
Figure 3.
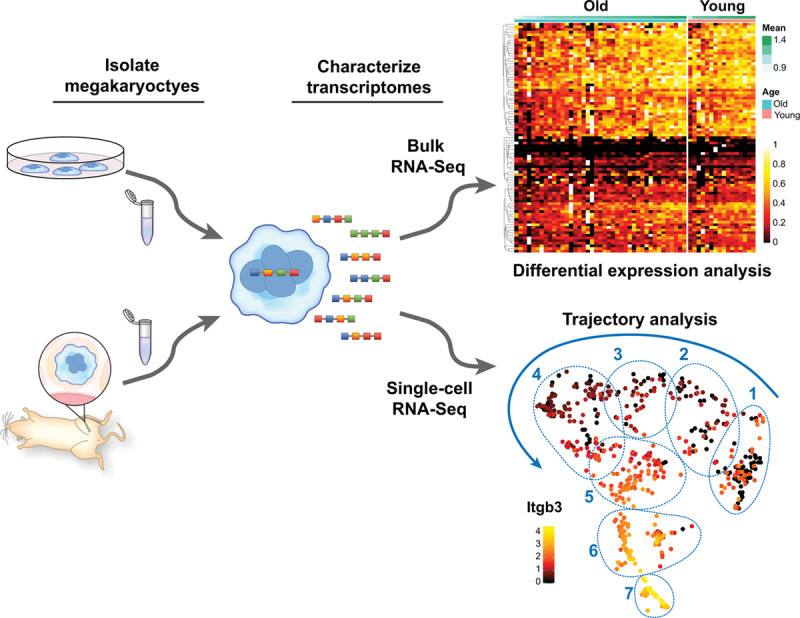
Transcriptomic analyses of megakaryocytes (MKs). Primary or cultured MKs can be subjected to different experimental conditions in vitro or in vivo before isolation for RNA sequencing. RNA-seq can be performed in bulk fashion (top right), allowing comparative analysis of MK populations under specific experimental conditions. Shown is a representative heat map of comparative, bulk, bone marrow MK transcriptomic analyses from older and younger mice. Recently, we optimized a protocol to perform single-cell RNA (scRNA-seq) sequencing of freshly isolated MKs from mice. Similar to bulk RNA-seq, scRNA-seq can be employed to compare large populations of MKs as a whole, or between clearly defined subpopulations of MKs (regions 1–7). However, scRNA-seq also affords the possibility to identify, track, and predict gene pathways involved in megakaryocyte a development and maturation. Shown is a representative trajectory analysis of 7 unique MK clusters at different maturation stages as evidenced by the expression levels of Itgb3 (β3 integrin)