreceptor cells.
Figure 9.

Proposed mechanisms showing atRAL-induced apoptosis in photoreceptor cells via activating JNK signaling. In photoreceptor cells, atRAL evokes collapsed ΔΨm and JNK activation. The latter is at least partially mediated by the ROS (including mitochondrial ROS) production. Phosphorylation of JNK results in decreased protein expression of Bcl2 and mitochondrial damage through increased Bak protein that may cause Bak activation. Cyt c is released from ruptured mitochondria into the cytosol where it stimulates the formation of apoptosomes consisting of Cyt c, caspase-9, and Apaf-1 (51, 52). Caspase-9 activation cleaves and activates caspase-3 to induce apoptosis. However, p-JNK, the active form of JNK, partially translocates into the nucleus where it phosphorylates the histone H2AX to form γH2AX, a sensitive marker for DNA double-strand breaks. Generation of γH2AX, which localizes to sites of DNA strand breaks, accounts for the onset of irreversible DNA damage correlated with apoptotic cell death (53). It should be mentioned that the ROS production caused by atRAL is one of the initiators to activate JNK signaling, and JNK activation is also not the only way for atRAL-stimulated DNA damage.
Experimental procedures
Materials
atRAL, 4′,6-diamidino-2-phenylindole (DAPI), Hoechst 33342, and anti-Flag (catalog no. F3165) were purchased from Sigma. NAC was obtained from Aladdin (Shanghai, China). Z-VAD-FMK was purchased from R&D Systems. JNK–IN-8 was obtained from Selleck Chemicals (Shanghai, China). H2DCFDA, rhodamine-123, MitoSOXTM Red mitochondrial superoxide indicator, and Lipofectamine® LTX and PLUSTM reagent were purchased from Thermo Fisher Scientific (Eugene, OR). Antibodies against cleaved caspase-3 (catalog no. 9664S and 9661S), caspase-9 (catalog no. 9508S), PARP (catalog no. 9542S), p-JNK (catalog no. 9255S), JNK (catalog no. 9252S), p-c-Jun (catalog no. 9261S), c-Jun (catalog no. 9165S), Bcl2 (catalog no. 3498S), Bak (catalog no. 12105S), COX IV (catalog no. 4844S), Cyt c (catalog no. 11940S), and GAPDH (catalog no. 5174S) were provided by Cell Signaling Technology (Danvers, MA). Anti-γH2AX (catalog no. 05-636) was purchased from Millipore (Billerica, MA). Anti-p-ATM (catalog no. NB100-306) was provided by Novus (Littleton, CO). p-JNK (catalog no. 4821) was purchased from Abcam. Horseradish peroxidase–conjugated goat anti-mouse (catalog no. 31430) and goat anti-rabbit IgG (H+L) (catalog no. 31460), Alexa Fluor 594-conjugated donkey anti-mouse (catalog no. A-21203), and donkey anti-rabbit IgG (H+L) (catalog no. A21207) secondary antibodies were purchased from Thermo Fisher Scientific (Rockford, IL). H&E staining kit (catalog no. P032IH) was obtained from Auragene Biotech (Changsha, HN, China). PrimeSTAR® HS (Premix) (catalog no. R040A) and pMDTM 19-T vector cloning kit (catalog no. 6013) were provided by Takara Biomedical Technology (Beijing, China). Escherichia coli DH5α was purchased from Transgene Biotech (Beijing, China).
Animals
C57BL/6J WT mice were purchased from the Xiamen University Laboratory Animal Center. Abca4−/−Rdh8−/− mice on a C57BL/6N genetic background were provided from the Krzysztof Palczewski Laboratory (Case Western Reserve University, Cleveland, OH) and contained the rd8 mutation in the Crb1 gene (54). However, Abca4−/−Rdh8−/− mice used in this study were maintained on a C57BL/6J genetic background (Fig. 8D), which were obtained by crossing Abca4−/−Rdh8−/− mice (C57BL/6N genetic background) with C57BL/6J WT mice. C57BL/6J WT and Abca4−/−Rdh8−/− mice (C57BL/6J genetic background) were free of the rd8 mutation in the Crb1 gene compared with C57BL/6N mice that carried the rd8 mutation of the Crb1 gene (Fig. 7D). Experiments with animals were approved by the Institutional Animal Care and Use Committee of Xiamen University School of Medicine. Mice were handled in strict accordance with good animal practice as defined by the Chinese Association for Research in Vision and Ophthalmology (CARVO), and maintained and bred under specific pathogen-free conditions with a 12-h light/12-h dark schedule at the Xiamen University Laboratory Animal Center. 48-h dark-adapted C57BL/6J WT and Abca4−/−Rdh8−/− mice at 4 weeks of age were exposed to LED light with an intensity of 10,000 lx for 2 h after their pupils were dilated with 1% tropicamide. Finally, the mice, 1, 3, and 5 days after light exposure, were euthanized, and their eyeballs were collected for subsequent studies. C57BL/6J WT and Abca4−/−Rdh8−/− mice raised normally in the dark for 7 days in the absence of 10,000 lx LED light served as controls. Alternatively, 48-h dark-adapted Abca4−/−Rdh8−/− mice at 4 weeks of age were administered intraperitoneally JNK–IN-8 or vehicle (DMSO) at a dose of 4 mg/kg body weight. 1-h later, pupils of mice were dilated with 1% tropicamide followed by exposure of JNK–IN-8- or vehicle (DMSO)-treated mice to LED light with the intensity of 10,000 lx for 2 h. After once-daily treatment with JNK–IN-8 or vehicle (DMSO) (4 mg/kg body weight) for 4 days, the mice, at day 5 after light exposure, were euthanized, and their eyeballs were collected for subsequent studies. Control Abca4−/−Rdh8−/− mice were injected intraperitoneally with JNK–IN-8 or vehicle (DMSO) (4 mg/kg body weight) in the absence of light exposure.
Cell culture
A murine photoreceptor cell line 661W, purchased from Shanghai Zishi Biotechnology (Shanghai, China), was routinely cultured in Dulbecco's modified Eagle's medium (Gibco, Shanghai, China) supplemented with 10% fetal bovine serum (HyClone, Beijing, China) and 1% penicillin/streptomycin (Thermo Fisher Scientific; Grand Island, NY) in a humidified incubator with 5% CO2 at 37 °C.
Treatment with atRAL
661W photoreceptor cells were seeded into 96-well plates at a density of 1.5 × 104 cells per well and cultured overnight. Cells were then incubated with serial concentrations of atRAL (2.5, 5, 10, and 20 μm) for 6 h.
Treatment with NAC, JNK–IN-8, and Z-VAD-FMK
661W photoreceptor cells seeded into 96-well plates at the density of 1.5 × 104 cells per well or 6-well plates at a density of 3 × 105 cells per well were cultured overnight, and pretreated with 2 mm NAC for 2 h, 1 μm JNK–IN-8 for 1 h, or 20 μm Z-VAD-FMK for 1 h. Cells were then treated for 6 h with or without 5 μm atRAL.
Construction of Jnk1−/−Jnk2−/− 661W photoreceptor cell line
To generate the Jnk1−/−Jnk2−/− 661W photoreceptor cell line, target sequences in the third exon of mouse Jnk1 (ACCGAGAACTAGTTCTTATG) and target sequences in the second exon of mouse Jnk2 (CAACTGAAACCGATCGGCTC) were designed at Massachusetts Institute of Technology (56). The oligonucleotides for specific guide RNAs (gRNAs) are displayed in Table 1 and synthesized by Sangon Biotech (Shanghai, China). The synthesized gRNA primers were annealed to form a gRNA duplex and cloned into the BsmbI site of pL-CRISPR.EFS.GFP vector purchased from Addgene (Watertown, MA). 661W photoreceptor cells seeded into 6-well plates were transfected with the targeting vector using Lipofectamine® LTX & PLUSTM reagent. 24 h hours post-transfection, GFP-positive single cells were sorted into 96-well plates by a MoFlo Astrios flow cytometry (Beckman Coulter; Brea, CA). Two weeks later, single colonies were expanded into 6-well plates. Jnk1−/−Jnk2−/− 661W photoreceptor cells were identified by Western blotting and genome sequencing. The primers for amplification of the genomic areas designed for Jnk1 and Jnk2 are shown in Table 2. Each PCR product was cloned with pMDTM19-T vector cloning kit, and transformed into E. coli DH5α. The colonies were sequenced using M13 reverse primers. WT and Jnk1−/−Jnk2−/− 661W photoreceptor cells seeded into 96- or 6-well plates were cultured overnight and exposed to 5 μm atRAL for 6 h, respectively.
Table 1.
Guide RNA sequences
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Jnk1 | CACCGAGCCTATAGGCTTTAAATTC | AAACGAATTTAAAGCCTATAGGCTC |
| Jnk2 | CACCGCAACTGAAACCGATCGGCTC | AAACGAGCCGATCGGTTTCAGTTG |
Table 2.
Primers for amplification of genomic DNA
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Jnk1 | CCTTGGTGAATATTTGGATGAAGCC | TTAGGACAAACCAGAAGAGGGCATC |
| Jnk2 | CCATTTTGACCATGCCATCTGAAGT | TAAAACGCCAAATTCCGGTTACACC |
Plasmids construction and overexpression
Full-length cDNAs encoding Bcl2, JNK1, and JNK2 were cloned into vector pcDNA3.1, respectively. Flag tag was added by PCR to all constructs using the oligonucleotides listed in Table 3. Jnk1−/−Jnk2−/− 661W photoreceptor cells were transfected with overexpression plasmids or control vector (pcDNA3.1) using Lipofectamine® LTX and PLUSTM reagent according to the manufacturer's protocol. Jnk1−/−Jnk2−/− 661W photoreceptor cells were seeded into 96- or 6-well plates and cultured overnight. Cells, 24 h after transfection, were treated with 5 μm atRAL or vehicle (DMSO) for 6 h. Protein expression of Flag-labeled Bcl2, JNK1, and JNK2 in Jnk1−/−Jnk2−/− 661W photoreceptor cells was analyzed by Western blotting using anti-Flag mAb.
Table 3.
Primers for PCR
| Primer | Sequences |
|---|---|
| pCDNA3.1-Bcl2-F | CTAGCTAGCATGGCGCAAGCCGGGAGAAC |
| pCDNA3.1-Bcl2-R | CCGCTCGAGCTACTTGTCATCGTCGTCCTTGTAATCCTTGTGGCCCAGGTATGCAC |
| pCDNA3.1-Jnk1 p46-F | CTAGCTAGCATGAGCAGAAGCAAACGTGA |
| pCDNA3.1-Jnk1 p46-R | CCGCTCGAGCTACTTGTCATCGTCGTCCTTGTAATCTTGCTGCACCTGTGCTAAAG |
| pCDNA3.1-Jnk1 p54-F | CTAGCTAGCATGAGCAGAAGCAAACGTGA |
| pCDNA3.1-Jnk1 p54-R | CCGCTCGAGCTACTTGTCATCGTCGTCCTTGTAATCTCTACAGCAGCCCAGAGGTC |
| pCDNA3.1-Jnk2 p46-F | CTAGCTAGCATGAGTGACAGTAAAAGCGAT |
| pCDNA3.1-Jnk2 p46-R | CCGCTCGAGCTACTTGTCATCGTCGTCCTTGTAATCCTGCTGCATCTGTGCTGAAG |
| pCDNA3.1-Jnk2 p54-F | CTAGCTAGCATGAGTGACAGTAAAAGCGA |
| pCDNA3.1-Jnk2 p54-R | CCGCTCGAGCTACTTGTCATCGTCGTCCTTGTAATCCCGGCAGCCTTCCAGGGGTC |
Fractionation of mitochondria and cytosol
Cell fraction was performed as described previously (55). Briefly, 661W photoreceptor cells were scratched and harvested in cold PBS. After washing twice with PBS, cells were resuspended in digitonin lysis buffer (75 mm NaCl, 1 mm NaH2PO4, 8 mm Na2HPO4, 250 mm sucrose, 190 μg/ml digitonin). After 5 min on ice, cells were centrifuged for 5 min at 14,000 rpm at 4 °C. The supernatants were used for detecting protein levels of Cyt c by immunoblotting. The pellets were resuspended in Triton lysis buffer (25 mm Tris-HCl (pH 8.0), 0.1% (v/v) Triton X-100), followed by analysis of mitochondrial Bak via Western blotting.
Cell viability
Cytotoxicity was assessed using MTs Assay, which was performed by adding 20 μl of the CellTiter 96 AQueous One Solution Reagent (Promega; Madison, WI) directly to 96-well plates, incubating for 1 h, and then recording absorbance at 490 nm using a 1510 Multiskan GO spectrophotometer (Thermo Fisher Scientific; Vantaa, Finland). Cells treated with DMSO served as vehicle controls.
H&E staining
Mouse eyes were harvested and fixed in 4% paraformaldehyde at 4 °C overnight prior to embedding in paraffin. Tissues were cut at a thickness of 8 μm. After drying at 60 °C for 1 h, sections were deparaffinized, rehydrated, and stained with H&E and visualized by a DM2500 microscope (Leica; Wetzlar, Germany).
Immunofluorescence
Cells were fixed with 4% paraformaldehyde for 15 min at 4 °C and permeabilized by 0.2% Triton X-100 in PBS for 20 min at room temperature. Subsequently, cells were blocked with 2% BSA and incubated with primary antibodies (1:100 dilution) overnight at 4 °C, followed by incubation with Alexa Fluor 594-conjugated secondary antibodies (1: 200 dilution) for 1 h at room temperature. Slides were mounted in Vectashield with DAPI (Vector Labs; Burlingame, CA). The epifluorescence images were performed using an Olympus FV1000 confocal microscope (Tochigi, Japan).
TUNEL assay
Tissue sections or cells were fixed with 4% paraformaldehyde at 4 °C for 15 min. Apoptosis was evaluated by TUNEL assay (Promega; Madison, WI). Slides were mounted in Vectashield with DAPI. Images were acquired using the confocal microscope, followed by counting and analysis of apoptotic nuclei.
Measurement of intracellular ROS
661W photoreceptor cells were treated with 5 μm atRAL for 6 h, incubated with 10 μm H2DCFDA for 10 min at 37 °C, and then measured using a CytoFLEX flow cytometer (Beckman Coulter; Brea, CA). As an alternative, 661W photoreceptor cells were incubated with 5 μm atRAL for 6 h, and stained with 10 μm H2DCFDA or 5 μm MitoSOX Red, a mitochondrial superoxide indicator, at 37 °C for 10 min. Nuclei were labeled by Hoechst 33342. After washing with PBS, the cells were examined using the confocal microscope. 661W photoreceptor cells were pretreated with antioxidant NAC (2 mm) for 2 h, followed by incubation with or without 5 μm atRAL for 6 h. The levels of intracellular ROS were detected by the Beckman Coulter flow cytometer. Control cells were treated with DMSO alone.
Rhodamine-123 staining
The fluorescent dye rhodamine-123, which accumulates within energized mitochondria, was used to assay ΔΨm in an apoptotic cell population. Live cells were stained with rhodamine-123 (10 μg/ml) for 20 min at 37 °C. After washing with PBS, the cells were imaged using the confocal microscope (Tochigi, Japan).
Western blotting
Cells and tissues were lysed in RIPA buffer supplemented with protease and phosphatase inhibitor mixture (Thermo Fisher Scientific; Rockford, IL). Protein concentrations were determined with BCA protein assay kit (Thermo Fisher Scientific; Rockford, IL). Equal quantities of protein were subjected to electrophoresis in 12% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Roche Applied Science, Mannheim, Germany). After blocking with 5% skim milk for 1 h at room temperature, the membranes were incubated with primary antibodies (1:1000 dilution) at 4 °C overnight, followed by incubation with the corresponding secondary antibodies (1:5000 dilution) for 1 h at room temperature. Membranes were developed using ECL Western blotting detection reagents (Advansta; Menlo Park, CA), and blots were analyzed using a ChemiDoc XRS+ Imaging system (Bio-Rad). Immunoreactive bands were quantified by densitometry using ImageJ software.
Genotyping
Genomic DNAs were isolated from mouse tail biopsies using TIANamp Genomic DNA kit (TIANGEN, Beijing, China) according to the manufacturer's instructions. DNA samples were amplified by PCR using specific primers (Table 4). To corroborate Abca4 deletion and Wt allele, samples 1 and 2 were amplified with the ABCR1 and ABCR2 primers, and samples 3 and 4 were amplified with the A0 and N1 primers. For the identification of Rdh8 deletion and Wt allele, samples 5 and 6 were amplified with the DMR5 and DMR6 primers, and samples 7 and 8 were amplified with the Neo1 and DMR11 primers. In contrast, genomic DNAs extracted from mouse tail biopsies were amplified for identifying Wt allele and rd8 mutation of the Crb1 gene using primers mCrb1 mF1, mCrb1 mF2, and mCrb1 mR.
Table 4.
Primers for genotyping
| Primer | Sequences |
|---|---|
| A0 | CCACAGCACACATCAGCATTTCTCC |
| N1 | TGCGAGGCCAGAGGCCACTTGTGTAGC |
| ABCR1 | GCCCAGTGGTCGATCTGTCTAGC |
| ABCR2 | CGGACACAAAGGCCGCTAGGACCACG |
| Neo1 | TGCGAGGCCAGAGGCCACTTGTGTAGC |
| DMR11 | TCCGCCTTGGAAACCTGAGCCAGAAG |
| DMR5 | GAAGGAGCCATTGGAGGCAGCTGC |
| DMR6 | CTGGAAGCACAGCTTTGACCAGAC |
| mCrb1 mF1 | GTGAAGACAGCTACAGTTCTGATC |
| mCrb1 mF2 | GCCCCTGTTTGCATGGAGGAAACTTGGAAGACAGCTACAGTTCTTCTG |
| mCrb1 mR | GCCCCATTTGCACACTGATGAC |
Statistical analyses
All data were analyzed using GraphPad Prism software (Version 5.0; La Jolla, CA). The results were averaged from at least three independent experiments to calculate the mean ± S.D. Statistical analyses were performed using one-way or two-way analyses of variance (ANOVA) followed by Tukey's or Sidak's multiple comparison test and Student's t test, as indicated in the corresponding figure legends. Significant levels retained were *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
Data availability
The data supporting the findings of this study are available within the article and the supporting information.
Author contributions
C. L., B. C., Y. F., J. C., Yiping Wu, J. Z., and Yalin Wu investigation; C. L. and Yalin Wu writing-original draft; Z. L. and Yalin Wu writing-review and editing; Yalin Wu conceptualization; Yalin Wu formal analysis; Yalin Wu supervision; Yalin Wu funding acquisition; Yalin Wu project administration.
Supplementary Material
This work was supported in part by China National Natural Science Foundation Grants 81870671 and 81570857 (to Yalin Wu), Basic Research Program of Shenzhen Grant JCYJ20180306173025004 (to Yalin Wu), Natural Science Foundation of Fujian Province Grant 2017J01148 (to Yalin Wu), and Sanming Project of Medicine in Shenzhen Grant SZSM201612022 (to Z. L. and Yalin Wu). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1 and S2.
- atRAL
- all-trans-retinal
- RPE
- retinal pigment epithelium
- AMD
- age-related macular degeneration
- STGD1
- Stargardt disease
- ANOVA
- analysis of variance
- TUNEL
- terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- DAPI
- 4,6-diamidino-2-phenylindole
- Z
- benzyloxycarbonyl
- FMK
- fluoromethyl ketone
- PARP
- poly-ADP-ribose polymerase
- ROS
- reactive oxygen species
- H2DCFDA
- 2′,7′-dichlorodihydrofluorescein diacetate
- NAC
- N-acetyl-l-cysteine
- JNK
- c-Jun N-terminal kinase
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt
- lx
- lux
- OS
- outer segment
- ONL
- outer nuclear layer
- H&E
- hematoxylin and eosin
- p-JNK
- phosphorylated JNK
- Cyt c
- cytochrome c
- p-c-Jun
- phosphorylated c-Jun
- ATM
- ataxia telangiectasia–mutated
- p-ATM
- phosphorylated ATM
- VEGF
- vascular endothelial growth factor
- H2AX
- histone H2A variant
- γH2AX
- phosphorylated H2AX.
References
- 1. Liu X., Chen J., Liu Z., Li J., Yao K., and Wu Y. (2016) Potential therapeutic agents against retinal diseases caused by aberrant metabolism of retinoids. Invest. Ophthalmol. Vis. Sci. 57, 1017–1030 10.1167/iovs.15-18429 [DOI] [PubMed] [Google Scholar]
- 2. Maeda A., Maeda T., Golczak M., and Palczewski K. (2008) Retinopathy in mice induced by disrupted all-trans-retinal clearance. J. Biol. Chem. 283, 26684–26693 10.1074/jbc.M804505200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maeda T., Golczak M., and Maeda A. (2012) Retinal photodamage mediated by all-trans-retinal. Photochem. Photobiol. 88, 1309–1319 10.1111/j.1751-1097.2012.01143.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liao Y., Zhang H., He D., Wang Y., Cai B., Chen J., Ma J., Liu Z., and Wu Y. (2019) Retinal pigment epithelium cell death is associated with NLRP3 inflammasome activation by all-trans retinal. Invest. Ophthalmol. Vis. Sci. 60, 3034–3045 10.1167/iovs.18-26360 [DOI] [PubMed] [Google Scholar]
- 5. Maeda A., Maeda T., Golczak M., Chou S., Desai A., Hoppel C. L., Matsuyama S., and Palczewski K. (2009) Involvement of all-trans-retinal in acute light-induced retinopathy of mice. J. Biol. Chem. 284, 15173–15183 10.1074/jbc.M900322200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen Y., Okano K., Maeda T., Chauhan V., Golczak M., Maeda A., and Palczewski K. (2012) Mechanism of all-trans-retinal toxicity with implications for Stargardt disease and age-related macular degeneration. J. Biol. Chem. 287, 5059–5069 10.1074/jbc.M111.315432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kiser P. D., Golczak M., Maeda A., and Palczewski K. (2012) Key enzymes of the retinoid (visual) cycle in vertebrate retina. Biochim. Biophys. Acta 1821, 137–151 10.1016/j.bbalip.2011.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Travis G. H., Golczak M., Moise A. R., and Palczewski K. (2007) Diseases caused by defects in the visual cycle: retinoids as potential therapeutic agents. Annu. Rev. Pharmacol. Toxicol. 47, 469–512 10.1146/annurev.pharmtox.47.120505.105225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Biswas-Fiss E. E., Affet S., Ha M., and Biswas S. B. (2012) Retinoid binding properties of nucleotide binding domain 1 of the Stargardt disease-associated ATP binding cassette (ABC) transporter, ABCA4. J. Biol. Chem. 287, 44097–44107 10.1074/jbc.M112.409623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen C., Thompson D. A., and Koutalos Y. (2012) Reduction of all-trans-retinal in vertebrate rod photoreceptors requires the combined action of RDH8 and RDH12. J. Biol. Chem. 287, 24662–24670 10.1074/jbc.M112.354514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maeda A., Maeda T., Imanishi Y., Kuksa V., Alekseev A., Bronson J. D., Zhang H., Zhu L., Sun W., Saperstein D. A., Rieke F., Baehr W., and Palczewski K. (2005) Role of photoreceptor-specific retinol dehydrogenase in the retinoid cycle in vivo. J. Biol. Chem. 280, 18822–18832 10.1074/jbc.M501757200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rattner A., Smallwood P. M., and Nathans J. (2000) Identification and characterization of all-trans-retinol dehydrogenase from photoreceptor outer segments, the visual cycle enzyme that reduces all-trans-retinal to all-trans-retinol. J. Biol. Chem. 275, 11034–11043 10.1074/jbc.275.15.11034 [DOI] [PubMed] [Google Scholar]
- 13. Adler L. 4th., Chen C., and Koutalos Y. (2014) Mitochondria contribute to NADPH generation in mouse rod photoreceptors. J. Biol. Chem. 289, 1519–1528 10.1074/jbc.M113.511295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Futterman S., Hendrickson A., Bishop P. E., Rollins M. H., and Vacano E. (1970) Metabolism of glucose and reduction of retinaldehyde in retinal photoreceptors. J. Neurochem. 17, 149–156 10.1111/j.1471-4159.1970.tb02195.x [DOI] [PubMed] [Google Scholar]
- 15. Li J., Cai X., Xia Q., Yao K., Chen J., Zhang Y., Naranmandura H., Liu X., and Wu Y. (2015) Involvement of endoplasmic reticulum stress in all-trans-retinal-induced retinal pigment epithelium degeneration. Toxicol. Sci. 143, 196–208 10.1093/toxsci/kfu223 [DOI] [PubMed] [Google Scholar]
- 16. Li J., Zhang Y., Cai X., Xia Q., Chen J., Liao Y., Liu Z., and Wu Y. (2016) All-trans-retinal dimer formation alleviates the cytotoxicity of all-trans-retinal in human retinal pigment epithelial cells. Toxicology 371, 41–48 10.1016/j.tox.2016.10.005 [DOI] [PubMed] [Google Scholar]
- 17. Sawada O., Perusek L., Kohno H., Howell S. J., Maeda A., Matsuyama S., and Maeda T. (2014) All-trans-retinal induces Bax activation via DNA damage to mediate retinal cell apoptosis. Exp. Eye Res. 123, 27–36 10.1016/j.exer.2014.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rosette C., and Karin M. (1996) Ultraviolet light and osmotic stress: activation of the JNK cascade through multiple growth factor and cytokine receptors. Science 274, 1194–1197 10.1126/science.274.5290.1194 [DOI] [PubMed] [Google Scholar]
- 19. Davis R. J. (2000) Signal transduction by the JNK group of MAP kinases. Cell 103, 239–252 10.1016/S0092-8674(00)00116-1 [DOI] [PubMed] [Google Scholar]
- 20. Johnson G. L., and Lapadat R. (2002) Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298, 1911–1912 10.1126/science.1072682 [DOI] [PubMed] [Google Scholar]
- 21. Lin A., and Dibling B. (2002) The true face of JNK activation in apoptosis. Aging Cell 1, 112–116 10.1046/j.1474-9728.2002.00014.x [DOI] [PubMed] [Google Scholar]
- 22. Sabapathy K., Hochedlinger K., Nam S. Y., Bauer A., Karin M., and Wagner E. F. (2004) Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun–dependent cell proliferation. Mol. Cell 15, 713–725 10.1016/j.molcel.2004.08.028 [DOI] [PubMed] [Google Scholar]
- 23. Dhanasekaran D. N., and Reddy E. P. (2008) JNK signaling in apoptosis. Oncogene 27, 6245–6251 10.1038/onc.2008.301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen Y. R., Wang X., Templeton D., Davis R. J., and Tan T. H. (1996) The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and γ radiation. Duration of JNK activation may determine cell death and proliferation. J. Biol. Chem. 271, 31929–31936 10.1074/jbc.271.50.31929 [DOI] [PubMed] [Google Scholar]
- 25. Hunot S., Vila M., Teismann P., Davis R. J., Hirsch E. C., Przedborski S., Rakic P., and Flavell R. A. (2004) JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 101, 665–670 10.1073/pnas.0307453101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhu X., Raina A. K., Rottkamp C. A., Aliev G., Perry G., Boux H., and Smith M. A. (2001) Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer's disease. J. Neurochem. 76, 435–441 10.1046/j.1471-4159.2001.00046.x [DOI] [PubMed] [Google Scholar]
- 27. Panaretakis T., Pokrovskaja K., Shoshan M. C., and Grandér D. (2002) Activation of Bak, Bax, and BH3-only proteins in the apoptotic response to doxorubicin. J. Biol. Chem. 277, 44317–44326 10.1074/jbc.M205273200 [DOI] [PubMed] [Google Scholar]
- 28. Zhang T., Inesta-Vaquera F., Niepel M., Zhang J., Ficarro S. B., Machleidt T., Xie T., Marto J. A., Kim N., Sim T., Laughlin J. D., Park H., LoGrasso P. V., Patricelli M., Nomanbhoy T. K., et al. (2012) Discovery of potent and selective covalent inhibitors of JNK. Chem. Biol. 19, 140–154 10.1016/j.chembiol.2011.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shiloh Y. (2006) The ATM-mediated DNA-damage response: taking shape. Trends Biochem. Sci. 31, 402–410 10.1016/j.tibs.2006.05.004 [DOI] [PubMed] [Google Scholar]
- 30. Blackford A. N., and Jackson S. P. (2017) ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol. Cell 66, 801–817 10.1016/j.molcel.2017.05.015 [DOI] [PubMed] [Google Scholar]
- 31. Mao X., Yu C. R., Li W. H., and Li W. X. (2008) Induction of apoptosis by shikonin through a ROS/JNK-mediated process in Bcr/Abl-positive chronic myelogenous leukemia (CML) cells. Cell Res. 18, 879–888 10.1038/cr.2008.86 [DOI] [PubMed] [Google Scholar]
- 32. Johnson-Cadwell L. I., Jekabsons M. B., Wang A., Polster B. M., and Nicholls D. G. (2007) 'Mild Uncoupling' does not decrease mitochondrial superoxide levels in cultured cerebellar granule neurons but decreases spare respiratory capacity and increases toxicity to glutamate and oxidative stress. J. Neurochem. 101, 1619–1631 10.1111/j.1471-4159.2007.04516.x [DOI] [PubMed] [Google Scholar]
- 33. de Jong P. T. (2006) Age-related macular degeneration. N. Engl. J. Med. 355, 1474–1485 10.1056/NEJMra062326 [DOI] [PubMed] [Google Scholar]
- 34. Ambati J., and Fowler B. J. (2012) Mechanisms of age-related macular degeneration. Neuron 75, 26–39 10.1016/j.neuron.2012.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Massé H., Wolff B., Bonnabel A., Bourhis A., Cornut P. L., De Bats F., Gualino V., Halfon J., Koehrer P., Souteyrand G., Streho M., Tick S., Zerbib J., and Chartier C. (2016) Overview of medical practices in wet AMD in France. J. Fr. Ophthalmol. 39, 40–47 10.1016/j.jfo.2015.10.001 [DOI] [PubMed] [Google Scholar]
- 36. Kulkarni A. D., and Kuppermann B. D. (2005) Wet age-related macular degeneration. Adv. Drug Deliv. Rev. 57, 1994–2009 10.1016/j.addr.2005.09.003 [DOI] [PubMed] [Google Scholar]
- 37. Du H., Sun X., Guma M., Luo J., Ouyang H., Zhang X., Zeng J., Quach J., Nguyen D. H., Shaw P. X., Karin M., and Zhang K. (2013) JNK inhibition reduces apoptosis and neovascularization in a murine model of age-related macular degeneration. Proc. Natl. Acad. Sci. U.S.A. 110, 2377–2382 10.1073/pnas.1221729110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guma M., Rius J., Duong-Polk K. X., Haddad G. G., Lindsey J. D., and Karin M. (2009) Genetic and pharmacological inhibition of JNK ameliorates hypoxia-induced retinopathy through interference with VEGF expression. Proc. Natl. Acad. Sci. U.S.A. 106, 8760–8765 10.1073/pnas.0902659106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bhutto I., and Lutty G. (2012) Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch's membrane/choriocapillaris complex. Mol. Aspects Med. 33, 295–317 10.1016/j.mam.2012.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu L., Nagasaki T., and Sparrow J. R. (2010) Photoreceptor cell degeneration in Abcr(−/−) mice. Adv. Exp. Med. Biol. 664, 533–539 10.1007/978-1-4419-1399-9_61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li P., Nijhawan D., Budihardjo I., Srinivasula S. M., Ahmad M., Alnemri E. S., and Wang X. (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479–489 10.1016/S0092-8674(00)80434-1 [DOI] [PubMed] [Google Scholar]
- 42. Jiang X., and Wang X. (2000) Cytochrome c promotes caspase-9 activation by inducing nucleotide binding to Apaf-1. J. Biol. Chem. 275, 31199–31203 10.1074/jbc.C000405200 [DOI] [PubMed] [Google Scholar]
- 43. Boulares A. H., Yakovlev A. G., Ivanova V., Stoica B. A., Wang G., Iyer S., and Smulson M. (1999) Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J. Biol. Chem. 274, 22932–22940 10.1074/jbc.274.33.22932 [DOI] [PubMed] [Google Scholar]
- 44. Modesti M., and Kanaar R. (2001) DNA repair: spot(light)s on chromatin. Curr. Biol. 11, R229–R232 10.1016/S0960-9822(01)00112-9 [DOI] [PubMed] [Google Scholar]
- 45. Burma S., Chen B. P., Murphy M., Kurimasa A., and Chen D. J. (2001) ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 276, 42462–42467 10.1074/jbc.C100466200 [DOI] [PubMed] [Google Scholar]
- 46. Matsuoka S., Ballif B. A., Smogorzewska A., McDonald E. R. 3rd., Hurov K. E., Luo J., Bakalarski C. E., Zhao Z., Solimini N., Lerenthal Y., Shiloh Y., Gygi S. P., and Elledge S. J. (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166 10.1126/science.1140321 [DOI] [PubMed] [Google Scholar]
- 47. Shimada K., Nakamura M., Anai S., De Velasco M., Tanaka M., Tsujikawa K., Ouji Y., and Konishi N. (2009) A novel human AlkB homologue, ALKBH8, contributes to human bladder cancer progression. Cancer Res. 69, 3157–3164 10.1158/0008-5472.CAN-08-3530 [DOI] [PubMed] [Google Scholar]
- 48. Kamata H., Honda S., Maeda S., Chang L., Hirata H., and Karin M. (2005) Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120, 649–661 10.1016/j.cell.2004.12.041 [DOI] [PubMed] [Google Scholar]
- 49. Nickell S., Park P. S., Baumeister W., and Palczewski K. (2007) Three-dimensional architecture of murine rod outer segments determined by cryoelectron tomography. J. Cell Biol. 177, 917–925 10.1083/jcb.200612010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Maeda A., Golczak M., Chen Y., Okano K., Kohno H., Shiose S., Ishikawa K., Harte W., Palczewska G., Maeda T., and Palczewski K. (2011) Primary amines protect against retinal degeneration in mouse models of retinopathies. Nat. Chem. Biol. 8, 170–178 10.1038/nchembio.759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li Y., Zhou M., Hu Q., Bai X. C., Huang W., Scheres S. H., and Shi Y. (2017) Mechanistic insights into caspase-9 activation by the structure of the apoptosome holoenzyme. Proc. Natl. Acad. Sci. U.S.A. 114, 1542–1547 10.1073/pnas.1620626114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hill M. M., Adrain C., Duriez P. J., Creagh E. M., and Martin S. J. (2004) Analysis of the composition, assembly kinetics and activity of native Apaf-1 apoptosomes. EMBO J. 23, 2134–2145 10.1038/sj.emboj.7600210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lu C., Zhu F., Cho Y. Y., Tang F., Zykova T., Ma W. Y., Bode A. M., and Dong Z. (2006) Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol. Cell 23, 121–132 10.1016/j.molcel.2006.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhao J., Liao Y., Chen J., Dong X., Gao Z., Zhang H., Wu X., Liu Z., and Wu Y. (2017) Aberrant buildup of all-trans-retinal dimer, a nonpyridinium bisretinoid lipofuscin fluorophore, contributes to the degeneration of the retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 58, 1063–1075 10.1167/iovs.16-20734 [DOI] [PubMed] [Google Scholar]
- 55. Heibein J. A., Barry M., Motyka B., and Bleackley R. C. (1999) Granzyme B-induced loss of mitochondrial inner membrane potential (Δψm) and cytochrome c release are caspase independent. J. Immunol. 163, 4683–4693 [PubMed] [Google Scholar]
- 56. Perez A. R., Pritykin Y., Vidigal J. A., Chhangawala S., Zamparo L., Leslie C. S., and Ventura A. (2017) GuideScan software for improved single and paired CRISPR guide RNA design. Nat. Biotechnol. 35, 347–349 10.1038/nbt.3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available within the article and the supporting information.