

Summary
The lung pathology seen in patients with coronavirus disease 2019 (COVID-19) shows marked microvascular thrombosis and haemorrhage linked to extensive alveolar and interstitial inflammation that shares features with macrophage activation syndrome (MAS). We have termed the lung-restricted vascular immunopathology associated with COVID-19 as diffuse pulmonary intravascular coagulopathy, which in its early stages is distinct from disseminated intravascular coagulation. Increased circulating D-dimer concentrations (reflecting pulmonary vascular bed thrombosis with fibrinolysis) and elevated cardiac enzyme concentrations (reflecting emergent ventricular stress induced by pulmonary hypertension) in the face of normal fibrinogen and platelet levels are key early features of severe pulmonary intravascular coagulopathy related to COVID-19. Extensive immunothrombosis over a wide pulmonary vascular territory without confirmation of COVID-19 viraemia in early disease best explains the adverse impact of male sex, hypertension, obesity, and diabetes on the prognosis of patients with COVID-19. The immune mechanism underlying diffuse alveolar and pulmonary interstitial inflammation in COVID-19 involves a MAS-like state that triggers extensive immunothrombosis, which might unmask subclinical cardiovascular disease and is distinct from the MAS and disseminated intravascular coagulation that is more familiar to rheumatologists.
Introduction
The coronavirus disease 2019 (COVID-19) pandemic and earlier coronavirus outbreaks have been associated with adult respiratory distress syndrome (ARDS) and worse outcomes in older patients.1, 2 The severity of systemic inflammation in response to human coronavirus family members has features reminiscent of a cytokine storm or macrophage activation syndrome (MAS), also known as secondary haemophagocytic lymphohistocytosis (sHLH).3, 4 This response has inspired use of directed anticytokine therapies for severe COVID-19 pneumonia, as these agents are known to be useful in diseases on the MAS spectrum.4, 5 A key feature of sHLH or MAS is haemophagocytosis and an acute consumptive coagulopathy, leading to disseminated intravascular coagulation. Disseminated intravascular coagulation has also been reported in COVID-19 pneumonia, but usually as a pre-terminal event.6, 7 The hypercytokinaemia with extreme hyperferritinaemia that is typically seen with sHLH is also evident in some patients with COVID-19 pneumonia.4
COVID-19 pneumonia is distinct from MAS
MAS-like pulmonary immunopathology characteristic of COVID-19 pneumonia is distinct from classical sHLH.8 Haemphagocytosis is a cardinal feature of MAS9, 10 and has been reported in patients with severe acute respiratory syndrome (SARS).11, 12 In SARS, this process might involve phagocytosis of extravascular red blood cells consequent to severe lung microvascular damage, microhaemorrhage with physiological haemophagocytosis of extravascular red blood cells, or possibly very advanced disease with frank MAS-like pathology and disseminated intravascular coagulation (figure 1 ). The hypercytokinaemia characteristic of sHLH or MAS is often associated with extremely high serum ferritin concentrations (≥10 000–100 000 ng/mL), whereas in patients with COVID-19, serum ferritin concentrations are typically in the 500–3000 ng/mL range, at least early in the disease course. Another clear distinguishing feature of sHLH or MAS is liver function derangement, which can contribute to coagulopathy secondary to loss of liver synthetic function and is not typically seen in patients with COVID-19 (figure 1).
Figure 1.
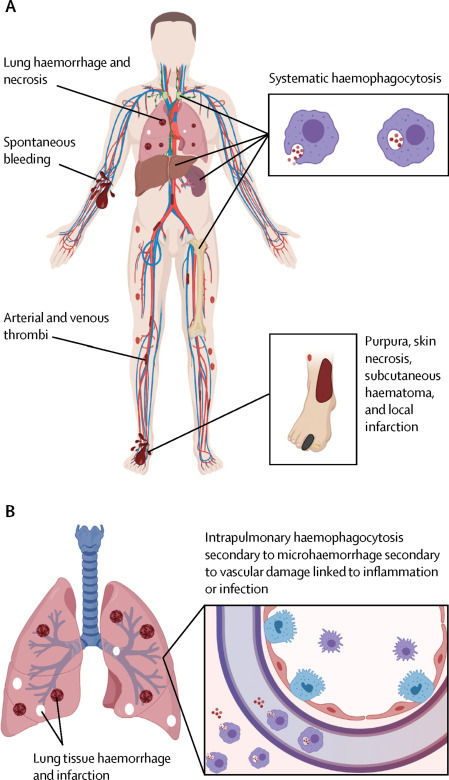
Early macrophage activation syndrome versus early COVID-19
(A) Secondary haemophagocytic lymphohistiocytosis or macrophage activation syndrome is associated with organomegaly, thrombocytopenia, haemophagocytosis, and disseminated intravascular coagulation with pulmonary involvement in half of cases.13 Activation of bone marrow, lymphoid organ, hepatic Kupffer cells, and circulating mononuclear cells lead to a severe consumptive coagulopathy with low fibrinongen levels and increased fibrinogen degradation. Additionally, liver dysfunction exacerbates the consumptive coagulopathy. A rapid onset disseminated intravascular coagulation pattern with hyperferritinaemia reflects generalised haemophagocytosis with erythrocyte degradation, sequestration, and export with diffuse clotting and bleeding. (B) Pulmonary involvement without generalised lymphoid organ hyperplasia is typical of COVID-19 pneumonia. Haemophagocytosis, albeit intrapulmonary, has also been reported in coronavirus family infection.12 However, in the early stages systemic coagulopathy is not a feature. Such intrapulmonary haemophagocytosis, which then drains to regional nodes, indicates removal of extravascular red blood cells mediated by activated macrophages, secondary to vascular injury. A disseminated intravascular coagulation picture might also develop late in the course of COVID-19 pneumonia in patients who develop acute respiratory distress syndrome. COVID-19=coronavirus disease 2019.
Extensive lung infiltration by macrophages and other immune cells leading to diffuse alveolar damage has been reported in SARS pneumonia, with similar findings emerging in patients with COVID-19 pneumonia.12, 14, 15, 16 The extensive nature of viral infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) results in diffuse lung inflammation that involves the large juxtaposed pulmonary vascular network.8 The diffuse, slowly evolving COVID-19 pneumonia has similarities to a MAS-like syndrome with regard to both clinical and laboratory features. These clinical findings suggest that an initial pulmonary intravascular coagulopathy occurs in patients with COVID-19 pneumonia that is distinct from disseminated intravascular coagulation.8 Herein, we propose a model for the pathophysiology of this pulmonary intravascular coagulopathy and describe how extensive coronavirus infection and age-related changes in immunity, combined with diffuse pulmonary immunothrombosis, explain the cardiovascular mortality in these patients (table 1 ).
Table 1.
Differences and similarities between DIC and PIC
| DIC linked to HLH or MAS | PIC linked to COVID-19 | |
|---|---|---|
| Clinical features | ||
| Onset | Acute | Subacute |
| Hepatosplenomegaly | +++ | .. |
| Adenopathy | ++ | .. |
| Pulmonary involvement (%) | 50% | 100% |
| Thrombosis | Multi-organ clotting | Mainly lung (occasional CNS and peripheral thrombosis reported; related to DIC evolution?) |
| Bleeding | Generalised | Intrapulmonary microhaemorrhage |
| Active infection considerations | Yes usually for primary HLH; secondary HLH might not have driving infection | Thought to be ongoing alveolar infection |
| Laboratory parameters | ||
| Liver function | Decreased synthetic function including fibrinogen and other clotting factors; raised transaminase +++ | Preservation of liver synthetic function; +/− |
| Anaemia | +++ | − |
| Thrombocytopenia | +++ | Normal or low |
| Immune cell cytopenia | ++ | No but lymphopenia is a feature of COVID-19 in general |
| Creatine kinase | + (skeletal and cardiac origin) | + (worse prognosis) |
| Troponin T | + | ++ with high levels associated with worse outcome |
| Haemophagocytosis | Generalised to marrow, liver, and other sites detectable in >80% | Occasional intrapulmonary and regional lymph node haemophagocytosis reported |
| Evolution | DIC secondary to MAS | PIC might evolve into DIC; PIC might occur without MAS |
| Coagulation and immunology markers | ||
| Elevated prothrombin time or activated partial thromboplastin time | +++/+++ | + or normal |
| Fibrinogen levels | Decreased | Normal or slight increase |
| Fibrin degradation products or D-dimer | Increased | Increased |
| C-reactive protein | Elevated | Elevated |
| Ferritin elevation | +++ | Elevated |
| Hypercytokinaemia | +++ | ++ |
Present (+), usually present (++), frequently present (+++), or absent (−) clinical features, laboratory parameters, and coagulation and immunology markers. DIC=disseminated intravascular coagulation. HLH=haemophagocytic lymphohistiocytosis. MAS=macrophage activation syndrome. PIC=pulmonary intravascular coagulopathy. COVID-19=coronavirus disease 2019.
Pulmonary vascular pathology patterns in SARS and COVID-19
Acute respiratory infections are associated with a high risk of cardiovascular-related death, especially in the weeks immediately after infection, and particularly in older patients and those with pre-existing cardiovascular disease. Severity of pneumonia in these patients is linked to an increased risk of death.17, 18 Of particular note, one pathology study showed similar vascular changes in post-mortem tissue from patients with bronchopneumonia not related to SARS as was found in individuals who died from SARS,20 indicating that vascular thrombosis triggered by infection, rather than the infection itself, could be key (figure 2 ).
Figure 2.
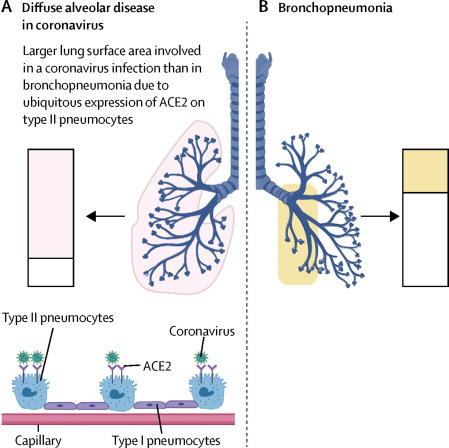
Extent of alveolar lung surface involved in COVID-19 and bronchopneumonia
(A) Some coronavirus family members gain access to the lungs via the ACE2 receptor that is expressed most abundantly on a subpopulation of type II pneumocytes. Shaded boxes indicate the much greater capability for immunothrombosis given the alveolar tropism of SARS-CoV-2. (B) Segmental bronchopneumonia (eg, bacterial and influenza) typically has a different lung distribution, with prominent bronchial tree involvement, including haemorrhagic destruction of trachea and large airways,20 and generally patchy alveolar network disease. There might be large areas with normal perfusion. The slow evolution of COVID-19 with alveolar hypoxia and microthrombosis might result in pulmonary arterial hypertension and the cardiac picture that mostly occurs with hypoxaemia and raised D-dimers. The scale of alveolar and microvascular inflammation, rather than systemic viral infection per se, determines the cardiovascular pattern of disease. Nonetheless, segmental bronchopneumonia could result in a degree of immunothrombosis sufficient to cause a cardiac event, especially in older patients with known or silent cardiac disease. ACE2=angiotensin-converting enzyme 2. COVID-19=coronavirus disease 2019. SARS-CoV-2=severe acute respiratory syndrome coronavirus 2.
Coronavirus family members show tropism for angiotensin-converting enzyme 2 (ACE2) on type II pneumocytes. This tropism, along with the close anatomical juxtaposition of type II pneumocytes and the pulmonary vascular network, and a severe multifaceted inflammatory reaction, is likely to drive the generalised pulmonary hypercoagulable state seen in patients with COVID-19 (figure 2).8, 21 Single-cell sequencing analysis has recently shown that most pulmonary gene expression is confined to a small population of type II pneumocytes, and ACE2 is largely absent from endothelial cells and alveolar macrophages.22 From a clinical perspective, the diffuse alveolar changes (determined by CT scan) seen in patients with COVID-19 are distinct from bronchopneumonia and show how SARS-CoV-2 interfaces with a large area of the pulmonary microvasculature. This finding probably explains the propensity for diffuse pathology in patients with COVID-19 pneumonia. In these patients, extensive air space changes on CT are associated with a worse prognosis compared with patients not showing these changes.23 Diffuse disease on CT is also a feature of patients who died from SARS-CoV infection (figure 2).23
ACE2 has been shown to regulate innate immunity, and mice with a genetic deletion of ACE2 developed more severe pulmonary inflammation after acid inhalation than did wild-type mice.24 These findings suggest that ACE2 downregulation could aggravate inflammation over a wide alveolar capillary network. It was subsequently shown that injecting the SARS spike protein led to similar lung pathology in mice, probably due to internalisation of the ACE2 receptor and subsequent inhibition of ACE2-mediated generation of the immunoregulatory angiotensin (1–7) peptide, which signals via the MAS1 receptor.24 Mice lacking ACE2 also developed more severe pneumonitis and had higher mortality after influenza infection compared with wild-type mice.25 Theoretically, a similar mechanism could increase the proclivity towards immunothrombosis in humans. In humans, the Middle East respiratory syndrome coronavirus (MERS-CoV) principally uses the dipeptidyl peptidase 4 (DPP4) receptor, which is not restricted to type II pneumocytes; nonetheless, this virus results in clinical features and pathology similar to SARS-CoV and SARS-CoV-2, including a high mortality rate.26 Analogous to ACE2, in some experimental systems the DPP4 receptor might negatively regulate lymphocyte function, which could indicate a shared coronavirus-specific mechanism that contributes to pulmonary intravascular coagulopathy (panel ).27
Panel. Immune factors contributing to pulmonary intravascular coagulopathy.
-
•
Diffuse alveolar damage and inflammation
-
•
Diffuse interstitial inflammation
-
•
Extensive pulmonary macrophage activation (MAS-like)
-
•
Dysregulation of pulmonary innate immune responses (eg, ACE2 receptor expression downregulation)
-
•
Adaptive immune responses to COVID-19
-
•
Activation of innate immunity with older age
-
•
Age-related coagulation cascade changes
-
•
Mechanical ventilation forcing viral immunostimulatory molecules into microvasculature, increasing the propensity towards immunothrombosis
MAS=macrophage activation syndrome. ACE2=angiotensin-converting enzyme 2. COVID-19=coronavirus disease 2019.
Pulmonary vascular disease in SARS and COVID-19
The SARS-CoV-2 and SARS-CoV virus genomes are highly homologous, and patients infected with these viruses appear to share common clinical and pathological features.28, 29 Initial post-mortem reports from three patients with SARS indicated not only diffuse alveolar damage and small pulmonary vessel thrombosis and haemorrhage, but also a more generalised small vessel thrombosis.30 A second post-mortem study of six patients reported vascular pathology in two cases.12 A study of pathological specimens from 20 patients with SARS showed the presence of not only diffuse alveolar damage, but also fibrin thrombi, small vessel occlusion, and pulmonary infarction in upwards of 80% of cases.19 A review of aggregated SARS pathology reports showed evidence for vessel wall oedema, inflammatory cell infiltration into the walls of the pulmonary microvasculature, marked haemorrhagic necrosis, and vessel microthrombi mostly confined to the lung and pulmonary tissue infarction, in the context of septal inflammation and diffuse alveolar damage.31 Some data have emerged from patients with COVID-19 pneumonia that show a similar pulmonary vascular picture with blood vessel wall oedema, modest vessel wall immune cell infiltration, hyaline thrombosis, haemorrhagic change, and infarction.15, 32, 33 Emergent data have also emphasised prominent capillary thrombosis,33 and one study reported cardiomegaly and right ventricular dilatation, pointing toward the expected development of pulmonary artery hypertension.33
Insufficient evidence for coronavirus myocarditis or coronary vasculitis
Myocarditis might occur after severe respiratory viral pneumonia. Although this outcome is relatively rare, it is well documented, especially in younger women following influenza infection (table 2 ). Understandably, increased cardiac enzymes in patients with COVID-19 has been taken to potentially represent myocarditis or cardiac vasculitis associated with viral infection. In SARS-CoV infection, however, most evidence points away from cardiac involvement.40, 41 The ACE2 receptor is expressed on endothelial cells, and one in situ hybridisation study suggested that pulmonary endothelial cells are infected with SARS-CoV.42 Other studies showed a complete absence or very low level of potential endothelial cell infection.40, 41
Table 2.
Findings of cardiovascular disease, myocarditis, and infection
| Findings | Studies | |
|---|---|---|
| SARS-CoV-2 viral access to heart | RNAaemia in only six (15%) of 41 COVID-19 cases; cardiac injury linked to intensive care unit admission and not RNAaemia | Huang at al3 |
| Community-acquired pneumonia and bronchopneumonia | Increased risk of cardiovascular death | Smeeth et al,17 Corrales-Medina et al18 |
| Human cardiac myocytes | No cytopathic change with SARS; no SARS viral protein detection | Hwang et al19 |
| Viral myocarditis | 47 cases of fatal influenza virus; fatalities predominantly in children and women; myocarditis deemed to be cause of death in five cases | Guarner et al20 |
| Cardiac injury not specific to COVID-19 | Cardiac injury in >60% with avian influenza H7N9 infection suggesting that cardiac disease linked to infection; also linked to history of cardiovascular disease, but not male sex or diabetes (myocarditis is well documented with influenza) | Gao et al34 |
| Cardiac injury not specific to COVID-19 | Pandemic (H1N1) 2009 virus; cardiac injury in 46% of cases; mean age 34 years; more common in women than in men; presumed myocarditis (no histology or post-mortem data) | Chacko et al35 |
| COVID-19 viral access to heart | No viraemia in nine cases of COVID-19 | Wölfel et al36 |
| Endothelial cells | Express ACE2 but cytopathic changes seen in pneumocytes not reported | Oudit et al37 |
| Other factors linked to cardiac pathology | Older age, male sex, hypertension, obesity, diabetes; hypoxia from acute respiratory distress syndrome development; emergent disseminated intravascular coagulation late in COVID-19 disease course | Guzik et al38 |
| Viral myocarditis | H1N1 influenza A virus; rare link to fulminant myocarditis reported | Bratincsák et al39 |
SARS-CoV-2=severe acute respiratory syndrome coronavirus 2. COVID-19=coronavirus disease 2019. SARS=severe acute respiratory syndrome. ACE2=angiotensin-converting enzyme 2.
Furthermore, a study in patients with SARS suggested a close link between infection of pneumocytes and inflammatory cytokine expression in the same cells;43 however, this link was not reported for endothelium. In SARS-CoV-2 infection, publications have reported that viral RNA was undetectable in the blood36 or was detected in only six (15%) of 41 patients,3 again arguing for a pathology centred around pneumocytes and adjacent tissue pathology, rather than systemic viral infection (figure 2).36, 44
A role for the ACE2 receptor in cardiovascular biology predates the link between pulmonary pathology and COVID-19 pneumonia. Genetic deletion of ACE2 in rodents was associated with ventricular contractility defects, but there was no evidence of cardiomyopathy or fibrosis.45 The link between ACE2 downregulation in lung tissue (either in genetically manipulated mice or mice treated with SARS spike protein) and increased inflammation suggests a relatively simple model, in which coronavirus infection of ACE2-expressing cardiomyocytes or cardiac endothelium might trigger a similar inflammatory pathology. In the most comprehensive study of cardiac involvement in patients who died from SARS, viral RNA was detectable in a third of post-mortem cardiac tissues and was associated with both decreased ACE2 expression and increased macrophage infiltration.37 The same study showed no evidence of myocyte necrosis or lymphocytic infiltration, which are both hallmarks of viral myocarditis.37, 46 Overall autopsy reports, including those emerging in COVID-19, vary from showing no clinically significant pathology or infiltrating macrophages, and some reports of occasional infiltrating CD4 T cells.33
The cardiac pathology in COVID-19 pneumonia needs evaluation in view of known cardiovascular comorbidities, including hypertension and pulmonary intravascular coagulopathy, as well as the MAS-like state with associated hypoxaemia and secondary pulmonary artery hypertension. The hypercytokinaemia that is part of the MAS-like state could modify ACE2 expression independently of viraemia. One key cytokine characteristic of MAS-like pathology, interferon-γ, has been shown to downregulate ACE2 expression on an epithelial cell line; however, this finding has not been reported for heart cells.47 Therefore, reported changes in the heart tissue, including endothelium, could represent a cytokine and hypoxia effect rather than direct viral infection.
Laboratory data indicate early pulmonary intravascular coagulopathy
The key early laboratory observations in patients with COVID-19 pneumonia include elevated plasma D-dimer concentrations in conjunction with elevated cardiac markers, including brain natriuretic peptide, creatine kinase, and troponin T.48 Elevation of troponin T at hospitalisation is linked to a poor prognosis.48 Similarly, a 2020 study reported that elevated plasma levels of fibrin degradation products, including D-dimers, constitutes a significant independent biomarker of poor prognosis.6 For example, Zhou and colleagues23 reported that 117 (68%) of 172 patients presenting with COVID-19 had increased activation of coagulation, as indicated by elevated D-dimer concentrations at presentation (>0·5 ug/mL). Importantly, D-dimer concentrations above 1 μg/mL were associated with an 18 times increased odds ratio for fatal outcome.23 Furthermore, progressive elevation of D-dimer and fibrin degradation products were seen in non-survivors.23 However, despite this increase in D-dimers, patients with COVID-19 do not typically develop overt systemic disseminated intravascular coagulation. In rare cases of COVID-19 in which overt disseminated intravascular coagulation does develop, it tends to be restricted to late-stage disease. This finding is reflected in the consistent observation that platelet counts and fibrinogen concentration are not substantially reduced in patients with COVID-19, despite marked increases in D-dimer concentrations.49 Fibrinogen generally remains elevated in these patients, consistent with an ongoing acute phase response.
Extensive pulmonary inflammation and thrombosis in COVID-19 pathology
Severe COVID-19 sepsis is associated with a marked MAS-type picture, and increased inflammatory markers and ferritin concentrations that undoubtedly result in local activation of pulmonary vasculature endothelial cells. For example, interleukin (IL)-1, IL-6, and tumor necrosis factor have all been shown to trigger acute endothelial cell activation.50 Given the crucial roles played by endothelial cells in maintaining normal haemostasis, regulating fibrinolysis, and determining vessel wall permeability, local endothelial cell dysfunction in the pulmonary microvasculature is likely to play an important role in the thromboinflammatory processes that ultimately result in COVID-19 vasculopathy, ventilation perfusion mismatch, and a clinical phenotype of refractory ARDS.
Additionally, the MAS-like picture associated with COVID-19 pneumonia will trigger expression of active tissue factor, both on endothelial cells and on activated infiltrating macrophages and neutrophils.50 The net effect will be local presentation of blood-borne tissue factor within the lungs, which will further amplify activation of the coagulation cascade. Importantly, endothelial cell disruption, tissue factor expression, and activation of the coagulation cascade will all be progressively exacerbated by development of local hypoxia,51 establishing a deleterious positive thromboinflammatory feedback loop within the small vessels of the lungs with thrombosis and haemorrhage (figure 3 ). Beyond the coagulopathic changes occurring within the pulmonary vasculature, a study of bronchoalveolar lavage showed that both severe pneumonia and ARDS are associated with enhanced thrombin generation and fibrin deposition within the bronchoalveolar system.52 These changes correlate with severity of inflammation53 and are primarily driven by upregulation of tissue factor expression within the alveoli, coupled with a reduction in fibrinolysis induced by plasminogen activation inhibitor 1.54 The biological mechanisms responsible for the extremely elevated plasma D-dimer concentrations in patients with severe COVID-19, together with the marked variations observed between individuals, are unclear. Nonetheless, these data clearly suggest hyperactive fibrinolysis with increased plasmin generation. Collectively, these findings have led to the suggestion that elevated plasmin (and plasminogen) concentrations might represent a risk factor for COVID-19 susceptibility.55
Figure 3.
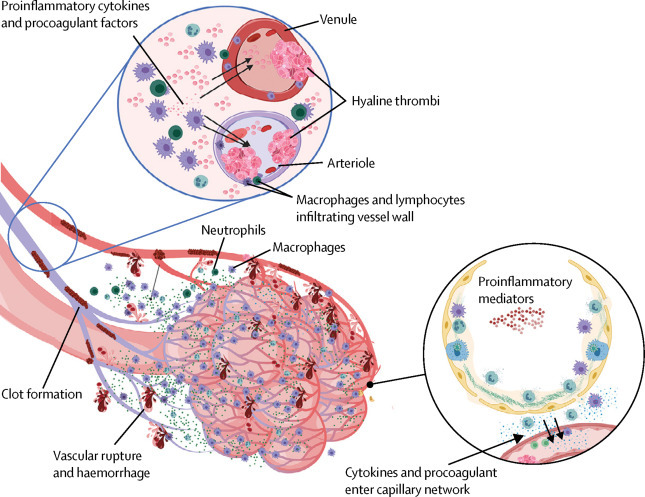
Pulmonary intravascular coagulopathy in COVID-19 pneumonia
Scheme showing how extensive COVID-19 lung involvement with large anatomical interface between infected type II pneumocytes, extensive interstitial immunocyte activation similar to macrophage activation syndrome, and the extensive pulmonary microvascular network, triggers diffuse pulmonary bed extrinsic inflammation with immunothrombosis. This inflammation causes microthrombotic immunopathology that leads to right ventricular stress and contributes to mortality. Diffuse type II pneumocyte centric pathology with extension into the interstitium leads to extensive pulmonary macrophage recruitment and activation, resulting in a clinical picture similar to local macrophage activation syndrome. Proinflammatory and procoagulants gain access to the capillary network (lower circle). The low pressure nature of the vascular system and thin vessel walls in and proximal to the alveolar network triggers immunothrombosis by various mechanisms (eg, local elevations in proinflammatory cytokines), vessel wall tissue damage with tissue factor production, and direct injury to small vessels. Vigorous fibrinolytic activity (detected early by D-dimer elevation) might not keep in check the extensive microthrombi formation, leading to the evolution of pulmonary infarction, haemorrhaging, and pulmonary hypertension induced by pulmonary intravascular coagulopathy, all of which are driven by COVID-19 inflammation. Thus, risk factors for cardiovascular disease might increase the likelihood of death in severe COVID-19 inflammation. COVID-19=coronavirus disease 2019.
Development of hypoxaemia, secondary to ARDS that is induced by COVID-19, might also activate the coagulation cascade and could be important in endothelial dysfunction beyond the capillary network.56, 57 Hypoxaemia might also play a role in adjacent small pulmonary vascular thrombosis.56, 57 Other factors, including mechanical ventilation in patients progressing to ARDS, might contribute to this picture. The role of vascular microthrombi formation, or immunothrombosis, in containment of bacterial infection and spread is well established, but its role in viral infection is not so well known.58 Likewise, the role of local pulmonary intravascular immunity and its effects on the phenotype of pulmonary intravascular coagulopathy is completely unknown.59 Nevertheless, access to the circulation in an artificial adenovirus model system triggers a MAS-like phenomenon with disseminated intravascular coagulation.60 The role of positive pressure ventilation as a factor driving release of viral nucleic acids and proteins across damaged alveolar endothelial cell barriers is another factor that needs consideration as a potential iatrogenic contributor to immunothrombosis and poor outcomes.
Experimental SARS models have shown that normal aged primates have a similar degree of viral replication as younger primates, but that aged primates have more pulmonary damage and lower activation of type 1 interferon responsive gene pathways.61 At the molecular level, aged primates showed exacerbated innate immune responses associated with NF-kB pathway activation, such as elevated IL-8 and expression of tissue factor, which activates the extrinsic clotting pathway protein.61 It is well established that type 1 interferon responses decrease with age in humans,62 and also in response to viral infection, but the age at which this transition occurs needs better definition.63 Viral or age-related impairment in type 1 interferon production appears to be associated with a second wave of inflammatory cytokine production and tissue factor expression that might substantially contribute to pulmonary intravascular coagulopathy.8
Implications of pulmonary intravascular coagulopathy
The role of anticoagulation in the setting of COVID-19 pulmonary intravascular coagulopathy is of considerable interest. Expert recommendations for the use of anticoagulants have already been published, reflecting the recognition of clotting dysregulation.64 The potential relevance of anticardiolipin antibodies in the critical care setting of COVID-19 is also recognised, but is of uncertain clinical significance.65 However, some data are available, mostly derived from prospective cohort studies. In a study of nearly 450 patients with COVID-19, low molecular weight heparin (mostly used in prophylactic doses rather than therapeutic doses) did not confer an overall survival advantage.49 However, the regimen was associated with improved survival in the group with a high sepsis-induced coagulopathy score and in patients with D-dimer concentrations that were more than six times the upper limit of normal.49 The role and timing of anticoagulation in this extensive virus-related immunothrombosis, especially where pulmonary haemorrhaging occurs, needs careful consideration. In disseminated intravascular coagulation, thrombosis and bleeding might occur simultaneously, and the same scenario also appears to arise in pulmonary intravascular coagulopathy (figure 3).
Given the MAS-like pathology of COVID-19 pneumonia, the question arises whether anticytokine therapy will ameliorate the diffuse immunothrombosis process associated with severe cases. In the translational setting, the use of the IL-1β blocker canakinumab is associated with a decreased risk of all-cause cardiovascular mortality,66 with an increased benefit in patients with the most dramatic reductions in serum IL-6.67 Unlike in low grade arterial inflammation in the absence of overt infection, it is still unclear whether severe COVID-19-associated MAS with pulmonary intravascular coagulopathy will be successfully targeted with these strategies; ongoing viral infection might represent a major hurdle in this context.8 In the past year, it has also emerged that the coagulation and immune systems are directly linked via thrombin cleavage of IL-1α from macrophages and platelets.68 This novel mechanism is of special interest with regard to anakinra, which blocks both the IL-1α and IL-1β pathways, whereas monoclonal antibodies (eg, canakinumab) selectively block IL-1β.68
The most crucial question for optimal therapeutic strategies is whether the emergent early activation of coagulopathy and fibrinolysis in patients with COVID-19 pneumonia is purely due to an appropriate immune response to the virus, or whether there is a degree of excessive inflammation that could be targeted to help prevent progression of pulmonary intravascular coagulopathy. The potential survival advantage of drugs pioneered to treat inflammatory and hyperinflammatory states needs to be viewed from the perspective of severe diffuse pulmonary immunothrombosis. The combination of immunomodulatory and anticoagulant strategies in patients with high D-dimer concentrations and evidence of myocardial stress warrants especially close attention. The use of Janus kinase (JAK) pathway inhibition needs careful scrutiny given the association between JAK inhibitors and thromboembolic disease.69
Some bleeding complications outside of the lung have been described in patients with COVID-19; this response is perhaps unsurprising in the few cases of pulmonary intravascular coagulopathy that progress to systemic disseminated intravascular coagulation. Reports of acute necrotising encephalopathy, which is associated with haemorrhage, are also emerging.70 However, this rare condition is well recognised in association with viral infections in general.71 Given that ACE2 is expressed on endothelial cells, the notion of thrombosis outside either pulmonary or disseminated intravascular coagulopathy needs further consideration.
This pulmonary intravascular coagulopathy model has implications for understanding cardiovascular mortality in the COVID-19 pandemic. This model shifts the focus away from COVID-19-related myocarditis or coronary vascular involvement, with cardiac injury secondary to viral infection of myocytes or endothelial cells expressing ACE2, and towards a scenario of multisite pulmonary vascular thrombosis with progressive myocardial ischaemia. The immunothrombotic pathology that evolves over several days in some patients with COVID-19 pneumonia is likely to manifest in individuals with underlying cardiovascular risk factors such as obesity, hypertension, and type 2 diabetes, and pathology will be compounded by the cardiac ischaemia that accompanies development of ARDS. Cardiovascular mortality is higher in men than in women aged 30–64 years in many different populations.72 These well known risk factors might also account for racial differences in COVID-19 mortality.73, 74 The COVID-19 pandemic, characterised by profound immunological changes that constitute a pulmonary MAS-like picture, is revealing a large burden of subclinical cardiovascular disease among patients predisposed to disease, by slowly triggering an extensive pulmonary immunothrombosis. Our model offers a different and robust alternative to the proposed effects of medications on ACE2 expression with resulting increased viral infectivity.75 It is also important to determine the degree to which COVID-19-mediated diffuse alveolar damage with development of ARDS in the absence of coagulopathy might also contribute to outcomes, because emerging post-mortem reports do not report this pathology in all cases.
Diffuse pulmonary intravascular coagulopathy is not unique to the MAS-like pattern of inflammation. Diffuse alveolar infection, activation of innate immune mechanisms, dysregulation of ACE2 protein expression, and marked adaptive antiviral immune responses could contribute to extensive pulmonary immunothrombosis without a clinically evident MAS (panel). Will this translate into differential effectiveness, if any, of immunosuppressive therapy between the MAS-like subgroup of patients and individuals without MAS? Ultimately, the tropism of COVID-19 for type II pneumocytes, along with evidence for extensive microvascular thrombosis during a global pandemic in patients without natural immunity, suggests an explanation for the increased cardiovascular mortality that has been recognised for some time in other settings of respiratory infection. We believe that this pulmonary intravascular coagulopathy, or pulmonary immunovascular coagulopathy, is the best explanation for the COVID-19 pneumonia risk factors for poor survival (eg, cardiovascular disease) in the present scenario, in which little evidence exists for systemic viraemia early in the COVID-19 disease course.36
Search strategy and selection criteria
We searched PubMed using the search terms “COVID-19 pneumonia”, “SARS-CoV-2”, “SARS” and “pulmonary thrombosis”, “embolism”. We also searched for D-dimer, fibrinogen, creatine kinase “CK“, and troponin and reviewed publications that reported data on these parameters. We also searched for articles on immunothrombosis and cytokines. We limited our search to articles that were published in English between Dec 22, 2019, and April 22, 2020.
Contributors
DM and CB developed the initial concepts for this Viewpoint. JSO and KS contributed to first draft writing. DM and CB wrote the final draft. DM, CB, and JSO did the literature review and critically revised the Viewpoint. KS made the figures. JSO and KS edited the Viewpoint. PE edited and approved the final draft. All authors have participated sufficiently in this work, take public responsibility for the content, and have made substantial contributions to this research. This manuscript has not been submitted to another journal and has not been published in whole, or in part, elsewhere previously.
Declaration of interests
DM has received honoraria and grant funding from Novartis and Sobi, and honoraria from Roche. All other authors declare no competing interests.
References
- 1.Chen N, Zhou M, Dong X. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–513. doi: 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Totura AL, Baric RS. SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon. Curr Opin Virol. 2012;2:264–275. doi: 10.1016/j.coviro.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang C, Wang Y, Li X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033–1034. doi: 10.1016/S0140-6736(20)30628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu X, Han M, Li T. Effective treatment of severe COVID-19 patients with tocilizumab. ChinaXiv. 2020;202003:v1. doi: 10.1073/pnas.2005615117. (preprint). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18:844–847. doi: 10.1111/jth.14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, Cao W, Xiao M. [Clinical and coagulation characteristics of 7 patients with critical COVID-2019 pneumonia and acro-ischemia] Zhonghua Xue Ye Xue Za Zhi. 2020;41:e006. doi: 10.3760/cma.j.issn.0253-2727.2020.0006. (in Chinese). [DOI] [PubMed] [Google Scholar]
- 8.McGonagle D, Sharif K, O'Regan A, Bridgewood C. The role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun Rev. 2020 doi: 10.1016/j.autrev.2020.102537. published online April 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.George MR. Hemophagocytic lymphohistiocytosis: review of etiologies and management. J Blood Med. 2014;5:69–86. doi: 10.2147/JBM.S46255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shoenfeld Y. Corona (COVID-19) time musings: our involvement in COVID-19 pathogenesis, diagnosis, treatment and vaccine planning. Autoimmun Rev. 2020 doi: 10.1016/j.autrev.2020.102538. published online April 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsueh PR, Chen PJ, Hsiao CH. Patient data, early SARS epidemic, Taiwan. Emerg Infect Dis. 2004;10:489–493. doi: 10.3201/eid1003.030571. [DOI] [PubMed] [Google Scholar]
- 12.Nicholls JM, Poon LL, Lee KC. Lung pathology of fatal severe acute respiratory syndrome. Lancet. 2003;361:1773–1778. doi: 10.1016/S0140-6736(03)13413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seguin A, Galicier L, Boutboul D, Lemiale V, Azoulay E. Pulmonary involvement in patients with hemophagocytic lymphohistiocytosis. Chest. 2016;149:1294–1301. doi: 10.1016/j.chest.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Franks TJ, Chong PY, Chui P. Lung pathology of severe acute respiratory syndrome (SARS): a study of 8 autopsy cases from Singapore. Hum Pathol. 2003;34:743–748. doi: 10.1016/S0046-8177(03)00367-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yao XH, Li TY, He ZC. [A pathological report of three COVID-19 cases by minimally invasive autopsies] Zhonghua Bing Li Xue Za Zhi. 2020;49:e009. doi: 10.3760/cma.j.cn112151-20200312-00193. (in Chinese). [DOI] [PubMed] [Google Scholar]
- 16.Tian S, Hu W, Niu L, Liu H, Xu H, Xiao SY. Pulmonary pathology of early-phase 2019 novel coronavirus (COVID-19) pneumonia in two patients with lung cancer. J Thorac Oncol. 2020 doi: 10.1016/j.jtho.2020.02.010. published online Feb 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smeeth L, Thomas SL, Hall AJ, Hubbard R, Farrington P, Vallance P. Risk of myocardial infarction and stroke after acute infection or vaccination. N Engl J Med. 2004;351:2611–2618. doi: 10.1056/NEJMoa041747. [DOI] [PubMed] [Google Scholar]
- 18.Corrales-Medina VF, Musher DM, Wells GA, Chirinos JA, Chen L, Fine MJ. Cardiac complications in patients with community-acquired pneumonia: incidence, timing, risk factors, and association with short-term mortality. Circulation. 2012;125:773–781. doi: 10.1161/CIRCULATIONAHA.111.040766. [DOI] [PubMed] [Google Scholar]
- 19.Hwang DM, Chamberlain DW, Poutanen SM, Low DE, Asa SL, Butany J. Pulmonary pathology of severe acute respiratory syndrome in Toronto. Mod Pathol. 2005;18:1–10. doi: 10.1038/modpathol.3800247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guarner J, Paddock CD, Shieh WJ. Histopathologic and immunohistochemical features of fatal influenza virus infection in children during the 2003-2004 season. Clin Infect Dis. 2006;43:132–140. doi: 10.1086/505122. [DOI] [PubMed] [Google Scholar]
- 21.Rivellese F, Prediletto E. ACE2 at the centre of COVID-19 from paucisymptomatic infections to severe pneumonia. Autoimmun Rev. 2020 doi: 10.1016/j.autrev.2020.102536. published April 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zuo W. Single-cell RNA expression profiling of ACE2, the receptor of SARS-CoV-2. bioRxiv. 2020 doi: 10.1101/2020.01.26.919985. published online Jan 26. (preprint) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou F, Yu T, Du R. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuba K, Imai Y, Rao S. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang P, Gu H, Zhao Z. Angiotensin-converting enzyme 2 (ACE2) mediates influenza H7N9 virus-induced acute lung injury. Sci Rep. 2014;4 doi: 10.1038/srep07027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arabi YM, Balkhy HH, Hayden FG. Middle East Respiratory Syndrome. N Engl J Med. 2017;376:584–594. doi: 10.1056/NEJMsr1408795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barreira da Silva R, Laird ME, Yatim N, Fiette L, Ingersoll MA, Albert ML. Dipeptidylpeptidase 4 inhibition enhances lymphocyte trafficking, improving both naturally occurring tumor immunity and immunotherapy. Nat Immunol. 2015;16:850–858. doi: 10.1038/ni.3201. [DOI] [PubMed] [Google Scholar]
- 28.Zhu N, Zhang D, Wang W. A novel coronavirus from patients with pneumonia in China, 2019. J N Engl J Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu X, Chen P, Wang J. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci China Life Sci. 2020;63:457–460. doi: 10.1007/s11427-020-1637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding Y, Wang H, Shen H. The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J Pathol. 2003;200:282–289. doi: 10.1002/path.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu J, Korteweg C. Pathology and pathogenesis of severe acute respiratory syndrome. Am J Pathol. 2007;170:1136–1147. doi: 10.2353/ajpath.2007.061088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo W, Yu H, Gou J. Clinical pathology of critical patient with novel coronavirus pneumonia (COVID-19) Preprints. 2020 published online Feb 27. 2020020407 (preprint). [Google Scholar]
- 33.Fox SE, Akmatbekov A, Harbert JL, Li G, Brown JQ, Vander Heide RS. Pulmonary and cardiac pathology in Covid-19: the first autopsy series from New Orleans. medRxiv. 2020 doi: 10.1101/2020.04.06.20050575. published online April 10. (preprint). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao C, Wang Y, Gu X. Association between cardiac injury and mortality in hospitalized patients infected with avian influenza A (H7N9) virus. Crit Care Med. 2020;48:451–458. doi: 10.1097/CCM.0000000000004207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chacko B, Peter JV, Pichamuthu K. Cardiac manifestations in patients with pandemic (H1N1) 2009 virus infection needing intensive care. J Crit Care. 2012;27:106e1–106e6. doi: 10.1016/j.jcrc.2011.05.016. [DOI] [PubMed] [Google Scholar]
- 36.Wölfel R, Corman VM, Guggemos W. Virological assessment of hospitalized patients with COVID-2019. Nature. 2020 doi: 10.1038/s41586-020-2196-x. published online April 1. [DOI] [PubMed] [Google Scholar]
- 37.Oudit GY, Kassiri Z, Jiang C. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur J Clin Invest. 2009;39:618–625. doi: 10.1111/j.1365-2362.2009.02153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guzik TJ, Mohiddin SA, Dimarco A. COVID-19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc Res. 2020 doi: 10.1093/cvr/cvaa106. published online April 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bratincsák A, El-Said HG, Bradley JS, Shayan K, Grossfeld PD, Cannavino CR. Fulminant myocarditis associated with pandemic H1N1 influenza A virus in children. J Am Coll Cardiol. 2010;55:928–929. doi: 10.1016/j.jacc.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 40.Nicholls JM, Butany J, Poon LL. Time course and cellular localization of SARS-CoV nucleoprotein and RNA in lungs from fatal cases of SARS. PLoS Med. 2006;3:e27. doi: 10.1371/journal.pmed.0030027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gu J, Gong E, Zhang B. Multiple organ infection and the pathogenesis of SARS. J Exp Med. 2005;202:415–424. doi: 10.1084/jem.20050828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ye J, Zhang B, Xu J. Molecular pathology in the lungs of severe acute respiratory syndrome patients. Am J Pathol. 2007;170:538–545. doi: 10.2353/ajpath.2007.060469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He L, Ding Y, Zhang Q. Expression of elevated levels of pro-inflammatory cytokines in SARS-CoV-infected ACE2+ cells in SARS patients: relation to the acute lung injury and pathogenesis of SARS. J Pathol. 2006;210:288–297. doi: 10.1002/path.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Corman VM, Landt O, Kaiser M. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 2020;25 doi: 10.2807/1560-7917.ES.2020.25.3.2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crackower MA, Sarao R, Oudit GY. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 46.Magnani JW, Dec GW. Myocarditis: current trends in diagnosis and treatment. Circulation. 2006;113:876–890. doi: 10.1161/CIRCULATIONAHA.105.584532. [DOI] [PubMed] [Google Scholar]
- 47.de Lang A, Osterhaus AD, Haagmans BL. Interferon-gamma and interleukin-4 downregulate expression of the SARS coronavirus receptor ACE2 in Vero E6 cells. Virology. 2006;353:474–481. doi: 10.1016/j.virol.2006.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo T, Fan Y, Chen M. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19) JAMA Cardiol. 2020 doi: 10.1001/jamacardio.2020.1017. published online March 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost. 2020 doi: 10.1111/jth.14817. published online March 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Levi M, van der Poll T. Coagulation and sepsis. Thromb Res. 2017;149:38–44. doi: 10.1016/j.thromres.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 51.Gupta N, Zhao YY, Evans CE. The stimulation of thrombosis by hypoxia. Thromb Res. 2019;181:77–83. doi: 10.1016/j.thromres.2019.07.013. [DOI] [PubMed] [Google Scholar]
- 52.Glas GJ, Van Der Sluijs KF, Schultz MJ, Hofstra JJ, Van Der Poll T, Levi M. Bronchoalveolar hemostasis in lung injury and acute respiratory distress syndrome. J Thromb Haemost. 2013;11:17–25. doi: 10.1111/jth.12047. [DOI] [PubMed] [Google Scholar]
- 53.Günther A, Mosavi P, Heinemann S. Alveolar fibrin formation caused by enhanced procoagulant and depressed fibrinolytic capacities in severe pneumonia. Comparison with the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2000;161:454–462. doi: 10.1164/ajrccm.161.2.9712038. [DOI] [PubMed] [Google Scholar]
- 54.Hofstra JJ, Haitsma JJ, Juffermans NP, Levi M, Schultz MJ. The role of bronchoalveolar hemostasis in the pathogenesis of acute lung injury. Semin Thromb Hemost. 2008;34:475–484. doi: 10.1055/s-0028-1092878. [DOI] [PubMed] [Google Scholar]
- 55.Ji HL, Zhao R, Matalon S, Matthay MA. Elevated plasmin(ogen) as a common risk factor for COVID-19 susceptibility. Physiol Rev. 2020;100:1065–1075. doi: 10.1152/physrev.00013.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ten VS, Pinsky DJ. Endothelial response to hypoxia: physiologic adaptation and pathologic dysfunction. Curr Opin Crit Care. 2002;8:242–250. doi: 10.1097/00075198-200206000-00008. [DOI] [PubMed] [Google Scholar]
- 57.Yan SF, Mackman N, Kisiel W, Stern DM, Pinsky DJ. Hypoxia/hypoxemia-induced activation of the procoagulant pathways and the pathogenesis of ischemia-associated thrombosis. Arterioscler Thromb Vasc Biol. 1999;19:2029–2035. doi: 10.1161/01.atv.19.9.2029. [DOI] [PubMed] [Google Scholar]
- 58.Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013;13:34–45. doi: 10.1038/nri3345. [DOI] [PubMed] [Google Scholar]
- 59.Hickey MJ, Kubes P. Intravascular immunity: the host-pathogen encounter in blood vessels. Nat Rev Immunol. 2009;9:364–375. doi: 10.1038/nri2532. [DOI] [PubMed] [Google Scholar]
- 60.Atasheva S, Yao J, Shayakhmetov DM. Innate immunity to adenovirus: lessons from mice. FEBS Lett. 2019;593:3461–3483. doi: 10.1002/1873-3468.13696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smits SL, de Lang A, van den Brand JM. Exacerbated innate host response to SARS-CoV in aged non-human primates. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1000756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shodell M, Siegal FP. Circulating, interferon-producing plasmacytoid dendritic cells decline during human ageing. Scand J Immunol. 2002;56:518–521. doi: 10.1046/j.1365-3083.2002.01148.x. [DOI] [PubMed] [Google Scholar]
- 63.Canaday DH, Amponsah NA, Jones L, Tisch DJ, Hornick TR, Ramachandra L. Influenza-induced production of interferon-alpha is defective in geriatric individuals. J Clin Immunol. 2010;30:373–383. doi: 10.1007/s10875-010-9374-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thachil J, Tang N, Gando S. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J Thromb Haemost. 2020 doi: 10.1111/jth.14860. published online April 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Y, Xiao M, Zhang S. Coagulopathy and antiphospholipid antibodies in patients with covid-19. N Engl J Med. 2020;382:e38. doi: 10.1056/NEJMc2007575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ridker PM, Everett BM, Thuren T. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 67.Ridker PM, Libby P, MacFadyen JG. Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: analyses from the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS) Eur Heart J. 2018;39:3499–3507. doi: 10.1093/eurheartj/ehy310. [DOI] [PubMed] [Google Scholar]
- 68.Burzynski LC, Humphry M, Pyrillou K. The coagulation and immune systems are directly linked through the activation of interleukin-1α by thrombin. Immunity. 2019;50:1033–1042. doi: 10.1016/j.immuni.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scott IC, Hider SL, Scott DL. Thromboembolism with Janus kinase (JAK) inhibitor for rheumatoid arthritis: how real is the risk? Drug Saf. 2018;41:645–653. doi: 10.1007/s40264-018-0651-5. [DOI] [PubMed] [Google Scholar]
- 70.Poyiadji N, Shahin G, Noujaim D, Stone M, Patel S, Griffith B. COVID-19-associated acute hemorrhagic necrotizing encephalopathy: CT and MRI Features. Radiology. 2020 doi: 10.1148/radiol.2020201187. published online March 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee Y-J, Smith DS, Rao VA. Fatal H1N1-related acute necrotizing encephalopathy in an adult. Case Rep Crit Care. 2011;2011 doi: 10.1155/2011/562516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bots SH, Peters SAE, Woodward M. Sex differences in coronary heart disease and stroke mortality: a global assessment of the effect of ageing between 1980 and 2010. BMJ Glob Health. 2017;2 doi: 10.1136/bmjgh-2017-000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baud D, Qi X, Nielsen-Saines K, Musso D, Pomar L, Favre G. Real estimates of mortality following COVID-19 infection. Lancet Infect Dis. 2020 doi: 10.1016/S1473-3099(20)30195-X. published online March 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yancy CW. COVID-19 and African Americans. JAMA. 2020 doi: 10.1001/jama.2020.6548. published online April 15. [DOI] [PubMed] [Google Scholar]
- 75.Ferrario CM, Jessup J, Chappell MC. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111:2605–2610. doi: 10.1161/CIRCULATIONAHA.104.510461. [DOI] [PubMed] [Google Scholar]