

Key Points
MYH9-RD mutations R702C, D1424N, and E1841K impair MK chemotaxis from the endosteum to the vasculature, causing thrombocytopenia.
These mutations suppress chemotaxis via distinct mechanisms, including defective cell adhesion and actomyosin cytoskeleton misalignment.
Abstract
Megakaryocytes (MKs), the precursor cells for platelets, migrate from the endosteal niche of the bone marrow (BM) toward the vasculature, extending proplatelets into sinusoids, where circulating blood progressively fragments them into platelets. Nonmuscle myosin IIA (NMIIA) heavy chain gene (MYH9) mutations cause macrothrombocytopenia characterized by fewer platelets with larger sizes leading to clotting disorders termed myosin-9–related disorders (MYH9-RDs). MYH9-RD patient MKs have proplatelets with thicker and fewer branches that produce fewer and larger proplatelets, which is phenocopied in mouse Myh9-RD models. Defective proplatelet formation is considered to be the principal mechanism underlying the macrothrombocytopenia phenotype. However, MYH9-RD patient MKs may have other defects, as NMII interactions with actin filaments regulate physiological processes such as chemotaxis, cell migration, and adhesion. How MYH9-RD mutations affect MK migration and adhesion in BM or NMIIA activity and assembly prior to proplatelet production remain unanswered. NMIIA is the only NMII isoform expressed in mature MKs, permitting exploration of these questions without complicating effects of other NMII isoforms. Using mouse models of MYH9-RD (NMIIAR702C+/−GFP+/−, NMIIAD1424N+/−, and NMIIAE1841K+/−) and in vitro assays, we investigated MK distribution in BM, chemotaxis toward stromal-derived factor 1, NMIIA activity, and bipolar filament assembly. Results indicate that different MYH9-RD mutations suppressed MK migration in the BM without compromising bipolar filament formation but led to divergent adhesion phenotypes and NMIIA contractile activities depending on the mutation. We conclude that MYH9-RD mutations impair MK chemotaxis by multiple mechanisms to disrupt migration toward the vasculature, impairing proplatelet release and causing macrothrombocytopenia.
Visual Abstract

Introduction
Megakaryocyte (MK) to platelet differentiation starts in the endosteal niche of bones, proceeding through intermediate steps until MKs migrate to the vasculature, where they form proplatelets, long protrusions from the cell body extending into the sinusoids. Here, the shear forces of blood flow release platelets from the proplatelets into the circulation. This cellular transformation requires DNA replication to amplify ploidy, transcription, and translation of MK lineage genes and profound changes in membrane and cytoskeletal structures.1-9 In an alternate model for thrombopoiesis, MKs transmigrate through endothelial junctions into bone marrow (BM) sinusoids and are delivered to lung capillaries via blood circulation, where they are fragmented into platelets.10-12 Thus, MK migration toward vasculature is a crucial step in various models of platelet biogenesis.
The actin cytoskeleton and numerous actin-binding proteins, like nonmuscle myosin IIA (NMIIA), Tmod3, filamin A, α-actinin1, and cofilin1 play crucial roles in proplatelet generation.13-17 NMIIA, a motor protein with actin-dependent ATPase activity,18 is a key player in thrombopoiesis, as mutations in the MYH9 gene coding for NMIIA result in macrothrombocytopenia, coagulation defects, and other disorders, which are grouped together as myosin-9–related disorders (MYH9-RDs).19-21 Cultured MKs from patients with MYH9-RD mutations have thicker proplatelets with significantly reduced secondary processes that release fewer proplatelets, a phenotype that can be rescued by treatment of MKs in vitro with blebbistatin, a NMIIA motor inhibitor. Furthermore, cultured MKs from MYH9-RD patients with motor mutations (R702H) tend to have fewer numbers of proplatelet-forming MKs than MKs with rod mutations (P1444L). It was also reported that MYH9-RD mutant MKs have enhanced force production in traction force microscopy,22 implicating increased NMIIA contractile activity in defective proplatelet biogenesis. In contradiction, another study using MK cell lines overexpressing NMIIA with MYH9-RD mutations observed that the production of larger platelets results from reduced force production in preplatelets.23 Thus, the underlying molecular basis for NMIIA dysfunction in MKs and how specific MYH9-RD mutations contribute to MK differentiation and maturation processes prior to proplatelet biogenesis remain unclear.
Structurally, NMIIA consists of 2 heavy chains (HCs), 2 regulatory light chains (RLC), and 2 essential light chains. Phosphorylation and dephosphorylation of the RLC and HC, respectively, regulate the motor enzymatic activity and ability to form bipolar filaments.24 NMII bipolar filaments interact with actin filaments (F-actin) to generate mechanical force, driving cell migration, shape changes, and adhesion.25-28 Since MKs migrate from the endosteal niche to the BM vasculature to release proplatelets, we hypothesized that an underlying cause of thrombocytopenia in MYH9-RD could be defective migration of MYH9-RD MKs to sinusoids, resulting in fewer proplatelets released into circulation. Furthermore, MK migration defects could also be accompanied or exacerbated by increased adhesion in the endosteal niche. This would result in proplatelet generation away from sinusoids under low shear conditions, ultimately leading to macrothrombocytopenia.
We studied how NMIIA regulates specific functions during stages of MK maturation prior to proplatelet extension and determined that mutations in different domains of the NMIIA molecule all impair MK chemotaxis. However, the mechanisms suppressing chemotaxis are unique for each of the point mutations located in different regions of the NMIIA molecule (Figure 1A). While the R702C mutation in the motor domain leads to a loss of contractile function phenotype, the D1424N mutation in the rod domain generates a gain of contractile function. E1841K, a second mutation in the rod domain, results in NMIIA bipolar filaments with altered dimensions that lead to misalignment of the actomyosin cytoskeleton, thereby also resulting in dysfunctional NMIIA contractile function. These results identify novel mechanisms explaining the macrothrombocytopenia phenotype observed in MYH9-RDs. Furthermore, we show that site-specific mutations in the NMIIA molecule regulate MK adhesion and migration functions differently. These findings indicate that development of multiple therapeutic strategies is required to enhance or reduce abnormal NMIIA contractility for treatment of MYH9-RDs.
Figure 1.
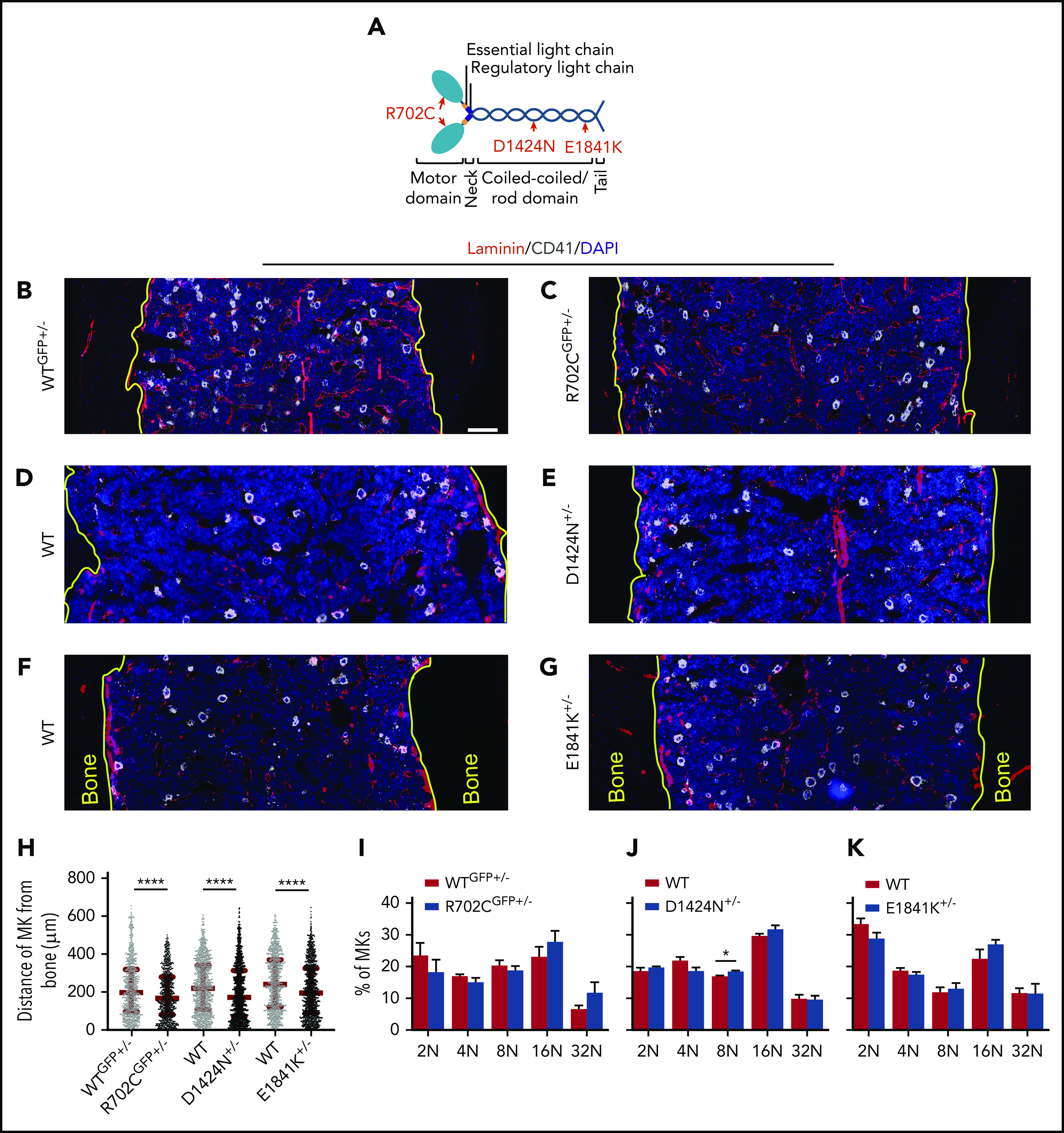
MKs with Myh9-RD mutations have aberrant distribution in BM. (A) Diagram depicting the domains of NMIIA molecule with MYH9-RD mutations highlighted in red. DAPI, 4′,6-diamidino-2-phenylindole. (B-G) Confocal images of femoral cryosections from Myh9-RD mice models and WT littermates following immunostaining for sinusoids (laminin) and MK (CD41). Yellow tracings represent the junction of bone and BM. Scale bar, 100 μm. (H) Scatterplots representing median distances of MKs from the endosteal niche (WTGFP+/−, n = 938; R702CGFP+/−, n = 777; WT, n = 1236; D1424N+/−, n = 1513; WT, n = 1101; E1841K+/−m N = 1165). Images were acquired with a Plan-Fluor ×63 objective (numerical aperture [NA], 1.4) of a Zeiss LSM 780 confocal microscope with Zeiss AxioCam. Images were processed and stitched together with ZEN software from Zeiss. (I-K) Ploidy levels in Myh9-RD mutant MKs harvested from mouse BM analyzed by flow cytometry, represented as mean ± standard error of the mean from ≥3 mice of each genotype.
Methods
Detailed descriptions of experimental methods are in supplemental Methods (available on the Blood Web site).
Mice
Wild-type (WT) and Myh9-RD mouse strains21 were obtained from Robert Adelstein (National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD). WT littermates were used as controls for the heterozygous mice from each mutant strain. To generate NMIIAGFP/NMIIAD1424N and NMIIAGFP/NMIIAE1841K mice, NMIIAGFP/NMIIAGFP mice were crossed with NMIIAD1424N+/− and NMIIAE1841K+/+ mice, respectively. Mice breeding and experiments were performed according to Institutional Animal Care and Use Committee guidelines at Scripps Research Institute and University of Delaware (28 1345-2018-0).
MK culture
Cells from mouse BM were differentiated into MKs as described previously.29
Immunofluorescence and confocal and superresolution microscopy
Fixed and decalcified bones were cryosectioned for immunofluorescence as described previously.30 Cultured MKs were fixed in 4% paraformaldehyde, followed by blocking and permeabilization with 0.3% Tween20. Cells or tissue sections were incubated overnight with primary antibodies at 4°C, followed by phalloidin/secondary antibody incubation at room temperature. Mounted samples were examined by confocal or superresolution fluorescence microscopy.
Cell migration assay
Cell migration assay protocols were modified from a method previously described.31-33 MKs were allowed to attach in the central well of ibidi chemotaxis slides and their displacement in a stromal-derived factor 1 (SDF-1) gradient was monitored by time-lapse imaging.
MK lysis and western blotting
MK cytoskeleton was fractionated as described previously.34 Protein levels in MK lysates were quantified by Bradford assay. Thirty micrograms was loaded on gradient sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels, subjected to western blotting, and labeled bands imaged with LiCor Odyssey.
Flow cytometry
MK ploidy analysis and CXCR4 surface expression were performed with FACSCalibur and FACSAria, respectively, modifying protocols as described elsewhere.33,35 Data analysis was performed with FlowJo.v10.
NMIIA assembly assays
NMIIA-HC with or without MYH9-RD mutations were coexpressed with human RLC and mouse essential light chains in Sf9 cells.24 Purified recombinant holomolecules were examined by electron microscopy to determine whether they were in a disassembled inactive state or assembled into bipolar filaments.
Statistics
Computation of mean, standard errors, and P values were performed by an unpaired Student t test or Mann-Whitney U tests using GraphPad Prism8.
Results
Myh9-RD MKs demonstrate defective migration patterns in SDF-1 gradients
MK chemotaxis toward sinusoids in BM is driven by SDF-1.1,36,37 To study in situ MK distribution in BM, we used mouse models of MYH9-RD generated by inserting the human mutations in the endogenous mouse Myh9 locus.21 We immunostained longitudinal femoral sections for CD41 (MK marker) and laminin (basement membrane of vasculature). We used tissues and cells from mice carrying one copy of the respective NMIIA mutations, as MYH9-RD patients are heterozygous for MYH9 mutations. All Myh9-RD heterozygous mice had increased numbers of MKs in close proximity to the endosteal surface as compared with WT littermate controls, where MKs were situated further from the endosteum, toward the central marrow region (Figure 1B-H). Moreover, fewer MKs in NMIIAR702C-GFP+/− and NMIIAE1841K+/− mice were in direct contact with the sinusoids as compared with WT controls (supplemental Figure 1A,C). Although the percentage of sinusoidally positioned MKs in NMIIAD1424N+/− mice did not reach statistical significance, the data trend was similar to the other mutants (supplemental Figure 1B). To eliminate the possibility that enhanced accumulation of Myh9-RD MKs in proximity to the endosteal regions resulted from MK maturation defects, we quantified ploidy levels. Barring a small increase of MKs with 8N ploidy in NMIIAD1424N+/− mice, we did not observe any differences in ploidy compared with control mice (Figure 1I-K). Hence, the data are consistent with the hypothesis that MYH9-RD mutations impair MK migration toward the BM sinusoids.
To extend these in vivo observations, we used an in vitro migration assay to track the motility of MKs in response to SDF-1 gradients using time-lapse imaging. Consistent with prior reports,38 a majority of MKs had relatively small net displacements, unlike other rapidly migrating BM cells (supplemental Videos 1-6). Plotting the trajectories of MKs showed that WT cells demonstrated a strong net movement toward the increasing SDF-1 gradient (Figure 2A,C,E), while a majority of mutant MKs appeared to migrate in random directions (Figure 2B,D,F). This is further evident on computing forward migration indices (Figure 2G). To determine whether the reduced chemotaxis is due to slower speed, we calculated MK velocities. Unlike a previous study that measured in vivo MK velocities of 0.38 ± 0.47 μm/min,38 in vitro migration rates of primary WT MKs are significantly higher (∼1.73 μm/min). Although the R702C motor domain mutation reduces MK velocity by ∼37%, the D1424N mutation has the opposite effect and significantly increases MK velocity by ∼27%. The E1841K mutation does not affect MK velocities (Figure 2H). Thus, loss of chemotaxis for the mutant MKs is coupled with a slower velocity in the case of R702C MKs but a faster velocity in the case of D1424N MKs and remains unchanged in the case of E1841K MKs, suggesting that the mechanism for chemotaxis defects is different for the 3 mutants. Since surface expression levels of CXCR4, the SDF-1 receptor, can regulate MK migration,33 we quantified CXCR4 levels on MK membranes to eliminate the possibility that the differences in chemotaxis are due to variations in receptor levels in the different mutants. However, there were no significant differences in the CXCR4 levels in the mutants compared with the WT controls (Figure 2I). Thus, in vitro assays corroborated in vivo data, demonstrating that motility defects in Myh9-RD mutant MKs can explain their altered distribution in mouse BM niches.
Figure 2.
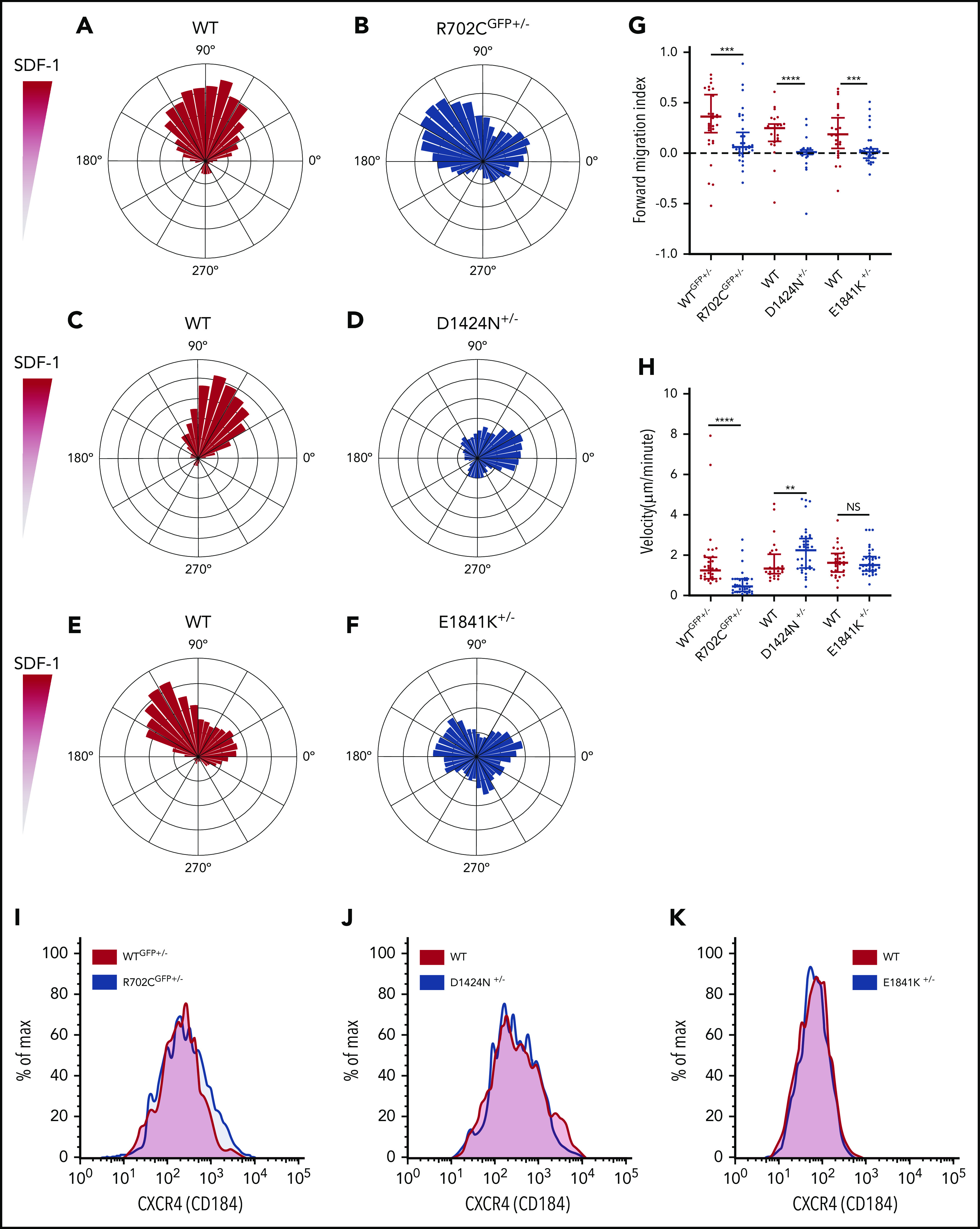
MKs with Myh9-RD mutations have defective migration patterns in SDF-1 gradients. (A-F) Wind-rose plots of WT and Myh9-RD mutant MKs toward SDF-1. The length of each leaflet represents the migration trajectory as percentage of cells (WTGFP+/−, n = 34; R702CGFP+/−, n = 40; WT, n = 30; D1424N+/−, n = 42; WT, n = 31; E1841K+/−, n = 36). For cell migration assays, live-cell imaging was performed at 37°C and 5% CO2 with an achromat ×10 (NA, 0.25) objective of a Nikon Eclipse TE200 microscope. Images were taken with a CoolSnap HQ2 camera with Metamorph software (Molecular Devices). (G) Scatterplots representing median and interquartile range of the distribution for forward migration indices of WT and mutant MKs up the SDF-1 gradient. Directional migration is indicated by values >0.1. (H) Scatterplots representing median distribution of MK velocities for WT and mutants. NS, not significant. (I-K) Representative histograms from 3 mice of each genotype showing the surface expression of CXCR4 on MKs by flow cytometry. **P ≤ .01; ***P ≤ .001; ****P ≤ .0001.
MKs with Myh9-RD mutations in NMIIA motor and rod domains differ in FA assembly and NMIIA activity
Apart from chemotaxis, NMIIA also plays crucial roles in cell adhesion.39,40 The sizes of focal adhesion (FA) sites have been shown to bear a biphasic correlation with migration rates. As FA sizes increase, there is a concomitant rise in velocity until a threshold speed is reached, followed by drop in cell speed as FA sizes increase further.41,42 Hence, an alternate explanation for the defective distribution of Myh9-RD MKs in the BM could be that the cells are more adherent to matrices constituting the osteoblastic niche, suppressing chemotaxis. Since the endosteum is enriched in collagen-I,43 we spread WT and mutant MKs on collagen-I–coated coverslips and measured cell-surface areas. Areas of all Myh9-RD mutant MKs on collagen-I were significantly reduced compared with WT controls (supplemental Figure 3A-G). As NMIIA activity regulates vinculin recruitment to FA structures,44 we next quantified the area of vinculin-positive adhesion structures in MKs attached to collagen-I. While NMIIAD1424N+/− MKs had significantly larger vinculin-positive adhesion sites compared with WT littermates (Figure 3C,D,G), the vinculin-positive sites in NMIIAR702C-GFP+/− MKs were smaller than their WT littermates (Figure 3A,B,G). NMIIAE1841K+/− MKs showed no significant differences in the sizes of adhesion structures compared with WT MKs (Figure 3E,F,G). With the exception of the R702C mutation, we did not observe any differences in the number of adhesion-structures per unit area (Figure 3H), although due to the reduced spreading area of mutant MKs (supplemental Figure 3G), there were fewer adhesion structures in each MK (supplemental Figure 3H). Together, these data suggest that the R702C and D1424N mutations in NMIIA have antagonistic effects on MK adhesion to collagen-I, with R702C decreasing and D1424N increasing adhesion. Since a recent study has reported that MKs cultured on matrices with enhanced stiffness vs liquid culture are more mature,45 we next compared the adhesion of MKs grown in liquid culture vs 2% methylcellulose. On quantifying the areas of MK spread on collagen-I (supplemental Figure 4A-C) and vinculin-positive FA sites (supplemental Figure 4D-F), we did not observe significant differences in MKs from the 2 different culture conditions.
Figure 3.
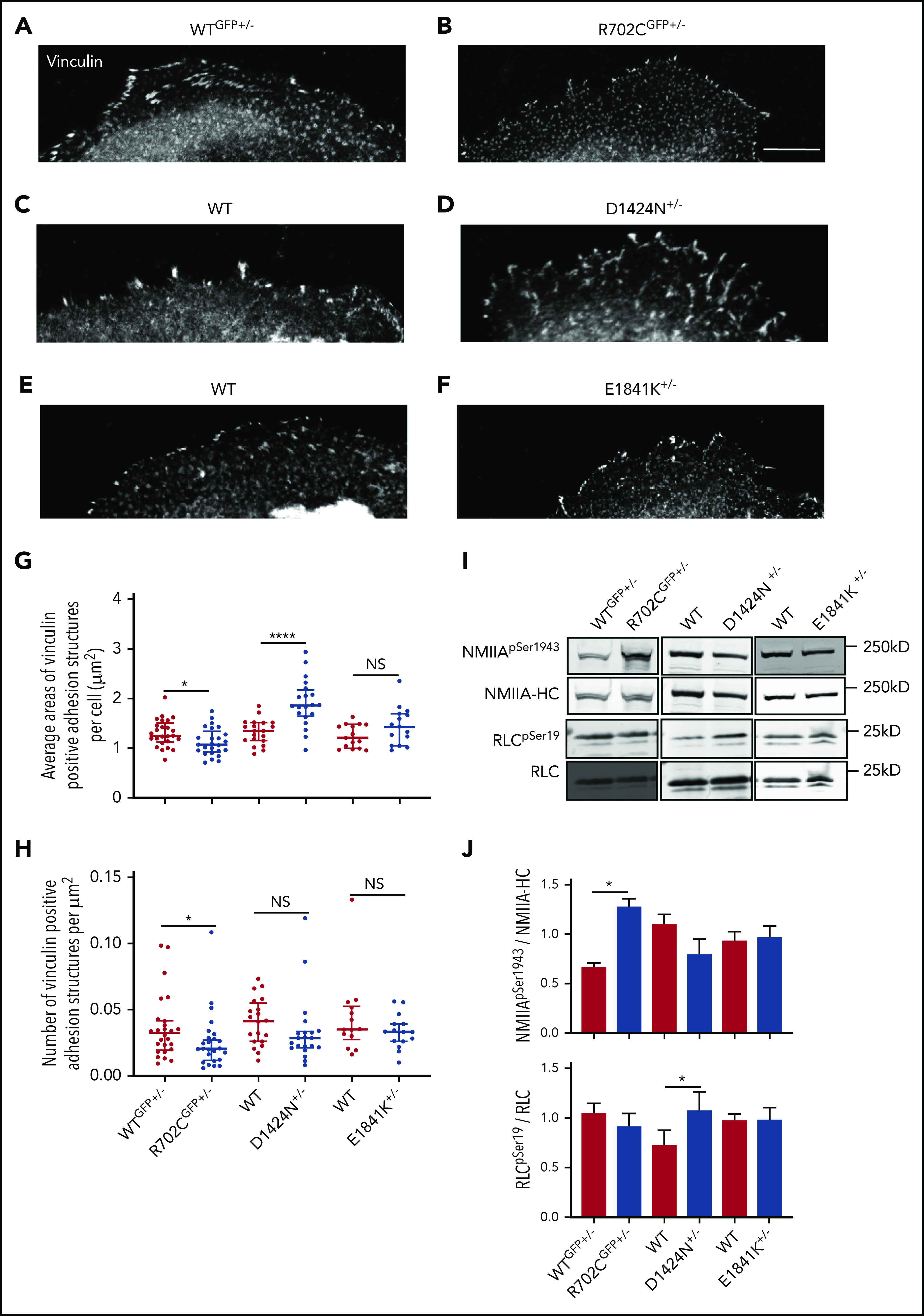
Motor and rod domain mutations associated with Myh9-RD differentially affect cell adhesion and NMIIA functions. (A-F) Vinculin staining of MKs spread on collagen-1 coverslips. Scale bar, 25μm. Images were acquired with a Plan-Fluor ×63 objective (N.A. 1.4) of a Zeiss LSM 780 confocal microscope with Zeiss AxioCam. Images were processed with ZEN software available from Zeiss. Analysis of vinculin-positive focal adhesion sites were performed with ImageJ. (G) Scatterplots representing median and interquartile range of the distribution for areas of vinculin-positive structures in WT and Myh9-RD mice MKs. (H) Scatterplots representing median and interquartile range of the distribution for vinculin-positive sites per unit area in MKs from Myh9-RD and WT littermate control mice (WTGFP+/−, n = 25; R702CGFP+/−, n = 25; WT, n = 20; D1424N+/−, n = 20; WT, n = 15; E1841K+/−, n = 15). (I) Representative immunoblots showing phosphorylated RLC (RLCSer19) and total RLC levels or showing phosphorylated myosin HC (NMIIASer1943) and total NMIIA-HC levels in WT and mutant MKs. (J) Graph represents quantification of immunoblots as mean ± standard error of the mean from ≥3 experiments
Since adhesion properties of platelets regulate stable thrombus formation, we next determined whether thrombin-activated platelets from Myh9-RD mice demonstrate defective spreading on fibrinogen. Areas of Myh9-RD platelets are significantly larger due to the larger resting platelet sizes conferred by the mutations (supplemental Figure 5A-D). However, upon classifying platelets as unspread, extending filopodia and/or lamellipodia (supplemental Figure 5E), we observed that the percentage of unspread platelets is elevated while that of platelets extending lamellipodia is reduced for the D1424N mutation (supplemental Figure 5G), indicating defective platelet spreading, similar to the D1424N MKs.
To investigate whether the differences in cell adhesion properties observed in R702C and D1424N mutant MKs are due to altered NMIIA contractility, we measured RLC phosphorylation. Phosphorylation and dephosphorylation of the RLC results in increased and decreased NMIIA contractile activity, respectively18,46-48. Immunoblots revealed that NMIIAD1424N+/− MKs have significantly higher levels of phosphorylated RLC as compared with WT controls, suggesting increased contractility (Figure 3I-J). On the contrary, MKs from NMIIAR702C-GFP+/− and NMIIAE1841K+/− mice did not show any differences in RLC phosphorylation (Figure 3I-J). NMIIA contractility in cells also depends on bipolar filament assembly, with NMIIA-HC phosphorylation at Ser1943 associated with reductions in assembled myosin.49 NMIIAR702C-GFP+/− MKs have significantly elevated HC-pSer1943, while neither NMIIAD1424N+/− nor NMIIAE1841K+/− MKs have any significant changes in HC-pSer1943 as compared with WT controls. This suggests that NMIIAR702C-GFP+/− may have impaired filament assembly or stability, while NMIIAE1841K+/− and NMIIAD1424N+/− MKs do not. Hence, we conclude that NMIIAD1424N+/− MKs have a contractile gain of function with increased phosphorylated RLC, whereas NMIIAR702C+/− MKs have impaired bipolar filament formation or turnover based on increased HC pSer1943, with a consequent loss of contractile function, consistent with decreased vinculin-positive FA sizes shown above.
In control experiments, to determine whether loss of contractile function for the R702C mutation could be due to impaired stability of the mutant protein, we compared NMIIAR702C-GFP protein levels vs NMIIAGFP and NMIIA proteins following translation inhibition with cycloheximide for 4 hours. Immunoblots showed that there was a minor increase in NMIIAR702C-GFP following cycloheximide treatment (supplemental Figure 6) rather than a decrease, indicating that the GFP tag did not compromise the stability of the protein. In addition, we performed immunoblots to determine if the loss of contractile function in NMIIA introduced by the R702C mutation resulted in a compensatory rise in expression of NMIIB or NMIIC (supplemental Figure 3I). However, other NMII protein isoforms in the Myh9-RD MK lysates were not detected.
Myh9-RD mutations in NMIIA motor and tail domains result in altered actomyosin assembly in MKs
Since NMIIA RLC and HC phosphorylation modulates bipolar filament assembly and alignment into continuous stacks along F-actin stress fibers in cells,50-53 we investigated whether NMIIA mutations with altered RLC/HC phosphorylation might affect actomyosin organization in MKs. To visualize endogenous NMIIA assembly in primary MKs, we crossed NMIIAD1424N+/− and NMIIAE1841K+/− mice with NMIIAGFP+/+ mice to generate NMIIAD1424N/NMIIAGFP and NMIIAE1841K/NMIIAGFP mice and confirmed HC heterodimerization by coimmunoprecipitation (supplemental Figure 8A). The MKs were immunostained for the NMIIA tail domain and visualized by Airyscan superresolution microscopy. Large, flat peripheral regions of MKs had a periodic pattern of NMIIA stripes associated with densely packed F-actin bundles in WT and all mutants, along with individual bipolar filaments visible at the extreme cell edges (Figure 4A; supplemental Figure 7A). All the Myh9-RD mutants formed bipolar filaments of similar apparent lengths (Figure 4B-C; supplemental Figure 7B-C). Strikingly, while NMIIAGFP+/− MKs formed prominent stacks aligned orthogonally to the F-actin bundles, stack length was shorter in NMIIAR702C-GFP+/− and NMIIAD1424N/NMIIAGFP MKs (Figure 4D). Stack length for NMIIAE1841K/NMIIAGFP MKs could not be computed, since actomyosin structures appeared misaligned (supplemental Figure 7A,D). Although stack lengths were significantly shorter in both NMIIAR702C-GFP+/− and NMIIAD1424N/NMIIAGFP MKs in comparison with NMIIAGFP+/− MKs, statistical analysis showed that stack lengths were somewhat longer in D1424N MKs compared with R702C MKs (Figure 4D). We measured the periodicity between bipolar filaments along F-actin bundles by line scans of the tail domain intensity for a series of repeating NMIIA bipolar filaments on the F-actin bundles (Figure 4E-F). Bipolar filament periodicities were significantly shorter in NMIIAR702C-GFP+/− and NMIIAD1424N/NMIIAGFP MKs as compared with NMIIAGFP+/− controls (Figure 4G), indicating aberrant NMIIA filament interactions with F-actin. However, NMIIAE1841K/NMIIAGFP bipolar filament periodicities were not significantly different from the control (supplemental Figure 7D-F). Together, these data indicate that MYH9-RD mutations disrupt NMIIA interactions with F-actin stress fibers, without appearing to affect bipolar filament formation itself. Moreover, the propensity of the three mutants to form stacks was different, with the R702C mutation interfering most severely with stack formation.
Figure 4.
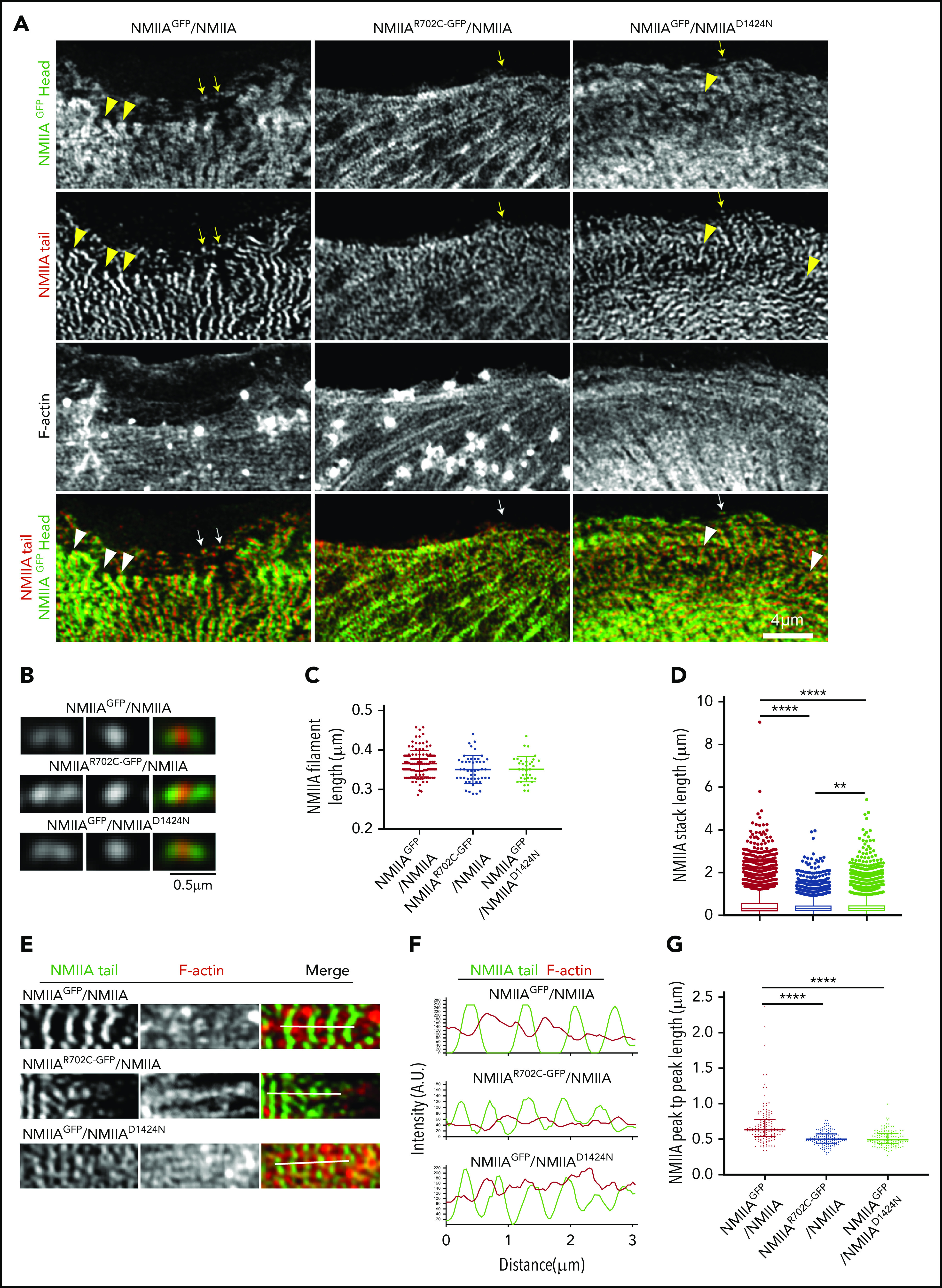
NMIIA mutations associated with Myh9-RDs alter stack assembly on actin stress fibers without affecting bipolar filament formation in MKs. (A) Confocal Airyscan images of NMIIA head, NMIIA tail, and F-actin in MKs spread on collagen I. Arrowheads indicate NMIIA stacks, and arrows indicate NMIIA bipolar filaments, which have been presented as zoomed images in panel B. Scale bars, 4 μm (A) and 0.5 μm (B). (C) Scatterplot showing the median and interquartile range of the distribution for bipolar filament lengths in WT and Myh9-RD mutant MKs (NMIIAGFP/NMIIA, n = 120; NMIIAR702C-GFP/NMIIA, n = 50; NMIIAGFP/NMIIAD1424N, n = 35). (D) Box and whisker plots representing the distribution of the lengths of NMIIA stacks (NMIIAGFP/NMIIA, n = 8614; NMIIAR702C-GFP/NMIIA; n = 5348; NMIIAGFP/NMIIAD1424N, n = 7360). (E) Representative confocal Airyscan images and (F) fluorescence intensity plots indicating the line scans used for graphical analysis of the arrangement of NMIIA filaments on actin stress fibers. (G) Scatterplots representing the median and interquartile range of the distribution for distance between 2 successive NMIIA filaments on actin stress fibers (NMIIAGFP/NMIIA, n = 142; NMIIAR702C-GFP/NMIIA, n = 142; NMIIAGFP/NMIIAD1424N, n = 144). Images were acquired with either a Plan-Apochromat ×63 or ×100 objective (NA, 1.46) of a Zeiss LSM 880 confocal Airyscan microscope with Zeiss AxioCam. Images were processed with ZEN software and quantifications were performed with Nikon Elements 5.1. **P ≤ .01; ***P ≤ .001; ****P ≤ .0001.
Comparison of F-actin staining in MKs indicated that the NMIIAD1424N+/− mutation enhanced stress fiber assembly in contrast to the NMIIAR702C-GFP+/− and NMIIAE1841K+/− mutations, which demonstrated stress fiber formation more similar to WT cells (Figure 4A; supplemental Figure 7). Biochemical fractionation of WT and Myh9-RD MKs to analyze the distribution of actin and NMIIA in the cytosolic (supernatant) and cytoskeletal (pellet) fractions,34 also showed increased accumulation of both proteins in the pellet (insoluble) fraction of NMIIAD1424N+/− MKs, consistent with enhanced contractility, as shown above. However, NMIIA and actin distribution in NMIIAR702C-GFP+/− or NMIIAE1841K+/− MKs did not differ significantly from their WT littermate controls (supplemental Figure 8B-D). These data suggest that the Myh9-RD mutations lead to differential effects on actomyosin contractility, F-actin stress fiber assembly and NMIIA stack formation in MKs.
The E1841K rod mutation impairs NMIIA bipolar filament dimensions in vitro
To test whether altered stack formation in MKs with MYH9-RD mutations is due to intrinsic defects in NMIIA associations, we performed in vitro assays with full-length recombinant NMIIA with the respective point mutations. We evaluated whether MYH9-RD mutations affected formation of the inactive, folded monomeric conformation by examining the structure of each protein in the presence of ATP and absence of RLC phosphorylation.24 Furthermore, we compared the ability of monomers to self-associate in an antiparallel fashion in the folded state (Figure 5A), to determine if mutations perturbed the strength of antiparallel tail domain interactions. All mutants were capable of forming the folded, monomeric state and the antiparallel dimer of the folded state. However, the E1841K mutation significantly enhanced the strength of the antiparallel interaction. Pie chart insets (Figure 5B-E) revealed that a majority (54%) of the E1841K molecules were in the antiparallel dimer state, while the dimeric state was comparatively rare for WT (21%), R702C (22%), or D1424N (22%) molecules. This propensity of the E1841K mutation to alter the rod-rod interactions between myosin molecules was also seen in the context of bipolar filament assembly. Although all mutant proteins were capable of forming bipolar filaments in conditions facilitating filament assembly (Figure 5F-I), E1841K filaments were significantly longer and wider than those formed by WT or the other mutants (Figure 5J-K). Strikingly, many E1841K bipolar filaments lacked a distinct bare zone, with heads projecting outwards along the length of the filament. In contrast, R702C and D1424N bipolar filaments typically had a distinct bare zone similar to WT. Importantly, filaments formed by E1841K had a greater variability in filament dimensions than WT, which could explain why MKs with this mutation lack regular NMIIA stacks, since stack formation is expected to depend partly on alignment of multiple equivalent-sized units into a larger superstructure.54 However, despite the alteration in NMIIA contractile activities in MKs introduced by the R702C or D1424N mutations, no changes in formation or structure of the bipolar filaments were observed in in vitro analyses.
Figure 5.
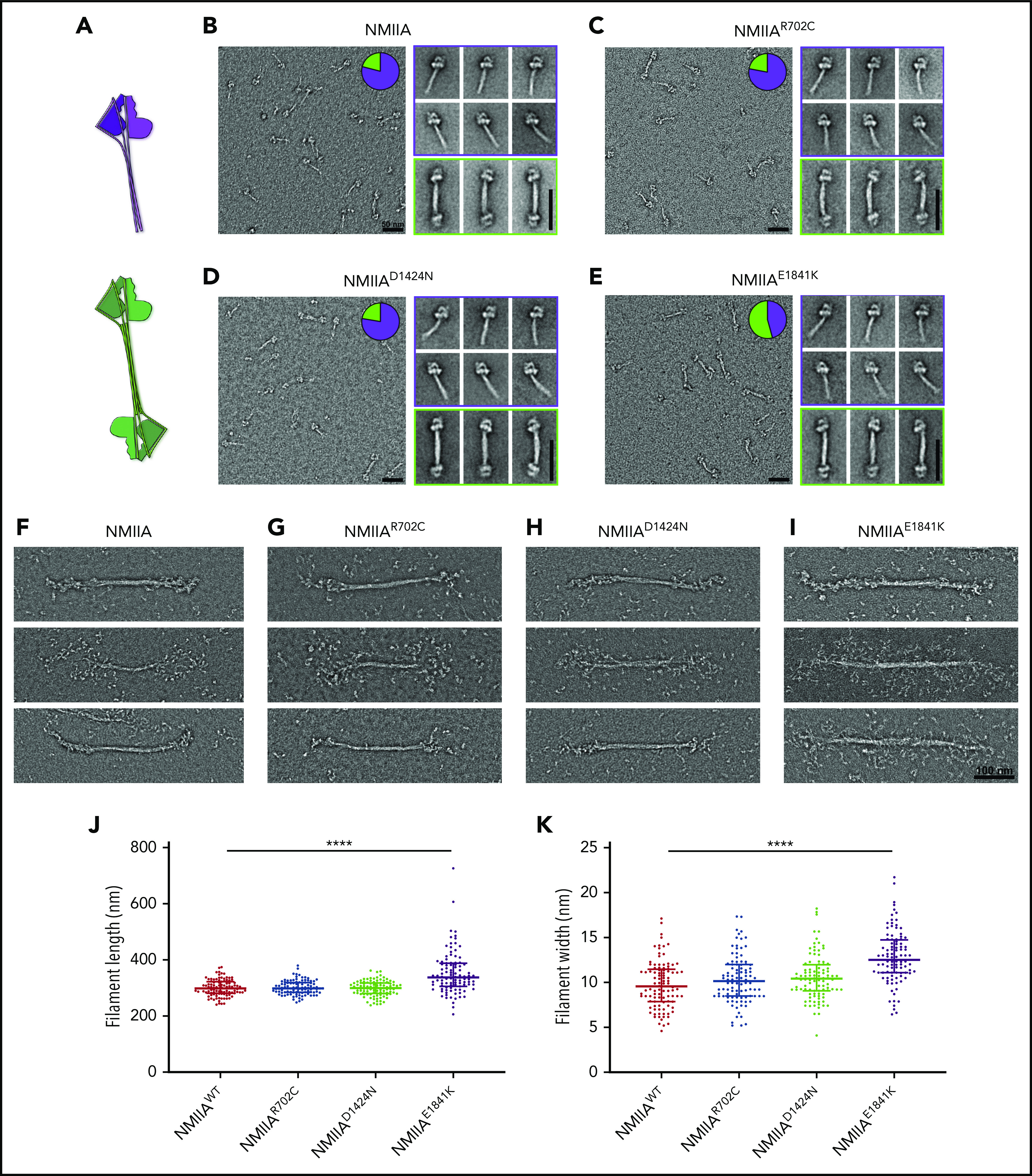
E1841K strengthens antiparallel interactions and results in overassembly of filaments in vitro. (A) Diagram depicting NMIIA folded monomer conformation and antiparallel association state. (B-E) Electron micrographs of WT and mutant NMIIA molecules in the disassembled state. Inset pie charts indicate the proportion of molecules in the folded monomer or antiparallel dimer state. Representative class averages of each type are shown alongside. Scale bars, 50 nm. (F-I) Representative electron micrographs of NMIIA bipolar filaments in the assembled state. Scale bar, 100 nm. Scatterplot representing median and interquartile range of filament length (J) and filament width (K). n = 100 filaments for each sample. Image acquisitions were performed with JEOL 1200EX electron microscope equipped with an AMT XR-60 CCD camera. Image alignments were done with SPIDER software, and quantifications were done with Fiji.
Discussion
NMII generated force production is known to regulate multiple aspects of MK differentiation, including ploidy, proplatelet formation, and release. In CD34+-derived MK cultures, prolonged pharmacological inhibition of NMII with blebbistatin introduced a 3- to 10-fold rise in ploidy by suppressing cytokinesis.55 This is consistent with an investigation reporting that exclusion of NMIIA from the cleavage furrow causes abnormal contractile ring formation resulting in endomitosis.56 Furthermore, ρ/ROCK cytoskeletal signaling pathways regulating RLC phosphorylation have also been implicated in proplatelet formation.57 Mice with MK-restricted genetic deletion of Myh9 show ultrastructural defects like a less reticular demarcation membrane system and abnormal organelle distribution in platelets.15,58
Here, we show that NMIIA additionally regulates MK chemotaxis from the endosteal niche to the BM vasculature. MKs with mutations in NMIIA have a compromised chemotaxis machinery that can explain the deficiency in thrombopoiesis and bleeding disorders associated with MYH9-RD. While MK migration in presence of SDF-1 leads to transendothelial migration during thrombopoiesis,37,59-62 the role of niche-specific MK migration in BM had been ignored as a possible mechanism underlying reduced platelet production in MYH9-RD. Comparative intravital imaging using NMIIAGFP+/− and NMIIAR702C-GFP+/− mice has demonstrated that R702C mutant MKs generated thicker and fewer proplatelets with reduced branching causing fewer circulating platelets.21 Although prior work using overexpression of MYH9-RD mutants in megakaryoblast cell lines has reported defective migration properties in an SDF-1 gradient,74 we not only demonstrate in situ MK migration anomalies in BM but also show that different point mutations associated with the disease inhibit chemotaxis by disparate mechanisms (supplemental Table 1).
For R702C MKs, defective migration is accompanied by decreased adhesion to collagen I and impaired actomyosin stress fiber assembly, likely due to increased Ser1943 phosphorylation of the HC and unstable NMIIA filaments. While the R702C mutation has no effect on bipolar filament morphology in vitro, it results in reduced NMIIA stack lengths on F-actin stress fibers in MKs. This is because NMIIA stack formation in cells requires normal NMIIA contractile activity and turnover.54,63 In previous in vitro studies with recombinant motor domain, the R702C mutation displayed reduced maximal ATPase activity of NMIIA, decreased F-actin gliding velocity, and slower ADP release leading to enhanced duty ratio and increased association with F-actin.50,64 Impaired NMIIA contractility may be a general mechanism underlying MK phenotypes of NMIIA motor domain mutations, based on other studies showing that another MYH9-RD motor domain mutation, N93K, also with low motor activity in vitro50-52 had reduced stack formation in HAP1 cells.54
On the contrary, impaired chemotaxis of D1424N MKs can be explained by excessive adhesion to collagen I due to increased RLC phosphorylation and contractility, resulting in enhanced actomyosin stress fiber formation. Interestingly, D1424N MKs have increased migration rates, whereas R702C MKs have decreased migration rates, consistent with enhanced NMIIA contractility in the former and loss of contractility in the latter. The D1424N rod domain mutation also resulted in a small but statistically significant reduction in NMIIA stack length, which may be due to defects in higher-order rod associations, since paracrystals generated from NMIIA tail fragments with the D1424N mutation demonstrated altered morphology.65 Since ρ/ROCK signaling regulates NMII activity in MKs,57 it is possible that feedback pathways may compensate for defective stack formation by activation of upstream kinases and increased RLC phosphorylation to enhance D1424N contractility. While instability of the D1424N protein and degradation has also been suggested to play a role in MYH9-RD MK phenotypes,66 western blotting did not reveal lower levels of NMIIA HC in this mutant compared with WT controls (supplemental Figure 3I).
A third mechanism explains the defective chemotaxis of E1841K mutant MKs; enhanced lateral associations of the mutant tail domains leading to aberrant E1841K NMIIA bipolar filament assembly. The variability in E1841K bipolar filament lengths and widths can explain an overall loss of order in NMIIA filament organization and misalignment of actomyosin stress fibers in MKs. Indeed, the E1841K mutation has no effect on MK migration velocity or adhesion, but it reduces directional chemotaxis, consistent with defects in cell polarity due to disordered actomyosin assembly. Thus, although the D1424N and the E1841K mutations are both in the coiled-coiled rod domain of the NMIIA molecule, the mechanisms regulating their defective MK chemotaxis phenotypes are dramatically different. While the D1424N mutation enhances NMIIA contractility, indicated by enhanced RLC phosphorylation levels and larger FAs, E1841K leaves RLC phosphorylation or FAs unaffected but instead alters the NMIIA filament dimensions, leading to misalignment of actomyosin cytoskeleton.
Thus, although NMIIA motor and rod domain mutations impair clotting, the mechanisms causing these overtly similar physiological phenotypes are different. Apart from providing insights into the basis for MYH9-RD defects in MK to platelet differentiation, these data reveal the roles of different NMIIA domains in regulating actomyosin assembly and contractility. MKs are a physiologically relevant, accessible cell type to study NMIIA-dependent functions in cell adhesion and migration. Impaired polarization of migrating MYH9-RD mutant MKs (expressing only NMIIA) in a chemotactic gradient, as shown in this investigation, may imply possible new NMIIA-based mechanisms regulating symmetry breaking in chemotaxis.67-69
This study may also suggest novel mechanisms leading to defective clotting observed in MYH9-RD. Indeed, activated platelets from D1424N mice displayed impaired spreading. Platelet shape change, spreading, mutual adhesion, and adhesion of the platelet plug to the endothelial wall are crucial for hemostasis.70 Since NMIIA activity regulates all of these phenomena, it is possible that prolonged bleeding times in these mouse models maybe partly due to impaired platelet adhesion and clot formation rather than solely due to macrothrombocytopenia. Since MYH9-RD mutations dysregulate cytoskeletal and cell adhesion signaling pathways in platelets,71 further investigations of platelet activation and clotting mechanisms are required.
Our findings that motor and rod domain NMIIA mutations all impair MK migration yet act via different mechanisms can be harnessed to develop customized therapeutic interventions for MYH9-RD patients by analogy to drugs developed for treatment of heart failure due to diverse mutations in cardiac myosin.72 Our findings identifying distinct consequences of different NMIIA mutations in MK physiology will also be relevant for cells in other tissues affected by MYH9-RDs (ie, inner ear, eye lens, and kidney).73
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Robert Adelstein from the National Institutes of Health (NIH)/National Heart, Lung, and Blood Institute (NHLBI) for the Myh9-RD mouse strains; Scott Henderson and Kersi Pestonjamasp from the Scripps Microscopy Core; Salvadore Loguercio for help with R Studio; and Richard West from the University of Delaware Flow Cytometry Core and the NHLBI Electron Microscopy Core for assistance.
This work was supported by the NIH/NHLBI (grant HL083464; V.M.F.) and the Judith Graham Pool National Hemophilia Foundation (postdoctoral fellowship; K.P.). J.R.S. acknowledges support for research from the NHLBI Division of Intramural Research (grant ZIAHL001786).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: V.M.F., K.P., N.B., and J.R.S. designed the study; K.P., R.N., N.B., R.L., and A.G. performed experiments; K.P., R.N., N.B., V.M.F., and J.R.S. analyzed the data; and K.P., V.M.F., N.B., and J.R.S. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Velia M. Fowler, Department of Biological Sciences, University of Delaware, 105 The Green, Newark, DE 19716; e-mail: vfowler@udel.edu.
REFERENCES
- 1.Hartwig J, Italiano J Jr. The birth of the platelet. J Thromb Haemost. 2003;1(7):1580-1586. [DOI] [PubMed] [Google Scholar]
- 2.Hartwig JH, Italiano JE Jr. Cytoskeletal mechanisms for platelet production. Blood Cells Mol Dis. 2006;36(2):99-103. [DOI] [PubMed] [Google Scholar]
- 3.Junt T, Schulze H, Chen Z, et al. . Dynamic visualization of thrombopoiesis within bone marrow. Science. 2007;317(5845):1767-1770. [DOI] [PubMed] [Google Scholar]
- 4.Machlus KR, Italiano JE Jr. The incredible journey: From megakaryocyte development to platelet formation. J Cell Biol. 2013;201(6):785-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Machlus KR, Thon JN, Italiano JE Jr. Interpreting the developmental dance of the megakaryocyte: a review of the cellular and molecular processes mediating platelet formation. Br J Haematol. 2014;165(2):227-236. [DOI] [PubMed] [Google Scholar]
- 6.Patel SR, Hartwig JH, Italiano JE Jr. The biogenesis of platelets from megakaryocyte proplatelets. J Clin Invest. 2005;115(12):3348-3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patel-Hett S, Richardson JL, Schulze H, et al. . Visualization of microtubule growth in living platelets reveals a dynamic marginal band with multiple microtubules. Blood. 2008;111(9):4605-4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patel-Hett S, Wang H, Begonja AJ, et al. . The spectrin-based membrane skeleton stabilizes mouse megakaryocyte membrane systems and is essential for proplatelet and platelet formation. Blood. 2011;118(6):1641-1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thon JN, Montalvo A, Patel-Hett S, et al. . Cytoskeletal mechanics of proplatelet maturation and platelet release. J Cell Biol. 2010;191(4):861-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis RE, Stenberg PE, Levin J, Beckstead JH. Localization of megakaryocytes in normal mice and following administration of platelet antiserum, 5-fluorouracil, or radiostrontium: evidence for the site of platelet production. Exp Hematol. 1997;25(7):638-648. [PubMed] [Google Scholar]
- 11.Levine RF, Eldor A, Shoff PK, Kirwin S, Tenza D, Cramer EM. Circulating megakaryocytes: delivery of large numbers of intact, mature megakaryocytes to the lungs. Eur J Haematol. 1993;51(4):233-246. [DOI] [PubMed] [Google Scholar]
- 12.Tavassoli M, Aoki M. Migration of entire megakaryocytes through the marrow–blood barrier. Br J Haematol. 1981;48(1):25-29. [DOI] [PubMed] [Google Scholar]
- 13.Begonja AJ, Pluthero FG, Suphamungmee W, et al. . FlnA binding to PACSIN2 F-BAR domain regulates membrane tubulation in megakaryocytes and platelets. Blood. 2015;126(1):80-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bender M, Eckly A, Hartwig JH, et al. . ADF/n-cofilin-dependent actin turnover determines platelet formation and sizing. Blood. 2010;116(10):1767-1775. [DOI] [PubMed] [Google Scholar]
- 15.Eckly A, Strassel C, Freund M, et al. . Abnormal megakaryocyte morphology and proplatelet formation in mice with megakaryocyte-restricted MYH9 inactivation. Blood. 2009;113(14):3182-3189. [DOI] [PubMed] [Google Scholar]
- 16.Kunishima S, Okuno Y, Yoshida K, et al. . ACTN1 mutations cause congenital macrothrombocytopenia. Am J Hum Genet. 2013;92(3):431-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sui Z, Nowak RB, Sanada C, Halene S, Krause DS, Fowler VM. Regulation of actin polymerization by tropomodulin-3 controls megakaryocyte actin organization and platelet biogenesis. Blood. 2015;126(4):520-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sellers JR. Regulation of cytoplasmic and smooth muscle myosin. Curr Opin Cell Biol. 1991;3(1):98-104. [DOI] [PubMed] [Google Scholar]
- 19.Palandri F, Zoli M, Polverelli N, et al. . MYH9-related thrombocytopenia and intracranial bleedings: a complex clinical/surgical management and review of the literature. Br J Haematol. 2015;170(5):729-731. [DOI] [PubMed] [Google Scholar]
- 20.Pecci A, Malara A, Badalucco S, et al. . Megakaryocytes of patients with MYH9-related thrombocytopenia present an altered proplatelet formation. Thromb Haemost. 2009;102(1):90-96. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Conti MA, Malide D, et al. . Mouse models of MYH9-related disease: mutations in nonmuscle myosin II-A. Blood. 2012;119(1):238-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, Boukour S, Milloud R, et al. . The abnormal proplatelet formation in MYH9-related macrothrombocytopenia results from an increased actomyosin contractility and is rescued by myosin IIA inhibition. J Thromb Haemost. 2013;11(12):2163-2175. [DOI] [PubMed] [Google Scholar]
- 23.Spinler KR, Shin JW, Lambert MP, Discher DE. Myosin-II repression favors pre/proplatelets but shear activation generates platelets and fails in macrothrombocytopenia. Blood. 2015;125(3):525-533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Billington N, Wang A, Mao J, Adelstein RS, Sellers JR. Characterization of three full-length human nonmuscle myosin II paralogs. J Biol Chem. 2013;288(46):33398-33410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conti MA, Adelstein RS. Nonmuscle myosin II moves in new directions. J Cell Sci. 2008;121(Pt 1):11-18. [DOI] [PubMed] [Google Scholar]
- 26.Dulyaninova NG, Bresnick AR. The heavy chain has its day: regulation of myosin-II assembly. Bioarchitecture. 2013;3(4):77-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dulyaninova NG, House RP, Betapudi V, Bresnick AR. Myosin-IIA heavy-chain phosphorylation regulates the motility of MDA-MB-231 carcinoma cells. Mol Biol Cell. 2007;18(8):3144-3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maupin P, Phillips CL, Adelstein RS, Pollard TD. Differential localization of myosin-II isozymes in human cultured cells and blood cells. J Cell Sci. 1994;107(Pt 11):3077-3090. [DOI] [PubMed] [Google Scholar]
- 29.Thon JN, Italiano JE. Visualization and manipulation of the platelet and megakaryocyte cytoskeleton. Methods Mol Biol. 2012;788:109-125. [DOI] [PubMed] [Google Scholar]
- 30.Zhou BO, Ding L, Morrison SJ. Hematopoietic stem and progenitor cells regulate the regeneration of their niche by secreting Angiopoietin-1. eLife. 2015;4:e05521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mazharian A. Assessment of megakaryocyte migration and chemotaxis. Methods Mol Biol. 2012;788:275-288. [DOI] [PubMed] [Google Scholar]
- 32.Mazharian A, Thomas SG, Dhanjal TS, Buckley CD, Watson SP. Critical role of Src-Syk-PLCgamma2 signaling in megakaryocyte migration and thrombopoiesis. Blood. 2010;116(5):793-800. [DOI] [PubMed] [Google Scholar]
- 33.Suraneni PK, Corey SJ, Hession MJ, et al. . Dynamins 2 and 3 control the migration of human megakaryocytes by regulating CXCR4 surface expression and ITGB1 activity. Blood Adv. 2018;2(23):3540-3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Breckenridge MT, Dulyaninova NG, Egelhoff TT. Multiple regulatory steps control mammalian nonmuscle myosin II assembly in live cells. Mol Biol Cell. 2009;20(1):338-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanaji T, Russell S, Cunningham J, Izuhara K, Fox JE, Ware J. Megakaryocyte proliferation and ploidy regulated by the cytoplasmic tail of glycoprotein Ibalpha. Blood. 2004;104(10):3161-3168. [DOI] [PubMed] [Google Scholar]
- 36.Avecilla ST, Hattori K, Heissig B, et al. . Chemokine-mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat Med. 2004;10(1):64-71. [DOI] [PubMed] [Google Scholar]
- 37.Niswander LM, Fegan KH, Kingsley PD, McGrath KE, Palis J. SDF-1 dynamically mediates megakaryocyte niche occupancy and thrombopoiesis at steady state and following radiation injury. Blood. 2014;124(2):277-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stegner D, vanEeuwijk JMM, Angay O, et al. . Thrombopoiesis is spatially regulated by the bone marrow vasculature. Nat Commun. 2017;8(1):127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009;10(11):778-790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang A, Ma X, Conti MA, Adelstein RS. Distinct and redundant roles of the non-muscle myosin II isoforms and functional domains. Biochem Soc Trans. 2011;39(5):1131-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gupton SL, Waterman-Storer CM. Spatiotemporal feedback between actomyosin and focal-adhesion systems optimizes rapid cell migration. Cell. 2006;125(7):1361-1374. [DOI] [PubMed] [Google Scholar]
- 42.Kim DH, Wirtz D. Focal adhesion size uniquely predicts cell migration. FASEB J. 2013;27(4):1351-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Semeniak D, Kulawig R, Stegner D, et al. . Proplatelet formation is selectively inhibited by collagen type I through Syk-independent GPVI signaling. J Cell Sci. 2016;129(18):3473-3484. [DOI] [PubMed] [Google Scholar]
- 44.Pasapera AM, Schneider IC, Rericha E, Schlaepfer DD, Waterman CM. Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J Cell Biol. 2010;188(6):877-890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aguilar A, Pertuy F, Eckly A, et al. . Importance of environmental stiffness for megakaryocyte differentiation and proplatelet formation. Blood. 2016;128(16):2022-2032. [DOI] [PubMed] [Google Scholar]
- 46.Komatsu S, Yano T, Shibata M, Tuft RA, Ikebe M. Effects of the regulatory light chain phosphorylation of myosin II on mitosis and cytokinesis of mammalian cells. J Biol Chem. 2000;275(44):34512-34520. [DOI] [PubMed] [Google Scholar]
- 47.Tan JL, Ravid S, Spudich JA. Control of nonmuscle myosins by phosphorylation. Annu Rev Biochem. 1992;61(1):721-759. [DOI] [PubMed] [Google Scholar]
- 48.Watanabe T, Hosoya H, Yonemura S. Regulation of myosin II dynamics by phosphorylation and dephosphorylation of its light chain in epithelial cells. Mol Biol Cell. 2007;18(2):605-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dulyaninova NG, Malashkevich VN, Almo SC, Bresnick AR. Regulation of myosin-IIA assembly and Mts1 binding by heavy chain phosphorylation. Biochemistry. 2005;44(18):6867-6876. [DOI] [PubMed] [Google Scholar]
- 50.Hu A, Wang F, Sellers JR. Mutations in human nonmuscle myosin IIA found in patients with May-Hegglin anomaly and Fechtner syndrome result in impaired enzymatic function. J Biol Chem. 2002;277(48):46512-46517. [DOI] [PubMed] [Google Scholar]
- 51.Shutova MS, Svitkina TM. Mammalian nonmuscle myosin II comes in three flavors. Biochem Biophys Res Commun. 2018;506(2):394-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Svitkina T. The actin cytoskeleton and actin-based motility. Cold Spring Harb Perspect Biol. 2018;10(1):a018267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Verkhovsky AB, Svitkina TM, Borisy GG. Myosin II filament assemblies in the active lamella of fibroblasts: their morphogenesis and role in the formation of actin filament bundles. J Cell Biol. 1995;131(4):989-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fenix AM, Taneja N, Buttler CA, et al. . Expansion and concatenation of non-muscle myosin IIA filaments drive cellular contractile system formation during interphase and mitosis. Mol Biol Cell. 2016;mbc.E15-10-0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shin JW, Swift J, Spinler KR, Discher DE. Myosin-II inhibition and soft 2D matrix maximize multinucleation and cellular projections typical of platelet-producing megakaryocytes. Proc Natl Acad Sci USA. 2011;108(28):11458-11463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lordier L, Jalil A, Aurade F, et al. . Megakaryocyte endomitosis is a failure of late cytokinesis related to defects in the contractile ring and Rho/Rock signaling. Blood. 2008;112(8):3164-3174. [DOI] [PubMed] [Google Scholar]
- 57.Chang Y, Auradé F, Larbret F, et al. . Proplatelet formation is regulated by the Rho/ROCK pathway. Blood. 2007;109(10):4229-4236. [DOI] [PubMed] [Google Scholar]
- 58.Pertuy F, Eckly A, Weber J, et al. . Myosin IIA is critical for organelle distribution and F-actin organization in megakaryocytes and platelets. Blood. 2014;123(8):1261-1269. [DOI] [PubMed] [Google Scholar]
- 59.Golfier S, Kondo S, Schulze T, et al. . Shaping of terminal megakaryocyte differentiation and proplatelet development by sphingosine-1-phosphate receptor S1P4. FASEB J. 2010;24(12):4701-4710. [DOI] [PubMed] [Google Scholar]
- 60.Hamada T, Möhle R, Hesselgesser J, et al. . Transendothelial migration of megakaryocytes in response to stromal cell-derived factor 1 (SDF-1) enhances platelet formation. J Exp Med. 1998;188(3):539-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang L, Orban M, Lorenz M, et al. . A novel role of sphingosine 1-phosphate receptor S1pr1 in mouse thrombopoiesis. J Exp Med. 2012;209(12):2165-2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang L, Urtz N, Gaertner F, et al. . Sphingosine kinase 2 (Sphk2) regulates platelet biogenesis by providing intracellular sphingosine 1-phosphate (S1P). Blood. 2013;122(5):791-802. [DOI] [PubMed] [Google Scholar]
- 63.Hu S, Dasbiswas K, Guo Z, et al. . Long-range self-organization of cytoskeletal myosin II filament stacks [published correction appears in Nat Cell Biol. 2017;19(3):258]. Nat Cell Biol. 2017;19(2):133-141. [DOI] [PubMed] [Google Scholar]
- 64.Ma X, Kovács M, Conti MA, et al. . Nonmuscle myosin II exerts tension but does not translocate actin in vertebrate cytokinesis. Proc Natl Acad Sci USA. 2012;109(12):4509-4514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Franke JD, Dong F, Rickoll WL, Kelley MJ, Kiehart DP. Rod mutations associated with MYH9-related disorders disrupt nonmuscle myosin-IIA assembly. Blood. 2005;105(1):161-169. [DOI] [PubMed] [Google Scholar]
- 66.Deutsch S, Rideau A, Bochaton-Piallat ML, et al. . Asp1424Asn MYH9 mutation results in an unstable protein responsible for the phenotypes in May-Hegglin anomaly/Fechtner syndrome. Blood. 2003;102(2):529-534. [DOI] [PubMed] [Google Scholar]
- 67.Thomas DG, Yenepalli A, Denais CM, et al. . Non-muscle myosin IIB is critical for nuclear translocation during 3D invasion. J Cell Biol. 2015;210(4):583-594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vicente-Manzanares M, Koach MA, Whitmore L, Lamers ML, Horwitz AF. Segregation and activation of myosin IIB creates a rear in migrating cells. J Cell Biol. 2008;183(3):543-554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vicente-Manzanares M, Newell-Litwa K, Bachir AI, Whitmore LA, Horwitz AR. Myosin IIA/IIB restrict adhesive and protrusive signaling to generate front-back polarity in migrating cells. J Cell Biol. 2011;193(2):381-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gibbins JM. Platelet adhesion signalling and the regulation of thrombus formation. J Cell Sci. 2004;117(Pt 16):3415-3425. [DOI] [PubMed] [Google Scholar]
- 71.Canobbio I, Noris P, Pecci A, Balduini A, Balduini CL, Torti M. Altered cytoskeleton organization in platelets from patients with MYH9-related disease. J Thromb Haemost. 2005;3(5):1026-1035. [DOI] [PubMed] [Google Scholar]
- 72.Malik FI, Hartman JJ, Elias KA, et al. . Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science. 2011;331(6023):1439-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pecci A, Ma X, Savoia A, Adelstein RS. MYH9: Structure, functions and role of non-muscle myosin IIA in human disease. Gene. 2018;664:152-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pecci A, Bozzi V, Panza E, et al. . Mutations responsible for MYH9-related thrombocytopenia impair SDF-1-driven migration of megakaryoblastic cells. Thromb Haemost. 2011;106(4):693-704. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.