

Abstract
Hexavalent chromium Cr(VI) is toxic and can be highly mobile in many aquifer systems. Redox reactions with naturally occurring minerals and organic compounds can reduce Cr(VI) to Cr(III), forming labile Cr(III) oxyhydroxide precipitates, which is a natural attenuation process. In fractured bedrock aquifers, reduction of Cr(VI) in the rock matrix can enhance attenuation beyond that from matrix diffusion only, and potentially reduce back diffusion if concentrations in fractures decline following source reduction via natural processes or engineered remediation. In this study, we develop an extraction method for labile Cr(III) precipitates from Cr(VI) reduction using 5% hydrogen peroxide (H2O2). Combining Cr(III) extractions with an established sodium hydroxide (NaOH) method for determination of Cr(VI) concentrations in rock porewater, a measure of the labile Cr(III) and Cr(VI) fractions in geologic samples is achieved. The methods were applied to cores from a contaminated groundwater system in fractured porous bedrock in order to assess the effectiveness of natural attenuation and whether Cr(VI) mass that diffused into the bedrock matrix was undergoing reduction. Detailed vertical distributions display two depth intervals with corresponding elevated concentrations of Cr(VI) in the porewater and extractable total Cr. The correspondence of Cr(VI) and labile Cr(III) provides evidence for reduction of Cr(VI) contamination in the bedrock matrix. Mineralogical analysis suggests that Fe(II)-bearing minerals, chlorite and biotite are the most likely candidates for natural reductants. This study provides evidence for the natural attenuation of anthropogenic Cr(VI) contamination in the porewater of a fractured bedrock aquifer, and it outlines a quantitative method for evaluating the effectiveness of natural attenuation in groundwater systems.
Keywords: Hexavalent chromium, Extraction, Natural attenuation, Fractured bedrock, ICP-MS
1. Introduction
The chromium concentration in Earth crust is about 100 mg/kg (Bordean, 2012), most of which is in the trivalent form, Cr(III), hosted by minerals such as olivine, pyroxene and chromite. Near Earth’s surface chromium also occurs in the toxic hexavalent form, Cr(VI), because redox reactions cause cycling of the oxidation state between Cr(III) and Cr(VI) (Lin, 2002, Schroeder and Lee, 1975, Zhao et al., 2016). Cr(VI) is soluble in aqueous solution across a wide range of pH, occurring in variably protonated forms of the oxyanion, chromate (H2CrO4, HCrO4−, CrO4− and Cr2O72 −) (Kumar and Riyazuddin, 2011, Unceta et al., 2010), whereas Cr(III) is soluble at low pH in the species Cr3 +, Cr(OH)2 + and Cr(OH)2+. In the near-neutral pH range of most natural waters Cr(III) has very low solubility because it forms Cr(III)-hydroxide precipitates (Rai et al., 1987), including crystalline Cr(OH)3 ∙ 3H2O, amorphous Cr(OH)3(am) and in the presence of Fe3 +, a mixed CrxFe(1-x)(OH)3, such as Cr0.25Fe0.75(OH)3 (Rai et al., 2004, Rai et al., 1987, Sass and Rai, 1987).
Elevated concentrations of Cr(VI) in natural waters are mainly caused by releases from human activities (Palmer and Wittbrodt, 1991), and Cr(VI) is a common contaminant at many sites (Kent et al., 2007, Palmer and Puls, 1994). Efforts to remediate Cr(VI) contamination have focused on reduction and precipitation (Hashim et al., 2011), adsorption (Mohan and Pittman, 2006) and bioremediation (Hasin et al., 2010, Kanmani et al., 2012) processes. Natural attenuation of Cr(VI) contamination is also possible (Kent et al., 2007), most effectively via reduction to Cr(III) and precipitation as Cr(III) hydroxide (Blowes, 2002, Kent et al., 2007). Naturally-occurring reductants include soil humic substances (humic acid and fulvic acid) (Palmer, 1996), aqueous Fe(II) (Pettine et al., 1998), pyrite (FeS2) (Lin and Huang, 2008), magnetite and ilmenite (White and Peterson, 1996), and Fe(II)-containing chlorite (Brigatti et al., 2000).
The viability of natural attenuation would be supported by evidence for the accumulation of Cr(III)-hydroxide on mineral surfaces in Cr(VI)-contaminated aquifers. These reaction products should be expected to accumulate in an aquifer where aqueous Cr(VI) is mobilized by flowing groundwater and is in contact with reductants. Aquifers in fractured porous bedrock are characterized by preferential pathways, with groundwater flow confined mostly within fractures, out of contact with the majority of reactive Fe(II)-mineral surface area in the rock matrix. However, diffusion of dissolved Cr(VI) from groundwater in fractures to the immobile porewater in the rock matrix can strongly attenuate plumes in fractured sedimentary rock (Parker et al., 2010), particularly for Cr(VI) which can subsequently be reduced to Cr(III) in the rock matrix (Fantoni et al., 2002, Friedly et al., 1995). The bedrock matrix porosity is typically orders of magnitude greater than the fracture porosity in sedimentary bedrock systems, and therefore the capacity for storage of Cr(VI) and reduction to Cr(III) in the matrix is much greater. Standard groundwater sampling and analytical methods are available to monitor contamination via conventional wells or multilevel monitoring systems, but these methods sample groundwater primarily from the fracture networks. To understand the natural attenuation of Cr(VI) in fractured porous bedrock, an appropriate sampling and analytical procedure is required for the determination of newly formed (labile) Cr(III) concentrations in the rock matrix.
In a previous study, a method was developed for determining the concentration of aqueous Cr(VI) in the porewater of core samples from contaminated fractured bedrock aquifers (Zhao et al., 2015). In an effort to investigate the reduction of Cr(VI) to Cr(III) in the natural environment, the objective of the present study was to develop a complementary method to quantify Cr(III)-hydroxides formed in-situ by reduction of Cr(VI) in the bedrock matrix. These Cr(III)-hydroxide species are herein defined as labile Cr(III) and one of the principal challenges is to distinguish them from the natural Cr background in the rock matrix.
2. Experimental methods
Preliminary tests with aggressive extraction methods, such as acid extraction, demonstrated that they were not sufficiently selective for labile Cr(III). Alternatively, oxidative dissolution of labile Cr(III) precipitates with hydrogen peroxide (H2O2) (reaction (1)):
| (1) |
was considered because the reaction rate is relatively low and can be controlled by adjusting experimental conditions, such as pH (Bokare and Choi, 2011). The reaction between H2O2 and different mineralogical forms of Cr in soil was investigated by Rock et al. (2001), who found that it was successful in extracting recently precipitated Cr(III) solids in electroplating waste, but was not effective in extracting Cr from serpentine-rich soil at a former Cr mine site. An additional benefit of the H2O2 approach is that aqueous Cr(VI) in porewater is extracted along with labile Cr(III), allowing for calculation of labile Cr(III) as the difference between total extractable Cr (H2O2 extraction) and Cr(VI) from the previous method (Zhao et al., 2015) (NaOH extraction).
2.1. Study site and materials
The study site is a former electroplating facility in New Jersey, U.S.A. where chromic acid was released approximately three decades ago from a ruptured storage tank to a groundwater system comprising a shallow unconsolidated aquifer with underlying fractured porous sandstone and siltstone of the Passaic Formation (Gregory C. Herman, 2001). A plume of Cr(VI)-contaminated groundwater moving toward a major river over a distance of approximately 800 m has since developed down gradient in overburden and bedrock. Bedrock core samples were collected within the plume using the Discrete Fracture Network (DFN) approach (Parker et al., 2012). Additional details on sampling and sample handling are provided by Zhao et al. (2015) who presented a detailed vertical distribution of Cr(VI) concentrations in the bedrock porewater. The present study utilizes 145 discrete depth bedrock core samples from the same borehole, positioned mid-plume between the source area and river, and collected over a depth interval from about 21 to 108 mbgs (meters below ground surface), where the ground surface elevation at the study site is 5.0 m ASL.
In an effort to test the general applicability to measure natural background values for H2O2-extractable Cr(III) in typical aquifer materials, samples of sand from two uncontaminated glacial-fluvial aquifers (Fredericton, New Brunswick, Canada and North Battleford, Saskatchewan, Canada) and a variety of rock samples, including Precambrian metagabbro and granitic gneiss from eastern Ontario, and red (oxidized) and green (unoxidized) Carboniferous sandstone from eastern New Brunswick, Canada, were analyzed. The data are not intended to be representative of geologic materials in general, but the glacial-fluvial aquifer sands were chosen with the view that they should reflect broad geographic sampling and a diversity of bedrock sources. Similarly, the sandstones were chosen to reflect broad geographic and geologic provenance, and the metagabbro and granitic gneiss were chosen to represent mafic and felsic endmembers for igneous and metamorphic rocks. An extraction was also conducted on a sample of pure chromite to define what might be the maximum expected Cr yield from the procedure. All materials were crushed (rocks) and sieved (< 2 mm mesh) to remove coarse fragments.
2.2. Extraction
Both Cr(VI) and labile Cr(III) are extracted with the H2O2 extraction procedure, using 1.5 mol/L H2O2 (plasma pure plus, SCP Science) in 0.1 mol/L ammonium acetate (NH4Ac) buffer at pH 7 (Sigma-Aldrich). For each sample, approximately 1 g of sand/crushed rock material and 10 mL of extractant were added to 15 mL centrifuge tubes, which were shaken for 30 min, centrifuged and the supernatant transferred to 50 mL tubes. The extraction was repeated on the same sample two more times and the extractant solutions were combined for a total of 30 mL. The extractant was then filtered sequentially with 0.45 μm and 0.1 μm nylon syringe filters (Fisher Scientific), and acidified to a final concentration of 0.16 mol/L HNO3 (plasma pure plus, SCP Science).
The NaOH extraction procedure targets Cr(VI) using approximately 1 g of rock material and 0.01 mol/L NaOH as reported by Zhao et al. (2015). The labile Cr(III) is operationally defined as the difference between the H2O2 and NaOH extractions, both of which are corrected for the background from rock matrix. The two extraction procedures are summarized in Table 1.
Table 1.
Summary of the H2O2 and NaOH extraction procedures for 1 g of sample.
| H2O2 Extraction | NaOH Extraction |
|---|---|
| Extract with 10 mL 1.5 mol/L H2O2 in 0.1 mol/L NH4Ac for 30 min | Extract with 10 mL 0.01 mol/L NaOH for 30 min Centrifuge |
| Repeat extraction twice more Centrifuge | Repeat extraction with 5 mL 0.01 mol/L NaOH Centrifuge |
| Combine extraction solutions | Combine extraction solutions |
| Sequential filtration (0.45 and 0.1 μm) | Sequential filtration (0.45 and 0.1 μm) |
| Acidify to 0.16 mol/L HNO3 | Acidify to pH 2–5 |
| Cation exchange separation/elute Cr(VI) from column with 10 mL of 0.00016 mol/L HNO3 | |
| Acidify to 0.16 mol/L HNO3 |
Total Cr concentrations were determined for each of the background materials using a heated HNO3-HCl-HF digestion followed by ICP-MS analysis. All acids used for digestion and evaporation are concentrated: HNO3 (16 mol/L), HCl (12 mol/L) and HF (29 mol/L). Approximately 25 mg of sand or pulverized rock sample were digested in 1.5 mL of HNO3, 0.5 mL HCl and 0.5 mL HF at 110 °C for about 4 h and then held at 90 °C overnight. The digestion mixture was evaporated to dryness at 90 °C, then 1.5 mL HNO3 and 0.5 mL HCl were added and evaporated twice to remove HF, and finally the sample was dissolved in 1.5 mL HNO3 and 0.5 mL HCl prior to ICP-MS analysis.
2.3. Quality control (QC) materials and analytical procedures
A laboratory reference material with low Cr(VI) concentration (LRS) was prepared from rock core collected at the study site from > 80 m depth, well below the Cr(VI) plume (Zhao et al., 2015). Crushed samples (< 2 mm) were pulverized, blended and sieved (< 180 μm). This reference material was used to prepare fresh Cr(III) precipitate for optimizing the extraction procedure. To monitor analytical precision for Cr(VI) and Cr(III) throughout the method development process, a second laboratory reference material with relatively high Cr(VI) concentration (HRS) was prepared by blending five crushed core samples (< 2 mm) from 37 to 48 mbgs, which is directly within the Cr(VI) groundwater plume (Zhao et al., 2015).
A reference material for fresh Cr(III) precipitate (FRS) was prepared by adding 40 μL of 1.9 × 10− 4 mol/L Cr(VI) (K2Cr2O7) in 10 mL 0.01 mol/L Na2SO3 solution acidified by HNO3 (~ pH 2) resulting in the reduction of Cr(VI) to Cr(III). The solution was neutralized using NaOH (1 mol/L), and about 1 g of LRS was immediately added and thoroughly mixed. The mixture was centrifuged and the resulting residue containing fresh Cr(III) precipitate was retained as the FRS. A second approach was also used wherein a 53Cr(III) labeled reference material (53Cr(III)-FRS) was prepared using Na53CrO4 (97.60%; Isoflex, US) as an isotopic tracer to improve the distinction between the spike and the background. In this case, 40 μL of 1.9 × 10− 4 mol/L 53Cr(VI) was reduced as described above and immediately mixed with 1 g of LRS.
To determine the extraction yields, 40 μL of 1.9 × 10− 4 mol/L Cr(III) (Cr standard, SCP Science) or Cr(VI) (K2Cr2O7) were spiked into about 1 g LRS (Cr(III)-LRS and Cr(VI)-LRS). To determine the yields for both Cr(VI) and Cr(III) simultaneously, 40 μL of 1.9 × 10− 4 mol/L 53Cr(VI) were spiked into about 1 g FRS (53Cr(VI)-FRS). All QC samples (Table 2) were tested in triplicate. All reference materials were prepared from rock powder.
Table 2.
Quality control materials (the ground surface elevation at the drill-hole location is 5.0 m ASL).
| QC material | Starting matrix | Spiked Cr |
|---|---|---|
| LRS | Rock (below 80 mbgs) | |
| HRS | Rock (37–48 mbgs) | |
| FRS | LRS | Cr(III)-fresh precipitated |
| Cr(III)-LRS | LRS | Cr(III) |
| Cr(VI)-LRS | LRS | Cr(VI) |
| 53Cr(III)-FRS | LRS | 53Cr(III)-fresh precipitated |
| 53Cr(VI)-FRS | FRS | Cr(III)-fresh precipitated + 53Cr(VI) |
Analyses for Cr were conducted by ICP-MS (Agilent 8800) in He gas mode. Scandium was used as internal standard to monitor the instrument stability. Matrix-matched calibration standards were used to account for possible matrix effects. Threshold values defining the upper limit of background concentrations for both Cr(VI) and labile Cr(III) were determined from the results of extractions on uncontaminated samples collected below 50 mbgs. The precision of the experimental procedures was tested with triplicate extractions for HRS and four bedrock samples from the contaminated zone where Cr(VI) and labile Cr(III) concentrations were greater than the respective threshold values.
Mineralogical investigations using scanning electron microscopy and energy dispersive X-ray spectroscopy (SEM-EDS) on polished thin sections were conducted for six samples at the University of Ottawa—Canadian Museum of Nature MicroAnalysis Laboratory on a JEOL 6610LV SEM. Powder X-ray diffraction analyses were conducted for seven samples with a Bruker D-8 Advance diffractometer at the University of New Brunswick.
3. Results and discussion
3.1. Extractable Cr in natural materials
The ranges of total H2O2– and NaOH-extractable Cr concentrations in the crushed rock samples and aquifer sands chosen to reflect a range of natural materials are reported in Table 3. The total Cr content varies from 1.0 to 10.6 × 10− 4 mol/kg (mean = 4.9 × 10− 4 mol/kg), and in all cases is approximately three orders of magnitude greater than the H2O2-extractable Cr and four orders of magnitude greater than the extractable Cr(VI). The H2O2-extractable Cr content varies from 1.5 to 11.5 × 10− 7 mol/kg (mean = 5.7 × 10− 7 mol/kg), except in the case of pure chromite which was approximately ten times higher (9.62 × 10− 6 mol/kg) than for the rock samples and aquifer sands. This may be close to a maximum expected yield for H2O2-extractable Cr(III) in geologic materials using this procedure. The NaOH-extractable Cr(VI) content is very low, ranging from 1.9 to 3.7 × 10− 8 mol/kg (mean = 2.7 × 10− 8 mol/kg). Values in Table 3 provide an indication of the respective Cr contents in natural geologic materials, but definition of background Cr values at any specific site should be assessed separately (see Section 3.3).
Table 3.
Total and extractable Cr (III and VI) concentrations for natural uncontaminated rock and aquifer sands (mean ± 1σ).
| Extractable Cr (× 10−8 mol/kg) | |||
|---|---|---|---|
| Total Cr (× 10−4 mol/kg) | H2O2 extraction (n = 3) | NaOH extraction (n = 3) | |
| LRS | 10.6 ± 0.4 (n = 3) | 77 ± 8 | 1.9 ± 1.2 |
| Meta gabbro | 5.1 ± 0.4 (n = 2) | 33 ± 8 | 2.3 ± 0.8 |
| Granitic gneiss | 1.0 ± 0.02 (n = 2) | 15 ± 4 | 1.9 ± 1.2 |
| Sand stone (unoxidized) | 1.0 ± 0.02 (n = 2) | 35 ± 4 | 3.7 ± 0.2 |
| Sand stone (oxidized) | 7.5 ± 0.2 (n = 2) | 58 ± 10 | 2.9 ± 0.8 |
| Sand (NB) | 5.5 ± 1.3 (n = 2) | 65 ± 2 | 3.3 ± 1.0 |
| Sand (SK) | 3.4 ± 0.1 (n = 2) | 115 ± 10 | 3.1 ± 0.6 |
| Mean | 4.9 | 57 | 2.7 |
| Chromite | 47,885 | 962 ± 33 | 23.1 ± 1.9 |
3.2. Quality control
In method development, 53Cr(III)-FRS was used to optimize the sequential H2O2 extraction. The recovery of extractable 53Cr (III) in each extraction step decreased as extraction was repeated and the maximum total yield was reached after three extraction steps (Fig. 1), indicating a complete extraction.
Fig. 1.
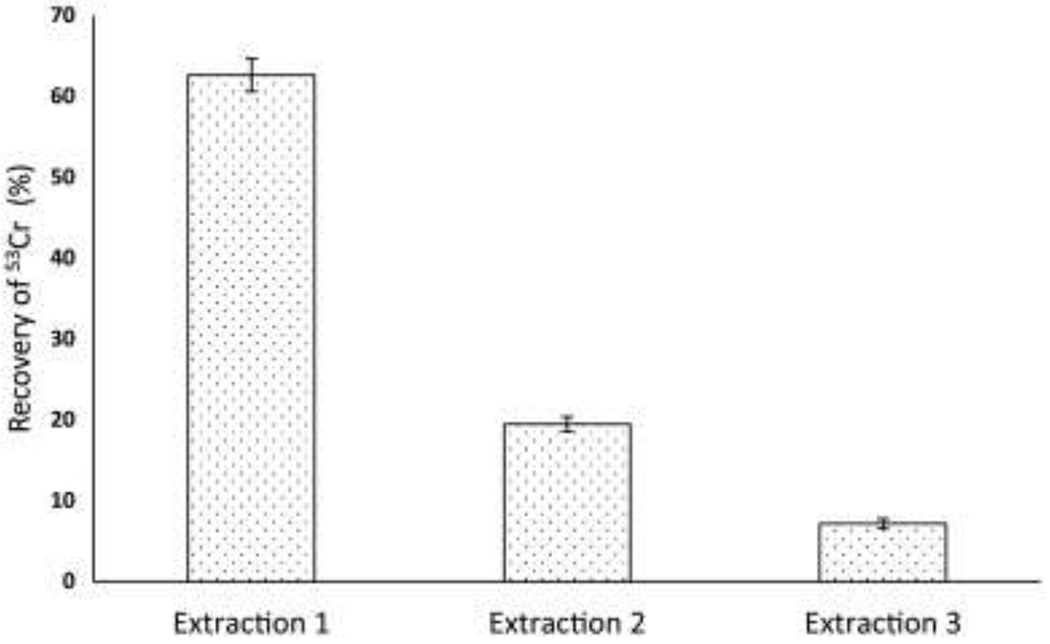
The recovery of 53Cr(III) in three sequential extractions with 1.5 mol/L H2O2 on 53Cr(III)-FRS.
Results for replicate analyses of reference materials spiked with Cr(III) or Cr(VI), including isotopically-traced species, are shown in Table 4. Recoveries of Cr(III) and Cr(VI), respectively, for spikes in the aqueous reagent blank, LRS and FRS were > 81% and > 89% for the H2O2 extraction and < 5% and > 83% for the NaOH extraction. The slightly lower recoveries for Cr(III) compared to Cr(VI) in the H2O2 extraction suggests that some fraction of the Cr(III) spike forms a non-labile solid species that is not fully oxidized during the 3 × 30 min extractions. Nevertheless, the extraction yields conform to commonly accepted standards, for example US EPA method 3060A defines 75 to 125% as the acceptance standard for yields of Cr(VI) on spiked samples (US EPA, 1996). The results indicate that the H2O2 extraction is quantitative for both Cr(VI) and labile Cr(III).
Table 4.
Yields of Cr for spiked materials.
| Material | Recovery for H2O2 extraction (%) | Recovery for NaOH extraction (%) | ||
|---|---|---|---|---|
| Cr(VI) | Cr(III) | Cr(VI) | Cr(III) | |
| Cr(III) spiked blank | 93 ± 2 | 5 ± 1 | ||
| Cr(VI) spiked blank | 93 ± 1 | 89 ± 2 | ||
| Cr(III)-LRS | 86 ± 1 | 1 ± 0.2 | ||
| Cr(VI)-LRS | 90 ± 1 | 85 ± 1.8 | ||
| FRS | 81 ± 1 | 1 ± 0.2 | ||
| 53Cr(III)-FRS | 84 ± 1 | 0.1 ± 0.1 | ||
| 53Cr(VI)-FRS | 89 ± 1 | 83 ± 2 | 83 ± 2 | 3 ± 0.1 |
By adding a 53Cr(VI) spike to the FRS which contains labile Cr(III), the experiments simulated the environment in contaminated bedrock, which may contain both Cr(VI) and labile Cr(III)-hydroxide precipitates. In these experiments, the spike recoveries for Cr(III) and 53Cr(VI) were 83 ± 2% and 89 ± 1% respectively, demonstrating a high level of selectivity for these species in bedrock samples.
The Cr concentrations in triplicate extractions of HRS and four additional bedrock samples from the contaminated groundwater zone are shown in Table 5. The relative standard deviations (RSDs) for triplicate extractions of HRS are 5% and 4% for H2O2 and NaOH extractions, respectively, and the RSD values for the four additional bedrock samples are slightly larger, ranging from 5 to 12% (mean 9%) and 2 to 18% (mean 13%) for the H2O2 and NaOH extractions, respectively.
Table 5.
Extractable Cr for HRS and samples from the contaminated groundwater zone (the ground surface elevation at the drill-hole location is 5.0 m ASL).
| Extractable Cr (× 10−8 mol/kg) | |||||
|---|---|---|---|---|---|
| Depth (mbgs) | H2O2 extraction | RSD (%) | NaOH extraction | RSD (%) | |
| HRS | 37–48 | 102 ± 6 | 5 | 21 ± 1 | 4 |
| Sample #9 | 22.5 | 492 ± 58 | 12 | 673 ± 119 | 18 |
| Sample #85 | 39.4 | 335 ± 37 | 11 | 98 ± 17 | 18 |
| Sample #86 | 39.8 | 160 ± 15 | 10 | 25 ± 1 | 2 |
| Sample #87 | 40.1 | 331 ± 17 | 5 | 73 ± 12 | 14 |
3.3. Cr(VI) and labile Cr(III) in the contaminated bedrock
Data for H2O2– and NaOH-extractable Cr in all samples from the field site are presented in Fig. 2. The Cr-contamination in the bedrock aquifer is most obvious at depths of about 22 mbgs and 41 mbgs, where elevated values are observed for both H2O2 and NaOH extractions, but an estimate of the threshold to background is required in order to fully define the contaminated zone. A variety of methods are available to estimate threshold values that distinguish contamination from the natural background (Reimann et al., 2005); some of these may be applied to datasets that contain both contaminated and uncontaminated samples, but definition of a threshold value for natural background is best done using data only from the uncontaminated material (Singh et al., 1994, US EPA, 2002). In many studies it can be difficult to identify a sample set that has been unaffected by contamination, but in the case of the study site, it is evident that there is no significant extractable Cr(VI) or labile Cr(III) at depths > 50 mbgs (Fig. 3). This visual assessment was used to support the selection of data from depths > 50 mbgs for threshold determination. Here we assume that the Cr(III) background is constant throughout the sampled section, above and below 50 mbgs, and this is supported by the observation that the borehole intersected similar red sandstone and siltstone from top to bottom. For a normally distributed dataset, the threshold value separating background from contaminated outliers can then be defined as the mean + 2 standard deviations (2σ). As for many geologic datasets, the data from the study site at depths > 50 mbgs are not normally distributed, but they conform reasonably well to a log-normal distribution (Fig. 3) so the threshold values (Table 6, Fig. 2) are calculated from log-transformed data as anti-log (meanlog + 2σlog), where meanlog and σlog are the average and standard deviation of log-transformed data, respectively.
Fig. 2.
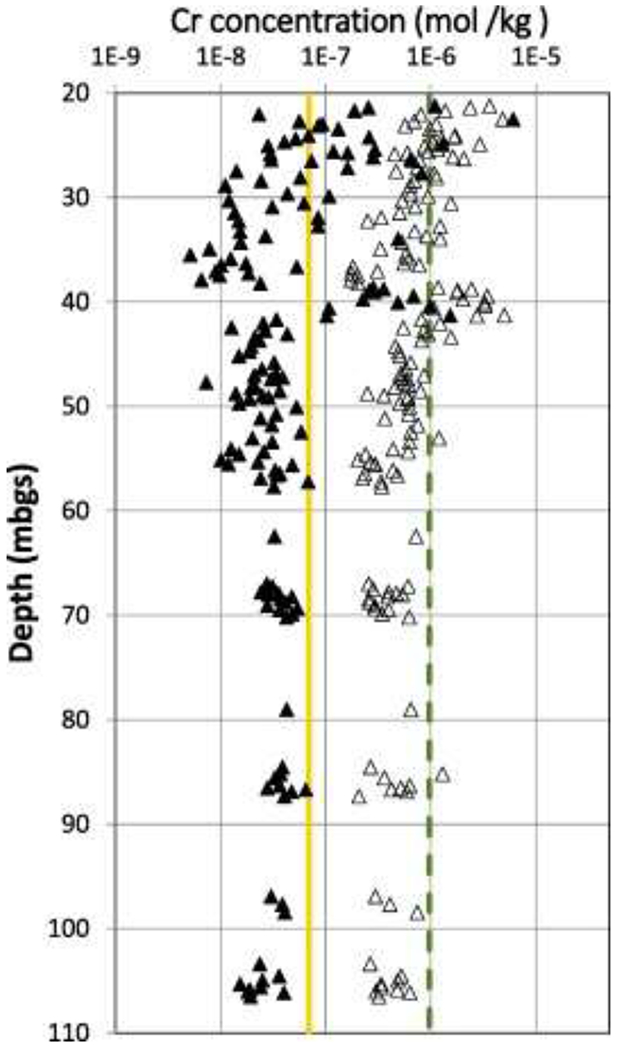
Raw data for extractable Cr by the H2O2 extraction (△) and the NaOH extraction (▲). The yellow solid line is the threshold for NaOH extraction and the green dashed line is the threshold for H2O2 extraction. The ground surface elevation at the drill-hole location is 5.0 m ASL. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 3.
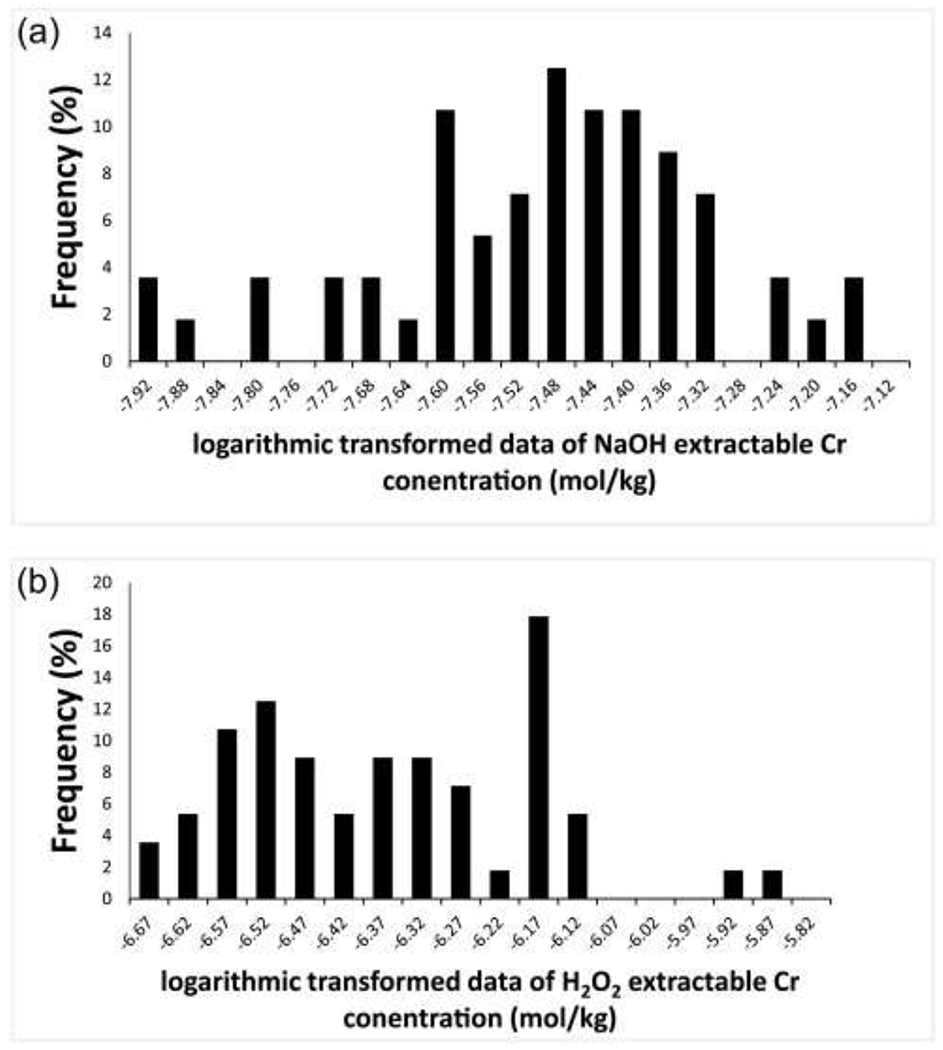
Frequency distribution of log-transformed extractable Cr concentration. a: NaOH extraction; b: H2O2 extraction.
Table 6.
Mean, standard deviation and threshold values calculated from log-transformed data for H2O2– and NaOH-extractable Cr (× 10−8 mol/kg).
| Extraction | Mean | Threshold |
|---|---|---|
| H2O2 | 41 | 97 |
| NaOH | 3 | 7 |
The thresholds were used to determine the concentrations of Cr(VI) and labile Cr(III) that are present in the bedrock aquifer due to the contamination event (Eqs. (2), (3)):
| (2) |
| (3) |
the H2O2 and NaOH extraction solutions, respectively, CrH2O2_Th and CrNaOH_Th are threshold values for the H2O2 (97 × 10− 8 mol/kg) and NaOH (7 × 10− 8 mol/kg) extractions (Table 6). The Cr(VI) and labile Cr(III) concentrations in the contaminated zone are shown in Fig. 4. The dark yellow horizontal bars represent Cr(VI) and the dark green horizontal bars represent labile Cr(III).
Fig. 4.
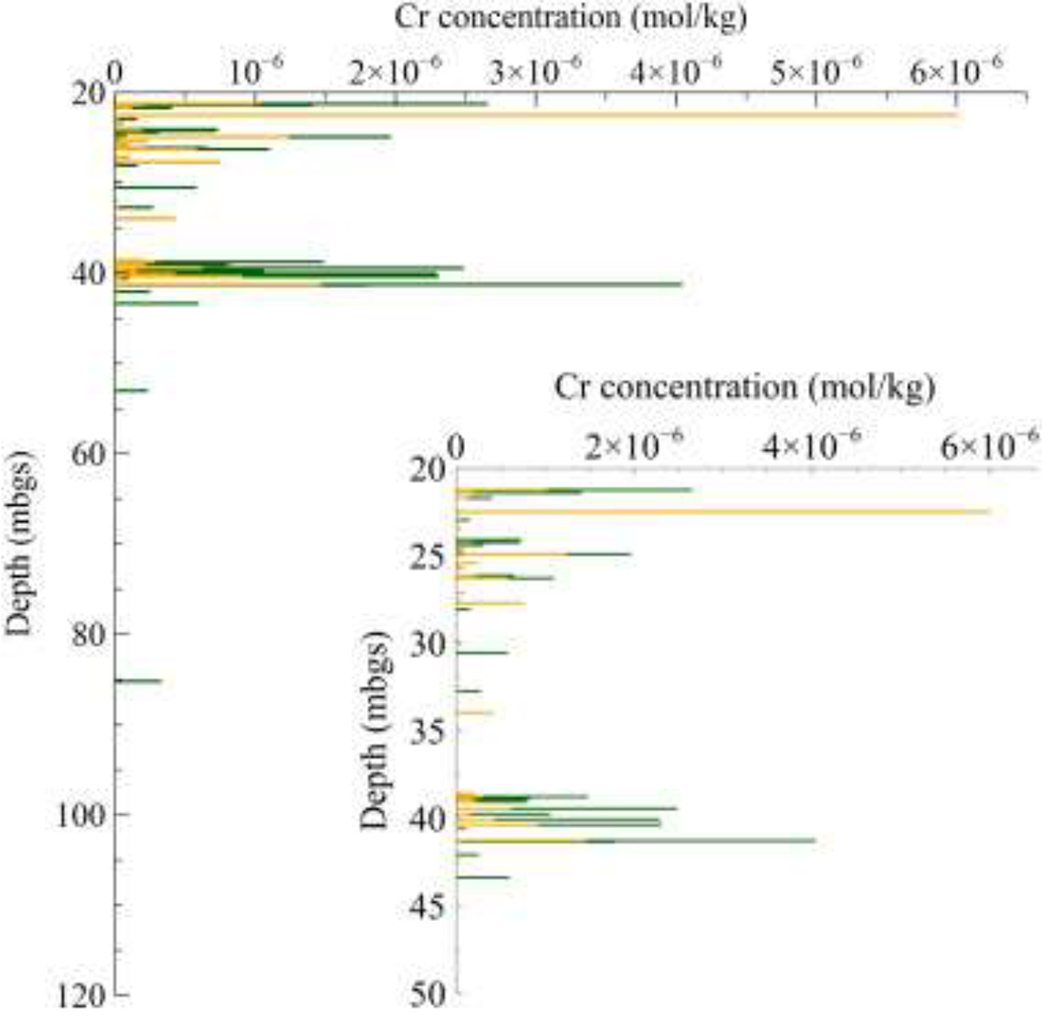
Stacked bars showing background-corrected data for extractable Cr(VI) and labile Cr(III). The dark yellow bars represent the concentration of Cr(VI), whereas the dark green bars represent labile Cr(III). The ground surface elevation at the drill-hole location is 5.0 m ASL. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The data presented in Fig. 2, Fig. 4 provide a detailed vertical distribution of Cr(VI) and labile Cr(III) in the bedrock matrix. Although the contaminant peaks are clearly evident at 22 and 41 mbgs, the distributions are very heterogeneous. Heterogeneity in Cr(VI) concentrations is thought to reflect the distribution of hydraulically active fractures that facilitate growth of the contaminant plume by advection (Zhao et al., 2015), and in timeframes for diffusion into and reduction reactions in the bedrock matrix varying with sample distances into the matrix off fractures. In general, the detection of significant amounts of labile Cr(III) in the core samples indicates that there are matrix reduction processes operating to limit the transport of Cr(VI), but the heterogeneous distributions of labile Cr(III) must be a reflection of spatial variations in the Cr(VI) residence time, pH, redox conditions, and the availability of natural solid-phase reductants. The initial contamination event occurred in 1983, indicating a long residence time for Cr(VI) in the groundwater system, but large variations in transport rates in fractures (advection) and the porous rock matrix (diffusion) are expected to result in corresponding spatial variations in Cr(VI) residence time. It is possible then that observed variability in Cr(VI) and labile Cr(III) in the bedrock matrix relates to the residence time control on reaction kinetics, but this cannot be verified. It does not appear that heterogeneous distributions of Cr(VI) and labile Cr(III) in the bedrock can be explained by variability in the aqueous geochemical conditions, because measurements in three multi-level groundwater sampling ports completed between 25 and 48 m depth in the same borehole (see Zhao et al., 2015) demonstrate that pH and EH conditions are stable (7.6 ± 0.1; 230 ± 10 mV; Wilkin, unpublished data).
A variety of natural reductants are known to react with Cr(VI), including aqueous Fe(II) and S(− II) species, minerals containing Fe(II), reduced forms of sulfur and organic carbon (Palmer and Puls, 1994). At the study site, the rock units containing the contaminated plume are hematite-rich red-bed sandstones and siltstones. The XRD results (Table 7) indicate that the major-mineral composition of the rocks is similar throughout the section, with quartz, muscovite, chlorite (clinochlore), feldspars and calcite occurring in variable proportions. Mineralogical analyses were also conducted on polished thin sections with scanning electron microscopy (SEM). Sulfide minerals were not detected, but Fe-oxide minerals are abundant throughout (Fig. 5). The common iron oxide minerals, magnetite and hematite are not easily distinguished with SEM-EDS analysis, but the red-bed character of the rocks suggests that the Fe oxides are dominated by hematite. Eary and Rai (1989) present evidence that Cr(VI) can be reduced by trace levels of Fe(II) in hematite. Mineralogical analyses also indicate the occurrence of fine-grained sheet silicates that partially infill small pores (Fig. 5). They occur as mixtures with other fine-grained phases such as Fe-oxides, so it is not possible to obtain discrete analyses, but the ubiquitous occurrence of Si, Al and Mg, and sporadic occurrence of K suggest the presence of chlorite, biotite and/or illite. Brigatti et al. (2000) demonstrate that Fe-bearing chlorite is an effective reductant for Cr(VI), and reduction of Cr(VI) by biotite is reported by Eary and Rai (1989), Ilton et al. (1997) and Ilton and Veblen (1994).
Table 7.
XRD analyses of bedrock samples (the ground surface elevation at the drill-hole location is 5.0 m ASL).
| Sample number | Depth (mbgs) | Mineralogical composition |
|---|---|---|
| 21 | 24.9 | Muscovite, Clinochlore, Albite, Quartz |
| 86 | 39.7 | Calcite, Albite, Quartz |
| 121 | 49.0 | Clinochlore, Quartz, Muscovite-2 M1 |
| 155 | 56.6 | Albite, Muscovite, Quartz |
| 204 | 67.9 | Quartz, Microcline, Clinochlore, Albite, Muscovite-2 M1 |
| 292 | 87.3 | Clinochlore, Muscovite-2 M1, Albite, Quartz |
| 392 | 106.1 | Microcline, Clinochlore, Muscovite, Quartz, Albite |
Fig. 5.

Backscattered-electron images from a sample within the contaminated zone (41.27 mbgs) showing (a) coarse grains of quartz and feldspar; (b) accumulations of sheet silicates filling small pores, and (c) dense accumulation of sheet silicates in a small pore. Bright, nano-scale mineral grains in (c) are Fe oxides. The scale bars are: (a) 200 μm, (b) and (c) 10 μm.
The reduction of Cr(VI) on the surface of minerals forms freshly precipitated Cr(III) that has been targeted for extraction by H2O2 in this study. In the presence of Fe(III), as would be the case where Cr(VI) reduction is coupled to Fe(II) oxidation, the precipitate is known to be a mixed Fe(III) and Cr(III) hydroxide phase with the general formula CrxFe(1-x)(OH)3 (Eary and Rai, 1989, Fendorf and Wielinga, 2000). According to Eary and Rai (1989), formation of this mixed hydroxide phase limits the solubility of Cr to < 10− 6 mol/L between pH 4 and 12. It is known that fresh Fe(III) and Cr(III) oxyhydroxide precipitates are relatively labile, and therefore amenable to extraction, but with aging the material aggregates, partially dehydrates and gradually crystallizes (Raiswell et al., 2010, Zhang et al., 2004). The labile nature of the fresh CrxFe(1-x)(OH)3 precipitate is key to obtaining contrast between contamination and natural background when applying extraction techniques.
4. Environmental implications
Reduction of Cr(VI) to form an immobile solid phase with low-solubility is a desirable result in aquifers affected by Cr(VI) contamination. Ideally these reduction reactions act on Cr(VI) that has diffused into the bedrock matrix, enhancing attenuation over that from diffusion processes only. The stability of the precipitate will increase as it ages and transforms from amorphous to crystalline structure, so this natural attenuation process should represent a long-term solution to the problem of contamination, i.e. if the reduction reaction is irreversible, the Cr should remain immobilized in the bedrock matrix as Cr(III), decreasing Cr(VI) concentrations in the bedrock matrix and leading to steeper concentration gradients to drive Cr(VI) diffusion from fractures into the matrix. Nevertheless, it is possible that the labile Cr(III) could be remobilized by oxidation, and Palmer and Puls (1994) note that effective natural attenuation of Cr(VI) requires that there be no net oxidation of Cr(III) to Cr(VI). In natural environments, dissolved O2 may oxidize Cr(III) to Cr(VI), but the reaction rate is very slow (Schroeder and Lee, 1975). The most common oxidant for Cr(III)-hydroxides is manganese oxide (Trebien et al., 2011), and adsorbed Mn(II) can act as a catalyst when it is oxidized to manganese oxide by dissolved O2 on the surface of Cr(OH)3(s) (Namgung et al., 2014). In fractured bedrock aquifers, any remobilization would be limited by availability and transport of O2 and Mn(II) so the risk should be very low. Overall, the methodology developed in this study allows for the determination of labile Cr(III) species produced by the reduction of Cr(VI) during reaction with natural reductants. Such measurements of labile Cr(III) at contaminated bedrock sites can be used as a new tool for confirming the viability and effectiveness of matrix diffusion and reduction reactions in the matrix as a contributor to natural attenuation of Cr(VI) plumes.
Acknowledgements
The work was funded by the U.S. EPA through the Regional Applied Research Effort (RARE) program under grant number RFQ-OH-14-00122, and by NSERC Discovery Grant number 194091-2011-RGPIN. CH2M Hill was the primary site consultant and provided field and logistical support for the core subsampling activities. Supplemental funding for development of the laboratory methods was provided by the University Consortium for Field-Focused Groundwater Contamination Research.
References
- Blowes D Tracking hexavalent Cr in groundwater Science, 295 (2002), pp. 2024–2025 [DOI] [PubMed] [Google Scholar]
- Bokare AD, Choi W Advanced oxidation process based on the Cr(III)/Cr(VI) redox cycle Environ. Sci. Technol, 45 (2011), pp. 9332–9338, 10.1021/es2021704 [DOI] [PubMed] [Google Scholar]
- Bordean D Chromium levels in soils and vegetables from Timis County Romania J. Hortic. Biotechnol, 16 (2012), pp. 106–111 [Google Scholar]
- Brigatti MF, Franchini G, Lugli C, Medici L, Poppi L, Turci E Interaction between aqueous chromium solutions and layer silicates Appl. Geochem, 15 (2000), pp. 1307–1316, 10.1016/S0883-2927(99)00120-1 [DOI] [Google Scholar]
- Eary LE, Rai D Kinetics of chromate reduction by ferrous ions derived from hematite and biotite at 25 °C Am. J. Sci (1989), 10.2475/ajs.289.2.180 [DOI] [Google Scholar]
- Fantoni D, Brozzo G, Canepa M, Cipolli F, Marini L, Ottonello G, Vetuschi M Zuccolini Natural hexavalent chromium in groundwaters interacting with ophiolitic rocks Environ. Geol, 42 (2002), pp. 871–882, 10.1007/s00254-002-0605-0 [DOI] [Google Scholar]
- Fendorf S, Wielinga B Chromium transformations in natural environments: the role of biological and abiological processes in chromium (VI) reduction Introd. Geol, 42 (2000), pp. 37–41 [Google Scholar]
- Friedly JC, Davis J. a, Kent DB Modeling hexavalent chromium seduction in groundwater in field- scale transport and laboratory batch experiments Water Resour. Res, 31 (1995), pp. 2783–2794 [Google Scholar]
- Hashim MA, Mukhopadhyay S, Sahu JN, Sengupta B Remediation technologies for heavy metal contaminated groundwater J. Environ. Manag, 92 (2011), pp. 2355–2388, 10.1016/j.jenvman.2011.06.009 [DOI] [PubMed] [Google Scholar]
- Hasin AAL, Gurman SJ, Murphy LM, Perry A, Smith TJ, Gardiner PHE Remediation of chromium(VI) by a methane-oxidizing bacterium Environ. Sci. Technol, 44 (2010), pp. 400–405, 10.1021/es901723c [DOI] [PubMed] [Google Scholar]
- Gregory C. Herman Hydrogeological framework of bedrock aquifers in the Newark Basin, New Jersey, Geol. Serv. to Public Heal. 18th Annu. Meet. Geol. Soc. New Jersey (2001), pp. 1–32 [Google Scholar]
- Ilton ES, Veblen DR Chromium sorption by phlogopite and biotite in acidic solutions at 25 °C: insights from X-ray photoelectron spectroscopy and electron microscopy Geochim. Cosmochim. Acta, 58 (1994), pp. 2777–2788 [Google Scholar]
- Ilton ES, Veblen DR, Moses CO, Raeburn SP The catalytic effect of sodium and lithium ions on coupled sorption-reduction of chromate at the biotite edge-fluid interface Geochim. Cosmochim. Acta, 61 (1997), pp. 3543–3563, 10.1016/S0016-7037(97)00185-3 [DOI] [Google Scholar]
- Kanmani P, Aravind J, Preston D Remediation of chromium contaminants using bacteria Int. J. Environ. Sci. Technol, 9 (2012), pp. 183–193, 10.1007/s13762-011-0013-7 [DOI] [Google Scholar]
- Kent DB, Puls RW, Ford RG Monitored Natural Attenuation of Inorganic Contaminants in Ground Water: Volume 2 Assessment for Non-radionuclides Including Arsenic, Cadmium, Chromium, Copper, Lead, Nickel, Nitrate, Perchlorate, and Selenium 2, 94 (2007) (doi:EPA/600/R-07/140) [Google Scholar]
- Kumar AR, Riyazuddin P Chromium speciation in a contaminated groundwater: redox processes and temporal variability Environ. Monit. Assess, 176 (2011), pp. 647–662, 10.1007/s10661-010-1610-5 [DOI] [PubMed] [Google Scholar]
- Lin CJ The chemical transformations of chromium in natural waters – a model study Water Air Soil Pollut, 139 (2002), pp. 137–158, 10.1023/A:1015870907389 [DOI] [Google Scholar]
- Lin YT, Huang CP Reduction of chromium(VI) by pyrite in dilute aqueous solutions Sep. Purif. Technol, 63 (2008), pp. 191–199, 10.1016/j.seppur.2008.05.001 [DOI] [Google Scholar]
- Mohan D, Pittman CU Activated carbons and low cost adsorbents for remediation of tri- and hexavalent chromium from water J. Hazard. Mater, 137 (2006), pp. 762–811, 10.1016/j.jhazmat.2006.06.060 [DOI] [PubMed] [Google Scholar]
- Namgung S, Kwon MJ, Qafoku NP, Lee G Cr(OH)3 (s) oxidation induced by surface catalyzed Mn(II) oxidation Environ. Sci. Technol, 48 (2014), pp. 10760–10768, 10.1021/es503018u [DOI] [PubMed] [Google Scholar]
- Palmer CD Effect of Temperature, Ionic Strength, Background Electrolytes, and Fe (III) on the Reduction of Hexavalent Chromium by Soil Humic Substances Vol. 30 (1996), pp. 2470–2477 [Google Scholar]
- Palmer CD, Puls RW EPA Ground Water Issue: Natural Attenuation of Hexavalent Chromium in Groundwater and Soils EPA (1994) [Google Scholar]
- Palmer CD, Wittbrodt PR Processes affecting the remediation of chromium contaminated sites Environ. Health, 92 (1991), pp. 25–40, 10.1289/ehp.919225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker BL, Chapman SW, Cherry JA Plume persistence in fractured sedimentary rock after source zone removal Ground Water, 48 (2010), pp. 799–803 [DOI] [PubMed] [Google Scholar]
- Parker BL, Cherry J. a, Chapman SW Discrete fracture network approach for studying contamination in fractured rock AQUAMundi J. Water Sci, 60 (2012), pp. 101–116, 10.4409/Am-052-12-0046 [DOI] [Google Scholar]
- Pettine M, D’Ottone L, Campanella L, Millero FJ, Passino R The reduction of chromium (VI) by iron (II) in aqueous solutions Geochim. Cosmochim. Acta, 62 (1998), pp. 1509–1519, 10.1016/S0016-7037(98)00086-6 [DOI] [Google Scholar]
- Rai D, Sass BM, Moore D. a Chromium (III) hydrolysis constants and solubility of chromium(III) hydroxide Inorg. Chem, 26 (1987), pp. 345–349, 10.1021/ic00250a002 [DOI] [Google Scholar]
- Rai D, Moore DA, Hess NJ, Rao L, Clark SB Chromium (III) hydroxide solubility in the system: a thermodynamic model J. Solut. Chem, 33 (2004) [Google Scholar]
- Raiswell R, Vu HP, Brinza L, Benning LG The determination of labile Fe in ferrihydrite by ascorbic acid extraction: methodology, dissolution kinetics and loss of solubility with age and de-watering Chem. Geol, 278 (2010), pp. 70–79, 10.1016/j.chemgeo.2010.09.002 [DOI] [Google Scholar]
- Reimann C, Filzmoser P, Garrett RG Background and threshold: critical comparison of methods of determination Sci. Total Environ, 346 (2005), pp. 1–16, 10.1016/j.scitotenv.2004.11.023 [DOI] [PubMed] [Google Scholar]
- Rock ML, James BR, Helz GR Hydrogen peroxide effects on chromium oxidation state and solubility in four diverse, chromium-enriched soils Environ. Sci. Technol, 35 (2001), pp. 4054–4059, 10.1021/es010597y [DOI] [PubMed] [Google Scholar]
- Sass BM, Rai D Solubility of amorphous chromium(III)-iron(III) hydroxide solid solutions Inorg. Chem, 26 (1987), pp. 2228–2232, 10.1021/ic00261a013 [DOI] [Google Scholar]
- Schroeder DC, Lee GF Potential transformations of chromium in natural waters Water Air Soil Pollut, 4 (1975), pp. 355–365 [Google Scholar]
- Singh A, Singh AK, Flatman G Estimation of background levels of contaminants Math. Geol, 26 (1994), pp. 361–388, 10.1007/BF02089229 [DOI] [Google Scholar]
- Trebien DOP, Bortolon L, Tedesco MJ, Bissani CA, Camargo FAO Environmental factors affecting chromium-manganese oxidation-reduction reactions in soil Pedosph. An Int. J, 21 (2011), pp. 84–89, 10.1016/S1002-0160(10)60082-3 [DOI] [Google Scholar]
- Unceta N, Seby F, Malherbe J, Donard OFX Chromium speciation in solid matrices and regulation: a review Anal. Bioanal. Chem, 397 (2010), pp. 1097–1111, 10.1007/s00216-009-3417-1 [DOI] [PubMed] [Google Scholar]
- US EPA Method 3060A: Alkaline Digestion for Hexavalent Chromium (1996) [Google Scholar]
- US EPA Guidance for Comparing Background and Chemical Concentrations in Soil for CERCLA Sites U.S. Environ. Prot. Agency - USEPA (2002), pp. 1–89 [Google Scholar]
- White AF, Peterson ML Reduction of aqueous transition metal species on the surfaces of Fe(II)-containing oxides Geochim. Cosmochim. Acta, 60 (1996), pp. 3799–3814, 10.1016/0016-7037(96)00213-X [DOI] [Google Scholar]
- Zhang Z, Rao L, Rai D, Clark SB Characterization of chromium(III) hydroxide solids and their oxidation by hydrogen peroxide MRS Proc, 824 (2004), pp. 1–6, 10.1557/PROC-824-CC6.5 [DOI] [Google Scholar]
- Zhao J, Al T, Chapman SW, Parker B, Mishkin KR, Cutt D, Wilkin RT Determination of hexavalent chromium concentrations in matrix porewater from a contaminated aquifer in fractured sedimentary bedrock Chem. Geol, 419 (2015), pp. 142–148, 10.1016/j.chemgeo.2015.10.034 [DOI] [Google Scholar]
- Zhao X, Sobecky PA, Zhao L, Crawford P, Li M Chromium(VI) transport and fate in unsaturated zone and aquifer: 3D sandbox results J. Hazard. Mater, 306 (2016), pp. 203–209, 10.1016/j.jhazmat.2015.12.004 [DOI] [PubMed] [Google Scholar]